 Original Article
Original Article

Affiliation:
Botany Department, Government Shrimant Madhavrao Scindia PG College, Shivpuri, Madhya Pradesh 473551, India
Email: nm_7582@rediffmail.com
ORCID: https://orcid.org/0000-0001-5208-3539
Explor Drug Sci. 2023;1:276–286 DOI: https://doi.org/10.37349/eds.2023.00018
Received: February 03, 2023 Accepted: May 22, 2023 Published: August 29, 2023
Academic Editor: Walter Filgueira de Azevedo Jr., Pontifical Catholic University of Rio Grande do Sul, Brazil
The article belongs to the special issue Machine Learning for Drug Science
Aim: The significance of β-amyloid protein as a key player in neuro-degenerative disorders viz. Alzheimer’s disease (AD), Parkinson’s disease (PD) has been extensively researched and reported. Glaucoma being another prominent form of neuro-degeneration involving the loss of retinal ganglion cells (RGCs) and human trabecular meshwork (HTM) cells, is also found to be similar to AD in many aspects, but its relation with β-amyloid has not been studied too far up to understanding its causation and pathogenesis where β-amyloid is expected to play important role. This study is an attempt to evaluate the chances of β-amyloid’s role in pathogenesis of retinal neurodegenerative disorder called glaucoma, in silico.
Methods: The study involved determination of feasibility of interaction between β-amyloid and well known glaucoma related proteins namely, myocilin and optineurin. The computational tool called Hex 8.0.0 has been used in this work.
Results: The docking score for β-amyloid and myocilin was found to be –724.1 kJ mol–1 while that for β-amyloid and wild-type optineurin pair was found to be –296.9 kJ mol–1 and that for β-amyloid and mutated optineurin was –607.1 kJ mol–1.
Conclusions: Interaction of β-amyloid with myocilin and optineurin in both forms (wild-type and mutated) is quite energetically favorable. The binding between β-amyloid and mutated optineurin is higher in comparison to that between β-amyloid and wild-type optineurin. Thus, functional significance of β-amyloid in glaucoma pathogenesis is fairly possible which should be studied and proved through in vitro and in vivo studies.
β-Amyloid protein is an amorphous 39–43 amino-acid containing peptide [1] formed from a ubiquitous transmembrane glycoprotein called amyloid precursor protein (APP) [2]. It is known to be involved in neurodegenerative disorders such as Alzheimer’s disease (AD). Neurodegenerative disorders are characterized by loss of neurons in the brain and/or spinal cord [3]. These disorders have in common a gradual loss of neurons and synaptic connections which occur generally in later life [4]. The loss of neurons in different neurodegenerative disorders is asynchronous with symptoms appearing as a result of cumulative loss of neurons which synthesize neurotransmitters required for propagation of signal through the particular brain circuitry associated with a particular disorder [5]. Glaucoma, the permanent vision loss disease, has also been categorized as a neurodegenerative disorder that is related to AD [6]. β-Amyloid accumulations have been recognized in the retina of neurodegenerative disorders such as age-related macular degeneration (AMD), AD, and glaucoma [7]. However, the relatedness has not been clearly evidenced as the mechanism and pathogenesis of this, are not fully understood, especially in relation to AD and β-amyloid.
There are many proteins associated with glaucoma causation with some knowledge about their normal physiological and pathological role. In a recent review done by Wang et al. [8], 22 loci have been presented with many of them being very closely associated to glaucoma genes. A complete understanding of their behavior, role, and mechanism of carrying out these functions is still lacking. For elucidating their mentioned aspects, one approach is to look closely into the possibility of their interaction with other proteins and also to work towards what outcomes do such interactions could bring about at the cellular level, if they do happen actually, in vivo. For this, one has to better understand these glaucoma associated proteins, major two of which are described below:
(a) Myocilin: Myocilin is a member of a protein family called olfactomedins that is, involved in the neuronal development and human diseases [9]. It was first reported about 2 decades ago, but its isolation in full length form for experimental work is still a technical challenge for the researchers [10]. Myocilin is a 504 amino acid protein that contains 2 folding domains: 1) an N-terminal coiled coil and 2) a C-terminal globular domain that is significantly homologous to olfactomedin module found in many different proteins [11, 12]. Myocilin has a unique tripartite architecture that is a Y-shaped parallel dimer of dimers with distinct tetramer and dimer regions, as observed in the molecular studies of its N-terminal coiled coil structure [9]. Full length myocilin is branched with two pairs of C-terminal olfactomedin domains [9].
The myocilin gene is found to get expressed in many ocular as well as non-ocular tissues with the highest levels detected in trabecular meshwork (TM) and sclera [13–17]. It is also expressed in regions namely retinal pigment epithelium (RPE) and choroid [16, 18]. Considering its expression in ocular tissue only, it is found to be expressed intracellularly in retinal ganglion cells (RGCs), RPE, and photoreceptors but not extracellularly in the retina [19, 20]. It is expressed in the TM cells intracellularly and also occurs extracellularly in the aqueous humor that bathes TM cells in vivo [17, 21, 22]. It is a secreted protein, occurring predominantly in dimeric form, found in the human aqueous humor, and mutations in its gene (myocilin) are commonly found in glaucoma patients [10].
(b) Optineurin: Optineurin is a cytosolic protein having 577 amino acid residues [23]. Even upon overexpression, it remains localized in the cytoplasm and does not get translocated to nucleus [23]. It is not a secreted protein that is ubiquitous and is expressed in many tissues like skeletal muscles, heart, liver, brain, and eyes [24, 25]. It is a multifunctional protein involved in many basic cellular activities like maintenance of NFκB pathway and Golgi apparatus, antibacterial and anti-viral signaling, etc. [23]. It has also been implicated in protein trafficking [26]. Park et al. [27] reported in human RPE and TM cells overexpress optineurin (wild-type). Optineurin is also associated with autophagy as it is found to be an autophagy receptor or adaptor [28]. It can induce autophagy and behave as a substrate for autophagic clearance when it is upregulated or mutated [28].
The idea behind the present work is that the localization of β-amyloid and two glaucoma associated proteins namely myocilin and optineurin in the ocular tissues coincides at some sites and hence there is a fair possibility of their interaction (between β-amyloid and myocilin and between β-amyloid and optineurin) at these sites in the ocular tissue. These interactions (and possibly aggregation also) may be contributing to the pathogenesis of glaucoma.
For long fibrous insoluble amyloids and for those whose atomic level structural resolution is difficult, computational methods have been preferred to study and predict their different aspects. Since β-amyloid is also a long fibrous insoluble protein, software based method for studying interaction is applied.
Few studies based on bioinformatics have been done for β-amyloid in reference to AD while the number is quite less for those in reference to glaucoma. Upon searching the terms “bioinformatics, beta amyloid protein, glaucoma” (date of searching: 12th January 2023), only 3 results are returned on the PubMed (NCBI) website indicating that very less bioinformatics-based analyses have been done so far for β-amyloid protein in relation to glaucoma. Thus, to the best of my knowledge, the present study is the first attempt to check for the possibility of interaction of β-amyloid protein with glaucoma associated proteins, myocilin and optineurin, using a freely available software called Hex 8.0.0.
This study is an attempt to evaluate the chances of β-amyloid’s role in pathogenesis of retinal neurodegenerative disorder called glaucoma, in silico. Hex 8.0.0 software is selected for this study as it is a readily available one for protein-protein docking and is validated by Critical Assessment of Predicted Interactions (CAPRI) blind docking experiments [29]. Also, it is a widely used software which runs independently on general-purpose computers with no requirement of expensive servers [30, 31]. It is also capable of running most analyses in a few minutes whereas others have longer turnaround times of a few days [31].
The freely available Hex 8.0.0 software was used for this work [32, 33]. The PDB files of the all the proteins of interest were also downloaded from www.rcsb.org. Hex 8.0.0 software was run for interaction of β-amyloid (PDB number: 2BEG [34]) with two significant glaucoma-associated proteins namely myocilin (PDB number: 4WXQ [35]) and optineurin (PDB number: 5B83 [36]). Interaction of β-amyloid with mutated optineurin (PDB ID: 5EOA [37]) was also done using the same software. Pre-docking, post docking, and post docking-harmonic structures were saved.
Interaction between β-amyloid and myocilin is feasible: Upon carrying out docking using Hex 8.0.0. for β-amyloid and myocilin, the docking score for the top solution was found to be –724.1 kJ mol–1. This indicates that interaction of these two proteins is energetically quite favorable. Since the localization of β-amyloid and myocilin in the ocular tissue coincides with each other, their interaction is quite possible. Considering normal healthy conditions, we know that myocilin protein gets secreted out of the cell and therefore due to its lowered concentration inside. Its interaction with β-amyloid is not expected to cause many problems, as the number of resultant aggregated products would be low due to low availability of myocilin molecules. If, however, there is mutation in myocilin which prevents its secretion or due to its overexpression induced by oxidative stress, there must be a constantly maintained high concentration of myocilin. Also, oxidative stress promotes β-amyloid production. Under these conditions, the possibility of interaction and aggregation of these two proteins is fairly high. A docking score of –724.1 kJ mol–1 further supports energetic feasibility of this interaction that may be leading to stable insoluble complex formation in vivo [intracellularly in retinal cells and both intra- and extracellularly in human trabecular meshwork (HTM) tissue] (Table 1, Figure 1, Table S1).
Top solutions for interactions between protein pairs on the basis of data generated using Hex 8.0.0.
| S. No. | Protein pair | Etotal (kJ mol–1) |
|---|---|---|
| 1 | Myocilin and β-amyloid | –724.1 |
| 2 | Optineurin and β-amyloid | –296.9 |
| 3 | Mutated optineurin and β-amyloid | –607.1 |
Etotal: total energy; S. No.: serial number
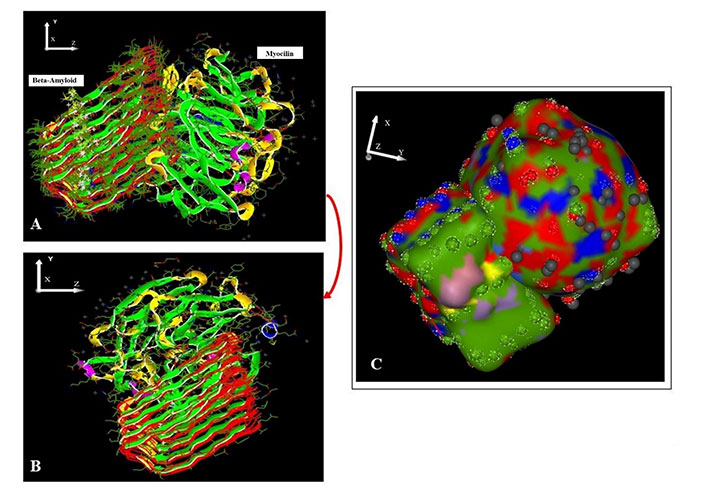
β-Amyloid (receptor) docked with myocilin (ligand) using Hex 8.0.0. A. Pre-docking; B. post-docking; C. harmonic surface figure of β-amyloid docked to myocilin
Interaction property of optineurin in mutated form is higher than that of its wildtype: the docking scores of both interactions (a) wild type optineurin with β-amyloid and (b) mutated optineurin (E50K) with β-amyloid are fairly high (Table 1, Figures 2 and 3). Further, the docking score/Etotal of mutated optineurin (E50K) with β-amyloid in comparison to that of wild-type optineurin interacting with β-amyloid is nearly 2 times higher indicating that mutated optineurin binds even more strongly with β-amyloid than its wild-type counterpart (Table 1, Figures 2 and 3). The docking scores of wildtype and mutant optineurin interacting with β-amyloid were found to be –296.9 kJ mol–1 and –607.1 kJ mol–1 while mutated optineurin interacting with itself gave a docking score of –643.9 kJ mol–1. These values indicate that the possibility of binding of mutated optineurin with β-amyloid is comparable to that of mutated optineurin interacting with itself.
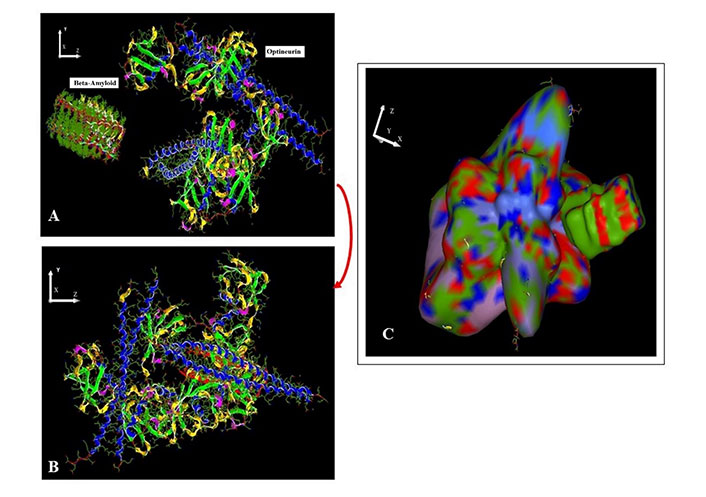
β-Amyloid (receptor) docked with optineurin (ligand) using Hex 8.0.0. A. Pre-docking; B. post-docking; C. harmonic surface figure of β-amyloid docked to optineurin
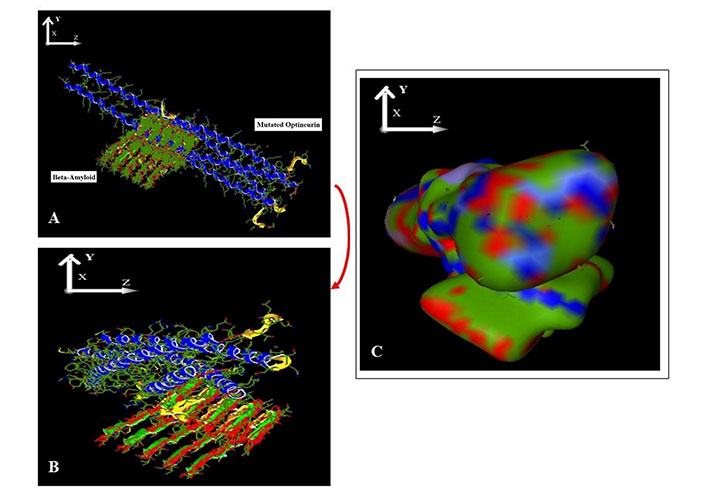
β-Amyloid (receptor) docked with mutated optineurin (ligand) using Hex 8.0.0. A. Pre-docking; B. post-docking; C. harmonic surface figure of β-amyloid docked to mutated optineurin
Myocilin and optineurin are both well-established glaucoma-associated proteins that contribute to its causation in their own respective ways. However, the complete understanding of their role in this pathological process is not there till now. Their aggregative property and ability to bind with other proteins are important features that may be responsible for their neurodegenerative effect. Even after knowing that myocilin is involved in glaucoma in mutated form, the normal physiological function of wild-type myocilin is still poorly understood [38]. Identification of proteins that interact with myocilin has been suggested to be a useful approach to determine its functions [39, 40]. It is reported that its absence does not cause the disease. Gain of function mutations in its olfactomedin domain lead to its toxic intracellular accumulation that hastens onset of open-angle glaucoma [41]. It is reported in vitro that myocilin gets overexpressed upon oxidative insult in the HTM cells [42]. It is well known that mutated myocilin does not get secreted and accumulates in the endoplasmic reticulum (ER) of the TM cells leading to ER stress, increased sensitivity of the TM towards oxidative stress [43]. Resultantly there is deterioration of TM functionality [44] due to apoptotic loss of TM cells. Since mutant myocilin aggregation is associated with inherited form of open-angle glaucoma, the comprehension of mutant myocilin aggregation is very important for glaucomatous pathogenesis [45]. This emphasizes the importance of unraveling the interaction and aggregation of myocilin with other proteins (like β-amyloid) to understand how these proteins are involved in glaucoma pathogenesis. It should be noted, however, that no previous studies in this regard have been done (to the best of author’s knowledge).
Considering the docking score results (–724.1 kJ mol–1) obtained for β-amyloid-myocilin pair, it is quite evident that this interaction is very much feasible (Table 1, Figure 1) and it may be a significant factor, not alone though, contributing to pathogenesis of glaucoma. Thus, it can be said on the basis of results obtained and previous reported studies, that if the cell does not get rid of myocilin through secretion due to its mutation or due to its overexpression (induced by factors like oxidative stress), it starts getting accumulating in the cell and once this accumulation begins, it interacts with other proteins (such as β-amyloid) and forms large aggregates which further hinder the normal physiological function of the cell ultimately leading to its death by apoptosis.
There are a few studies hinting towards how optineurin is involved in glaucoma. Mutations in optineurin are associated with neurodegenerative disorders like amyotropic lateral sclerosis and primary open-angle glaucoma (POAG) [46]. Liu and Tian [47] have hypothesized that optineurin may be a common link between normal tension glaucoma (NTG) and AD. A report suggested that the E50K mutant of optineurin has ability to induce death selectively in RGCs sparing other cells [48], while the wild-type optineurin is cytoprotective [49]. In human TM cells, overexpression of optineurin is reported to induce upregulation of endogenous myocilin and to result in prolonged turn-over rate of myocilin mRNA with no effect on the promoter activity of myocilin gene [50].
Optineurin interacts with many proteins to carry out its multiple functions such as myosin VI, transferrin receptor, Ras-related protein-8 (Rab8), TANK binding kinase 1 (TBK1), huntingtin, etc. [25]. Further, wild-type optineurin is reported to interact with itself to give rise to homo-hexamers and also with its binding partners to form super-molecular complexes [51]. Gao et al. [52] have also suggested that endogenous or ectopically expressing optineurin interacts with itself to form high molecular weight protein complexes in cells. Chalasani et al. [53] speculated that possibly it is the loss of interaction of mutated (E50K) optineurin with Rab8 or a gain of interaction with some other protein that is responsible for its cell death-inducing activity towards RGC-5 cells. It has been reported that in neurons obtained from induced pluripotent stem cells (iPSCs) from NTG patients carrying E50K mutation and in human embryonic kidney 293 (HEK293) cells overexpressing E50K mutation, there is accumulation of insoluble optineurin in the ER [46]. The group further suggested that E50K mutant strongly interacted with TBK1. Due to this, proper oligomerization and solubility of optineurin were prohibited which negatively affect the intracellular transition of optineurin and evokes POAG [46]. All these studies indicate that optineurin, whether wild-type or mutant, has tendency to form large complexes either with themselves or by combining with other protein molecules and accumulation of insoluble optineurin is one of the underlying processes of glaucomatous pathogenesis or neurodegeneration in a more generalized way. Thus, there is a possibility that the mutated optineurin or overexpressing optineurin (wild-type) strongly binds to β-amyloid in the RGCs and TM tissue and gets deposited in these cells leading to their death. It is fairly possible that optineurin forms large, stable, and insoluble aggregates with itself and with many other protein molecules present in its vicinity including β-amyloid; interfere with normal physiology of the RGCs and HTM cells and induce cytotoxicity and apoptosis of these cells.
It can be concluded that there is a significant possibility that β-amyloid interacts with different proteins to form large insoluble complexes that affect the normal physiology of different ocular tissues and contributes to glaucoma pathogenesis (Figure 4). Being a multifactorial disease, glaucoma is not caused only by β-amyloid interactions, but it is quite apparent that these interactions might be contributing substantially and their role needs to be further elucidated using other approaches.
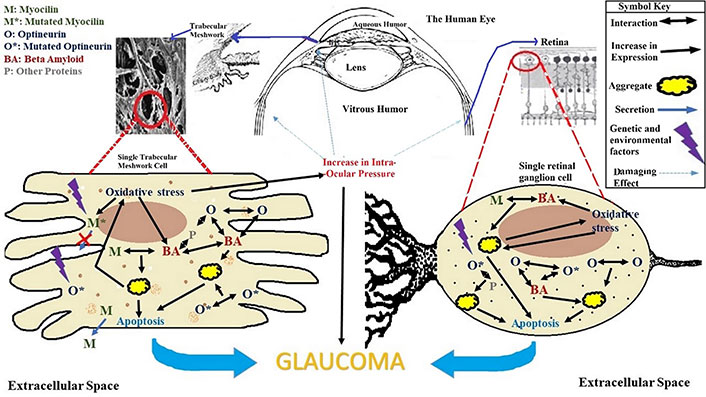
An overview of glaucoma progression and factors contributing to it with reference to β-amyloid, myocilin and optineurin. The pathogenesis of glaucoma is a slow process occurring over time due to increase in oxidative stress. Genetic and environmental factors cause mutations that alter the normal physiology and function of proteins. Formation of large insoluble protein aggregates, inability of myocilin to get secreted, interactions of molecules among themselves and with other molecules, increase in intra-ocular pressure, etc. cause cellular stress and apoptosis ultimately leading to glaucoma. The red cross means that mutated myocilin denoted by M* does not get secreted out of the cell unlike normal wildtype myocilin denoted by M
This will help in understanding glaucoma pathogenesis in a better way and may pave way to establishment of it being mechanistically related to other forms of neurodegenerative disorders such as AD and Parkinson’s disease (PD). Further, it will also help in finding new therapeutic avenues for glaucoma which are for now used for other well established neurodegenerative diseases.
The present work is an in silico study and is done using a single software tool. Its results need further proof and validation using other in silico tools and in vitro, in vivo studies. The study does not claim to be confirmatory and is only suggesting a possible way of involvement of myocilin, optineurin and β-amyloid in glaucomatous pathogenesis, which needs to be further confirmed using other studies and experimental methods.
AD: Alzheimer’s disease
ER: endoplasmic reticulum
HTM: human trabecular meshwork
RGCs: retinal ganglion cells
RPE: retinal pigment epithelium
TM: trabecular meshwork
The supplementary materials for this article is available at: https://www.explorationpub.com/uploads/Article/file/100818_sup_1.pdf.
The author acknowledges Prof. Mahendra Kumar, Principal, Government Shrimant Madhavrao Scindia PG College, Shivpuri, Madhya Pradesh 473551, India, for providing necessary facilities to conduct this work.
NM: Conceptualization, Data curation, Writing—original draft, Writing—review & editing.
The author declares that she has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Walter F. de Azevedo Jr.
Kevin Crampon ... Luiz Angelo Steffenel
Minyi Su, Enric Herrero
