Machine Learning for Drug Science
Guest Editor
Prof. Walter Filgueira de Azevedo Jr. E-Mail
Professor, School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre, Brazil
Research Keywords: drug discovery; protein; machine learning; protein-ligand interaction; computational models
About the Special lssue
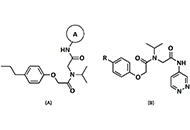
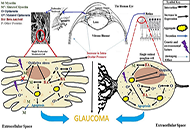
Computational methods to evaluate the binding of ligands against proteins have a beneficial impact on the early stages of drug discovery. Recent progress in this area has converged toward integrating systems approaches and machine learning methods. Within this view, it is possible to model protein-drug interactions as relationships between the protein space [1-10] and the chemical space [11-15]. We consider these sets a unique complex system where computational methodologies could contribute to understanding the structural basis for the specificity of ligands for proteins. Such computational approaches have the potential to create novel scoring functions to predict binding affinity with superior predictive power when compared with standard methodologies such as docking functions. Scoring functions employ atomic coordinates of protein-ligand complexes to predict binding energy [16]. We may obtain these protein-ligand complexes from experimental techniques (e.g., X-ray diffraction crystallography) [17, 18] or computational methodologies such as docking simulations [19].
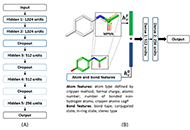
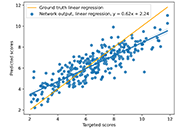
We use the abstraction of a mathematical space composing infinite computational models to predict ligand-binding affinity, named scoring function space [20, 21]. With this approach, we select an element of the protein space (e.g., cyclin-dependent kinase 2 (CDK2)) and define a subset of the chemical space composed of known CDK2 inhibitors [22-25]. Using machine learning regression methods, we scan the scoring function space. We search for a scoring function to predict protein-ligand energetics specifically for one protein system. This methodology generates a computational model targeted to one element of the protein space and shows superior predictive performance compared with classical scoring functions [22-25].
One key challenge in applying the concept of scoring function space to study protein-ligand interactions is the structural information of protein targets. In this field, we observe rapid progress due to the application of deep learning methods to predict the 3D structures of proteins [26-30]. Once the 3D model of a protein is available, we may generate protein-ligand complexes by applying docking simulations.
In this volume, we have papers on recent applications of machine learning methods to develop scoring functions targeted to one protein system and to generate 3D models of protein targets. Also, there are reviews of studies focused on specific protein targets and the impact of computational methods to analyze them.
References
[1] Smith, J.M. Natural selection and the concept of a protein space. Nature, 1970, 225(5232), 563-564.
[2] Hou, J.; Jun, S.R.; Zhang, C.; Kim, S.H. Global mapping of the protein structure space and application in structure-based inference of protein function. Proc. Natl. Acad. Sci. U.S A., 2005, 102(10), 3651-3656.
[3] Bepler, T.; Berger, B. Learning the protein language: Evolution, structure, and function. Cell Syst., 2021, 12(6), 654-669.e3.
[4] Kolodny, R. Searching protein space for ancient sub-domain segments. Curr. Opin. Struct. Biol., 2021, 68, 105-112.
[5] Vila, J.A. About the Protein Space Vastness. Protein J., 2020, 39(5), 472-475.
[6] Hecht, N.; Monteil, C.L.; Perrière, G.; Vishkautzan, M.; Gur, E. Exploring Protein Space: From Hydrolase to Ligase by Substitution. Mol. Biol. Evol., 2021, 38(3), 761-776.
[7] Ogbunugafor, C.B. A Reflection on 50 Years of John Maynard Smith's "Protein Space". Genetics, 2020, 214(4), 749-754.
[8] Narunsky, A.; Ben-Tal, N.; Kolodny, R. Navigating Among Known Structures in Protein Space. Methods Mol. Biol., 2019, 1851, 233-249.
[9] Ogbunugafor, C.B.; Hartl, D.L. A New Take on John Maynard Smith's Concept of Protein Space for Understanding Molecular Evolution. PLoS Comput. Biol., 2016, 12(10), e1005046.
[10] Rackovsky, S. Nonlinearities in protein space limit the utility of informatics in protein biophysics. Proteins, 2015, 83(11), 1923-1928.
[11] Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev., 1996, 16(1), 3-50.
[12] Dobson, C.M. Chemical space and biology. Nature, 2004, 432(7019), 824-828.
[13] Lu C, Liu S, Shi W, Yu J, Zhou Z, Zhang X, Lu X, Cai F, Xia N, Wang Y. Systemic evolutionary chemical space exploration for drug discovery. J. Cheminform., 2022, 14(1), 19.
[14] Grigalunas, M.; Brakmann, S.; Waldmann, H. Chemical Evolution of Natural Product Structure. J. Am. Chem. Soc., 2022, 144(8), 3314-3329.
[15] Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc., 2022, 17(3), 672-697.
[16] Wójcikowski, M.; Siedlecki, P.; Ballester, P.J. Building Machine-Learning Scoring Functions for Structure-Based Prediction of Intermolecular Binding Affinity. Methods Mol. Biol., 2019, 2053, 1-12.
[17] Canduri, F.; de Azevedo, W.F. Protein crystallography in drug discovery. Curr. Drug Targets, 2008, 9(12), 1048-1053.
[18] Veit-Acosta, M.; de Azevedo Junior, W.F. The Impact of Crystallographic Data for the Development of Machine Learning Models to Predict Protein-Ligand Binding Affinity. Curr. Med. Chem., 2021, 28(34), 7006-7022.
[19] Bitencourt-Ferreira, G.; de Azevedo. W.F. Jr. How Docking Programs Work. Methods Mol. Biol., 2019, 2053, 35-50.
[20] Heck, G.S.; Pintro, V.O.; Pereira, R.R., de Ávila, M.B.; Levin, N.M.B, de Azevedo, W.F. Supervised Machine Learning Methods Applied to Predict Ligand-Binding Affinity. Curr. Med. Chem., 2017; 24(23), 2459-2470.
[21] Bitencourt-Ferreira, G.; de Azevedo. W.F. Jr. Exploring the Scoring Function Space. Methods Mol. Biol., 2019, 2053, 275-281.
[22] Shimazaki, T.; Tachikawa, M. Collaborative Approach between Explainable Artificial Intelligence and Simplified Chemical Interactions to Explore Active Ligands for Cyclin-Dependent Kinase 2. ACS Omega, 2022, 7(12), 10372-10381.
[23] Bitencourt-Ferreira, G.; da Silva, A.D.; de Azevedo, W.F. Jr. Application of Machine Learning Techniques to Predict Binding Affinity for Drug Targets. A Study of Cyclin-Dependent Kinase 2. Curr. Med. Chem., 2021, 28(2), 253–265.
[24] Mohammadi, S.; Narimani, Z.; Ashouri, M.; Firouzi, R.; Karimi-Jafari, M.H. Ensemble learning from ensemble docking: revisiting the optimum ensemble size problem. Sci Rep., 2022, 12(1), 410.
[25] Veit-Acosta, M.; de Azevedo Junior, W.F. Computational Prediction of Binding Affinity for CDK2-ligand Complexes. A Protein Target for Cancer Drug Discovery. Curr. Med. Chem. 2022, doi: 10.2174/0929867328666210806105810.
[26] Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; Penedones, H.; Petersen, S.; Simonyan, K.; Crossan, S.; Kohli, P.; Jones, D.T.; Silver, D.; Kavukcuoglu, K.; Hassabis, D. Improved protein structure prediction using potentials from deep learning. Nature, 2020, 577(7792), 706-710.
[27] Singh, A. Deep learning 3D structures. Nat. Methods., 2020, 17(3), 249.
[28] Pakhrin, S.C.; Shrestha, B.; Adhikari, B.; Kc, D.B. Deep Learning-Based Advances in Protein Structure Prediction. Int. J. Mol. Sci., 2021, 22(11), 5553.
[29] Suh, D.; Lee, J.W.; Choi, S.; Lee, Y. Recent Applications of Deep Learning Methods on Evolution- and Contact-Based Protein Structure Prediction. Int. J. Mol. Sci., 2021; 22(11), 6032.
[30] Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; Millán, C.; Park, H.; Adams, C.; Glassman, C.R.; DeGiovanni, A.; Pereira, J.H.; Rodrigues, A.V.; van Dijk, A.A.; Ebrecht, A.C.; Opperman, D.J.; Sagmeister, T.; Buhlheller, C.; Pavkov-Keller, T.; Rathinaswamy, M.K.; Dalwadi, U.; Yip, C.K.; Burke, J.E.; Garcia, K.C.; Grishin, N.V.; Adams, P.D.; Read, R.J.; Baker, D. Accurate prediction of protein structures and interactions using a three-track neural network. Science, 2021, 373(6557), 871-876.
Keywords: Drug discovery, machine learning, protein target, simulation, docking, deep learning
Published Articles