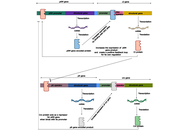





Today, researchers have already made great progress in finding drug-based clinical solutions against microbial infections. It has become an essential part of a healthy human lifestyle. High antibiotic consumption has accelerated antibiotic resistance in microbial species (multi-drug-resistant microbial strains, like Staphylococcus aureus and Mycobacterium tuberculosis, have already been reported). Loss of microflora is also associated with the heavy and unnecessary use of drugs. This review presents bacteriophage as an alternative to antibiotics. The supporting bacteriophage characteristics include bacteriophage lytic mode of replication, specificity towards its host, bacteriophage mass production, and bacteriophage genetic modification (BRED and CRISPR) to make it capable of degrading microbial biofilm. The author has also tried to inculcate previous work that has already been done with bacteriophages for some clinical therapies. Potential administration routes (oral, intravenous, and intraoperative) used in clinical therapies are discussed.
Today, researchers have already made great progress in finding drug-based clinical solutions against microbial infections. It has become an essential part of a healthy human lifestyle. High antibiotic consumption has accelerated antibiotic resistance in microbial species (multi-drug-resistant microbial strains, like Staphylococcus aureus and Mycobacterium tuberculosis, have already been reported). Loss of microflora is also associated with the heavy and unnecessary use of drugs. This review presents bacteriophage as an alternative to antibiotics. The supporting bacteriophage characteristics include bacteriophage lytic mode of replication, specificity towards its host, bacteriophage mass production, and bacteriophage genetic modification (BRED and CRISPR) to make it capable of degrading microbial biofilm. The author has also tried to inculcate previous work that has already been done with bacteriophages for some clinical therapies. Potential administration routes (oral, intravenous, and intraoperative) used in clinical therapies are discussed.
DOI: https://doi.org/10.37349/eds.2026.1008166
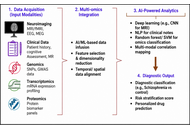
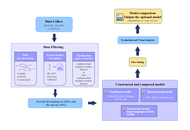
Schizophrenia affects approximately 24 million people worldwide, representing 0.3–0.7% of the global population, and remains a leading cause of years lived with disability. The disorder contributes to over 13 million disability-adjusted life years, underscoring its substantial global health and socioeconomic burden. This review critically examines the convergence of artificial intelligence (AI) and advanced oral drug delivery systems as an emerging strategy in schizophrenia management. Conventional diagnostic frameworks, reliant on subjective symptom assessment, often result in delayed or inaccurate diagnosis, while standard oral antipsychotics are limited by poor bioavailability, extensive first-pass metabolism, and inadequate brain targeting. Recent advances in AI, including natural language processing (NLP), neuroimaging analytics, electrophysiological modeling, and multi-omics integration, support diagnostic classification, risk prediction, symptom monitoring, and patient stratification. Simultaneously, nanotechnology-driven oral delivery platforms such as lipid nanoparticles, dendrimers, and proliposomes enhance pharmacokinetics, central nervous system targeting, and therapeutic adherence. The integration of AI with pharmacogenomics, wearable monitoring, and digital twin models further facilitates real-time dose optimization and personalized therapy. Despite promising preclinical and clinical outcomes, challenges related to data privacy, algorithmic bias, scalability, and regulatory translation persist. This review highlights a shift toward precision psychiatry, where AI-enabled diagnostics and smart oral therapeutics may support predictive, personalized, and adaptive care in schizophrenia.
Schizophrenia affects approximately 24 million people worldwide, representing 0.3–0.7% of the global population, and remains a leading cause of years lived with disability. The disorder contributes to over 13 million disability-adjusted life years, underscoring its substantial global health and socioeconomic burden. This review critically examines the convergence of artificial intelligence (AI) and advanced oral drug delivery systems as an emerging strategy in schizophrenia management. Conventional diagnostic frameworks, reliant on subjective symptom assessment, often result in delayed or inaccurate diagnosis, while standard oral antipsychotics are limited by poor bioavailability, extensive first-pass metabolism, and inadequate brain targeting. Recent advances in AI, including natural language processing (NLP), neuroimaging analytics, electrophysiological modeling, and multi-omics integration, support diagnostic classification, risk prediction, symptom monitoring, and patient stratification. Simultaneously, nanotechnology-driven oral delivery platforms such as lipid nanoparticles, dendrimers, and proliposomes enhance pharmacokinetics, central nervous system targeting, and therapeutic adherence. The integration of AI with pharmacogenomics, wearable monitoring, and digital twin models further facilitates real-time dose optimization and personalized therapy. Despite promising preclinical and clinical outcomes, challenges related to data privacy, algorithmic bias, scalability, and regulatory translation persist. This review highlights a shift toward precision psychiatry, where AI-enabled diagnostics and smart oral therapeutics may support predictive, personalized, and adaptive care in schizophrenia.
DOI: https://doi.org/10.37349/eds.2026.1008165
This article belongs to the special issue Nanoformulations for Non-Intravenous Drug Delivery
Brazil harbors remarkable biological and cultural diversity, reflected in a rich body of traditional knowledge regarding the medicinal use of plants. This study synthesized ethnobotanical evidence on plants traditionally used for skin and wound healing in Brazil and examined their convergence with available antibacterial data. An integrative literature review identified twenty ethnobotanical studies, mainly involving rural populations and local residents, reporting 51 plant species traditionally used for skin and wound healing across 22 genera, predominantly native and mainly documented in the Northeastern and Northern regions. The most frequently cited species included Aloe vera (L.) Burm.f. and Anacardium occidentale L., followed by Stryphnodendron adstringens (Mart.) Coville. Fabaceae and Anacardiaceae concentrated the highest number of species with confirmed antibacterial activity, followed by Piperaceae and Euphorbiaceae, which also showed a high proportional representation of active species. A meaningful convergence between ethnobotanical use and experimental antibacterial evidence was observed for more than half of the plants, frequently against Staphylococcus aureus, a key pathogen in wound infections. Antibacterial data were predominantly derived from in vitro assays using non-standardized extracts, and only a limited number of studies reported possible mechanisms of action, such as membrane disruption and biofilm inhibition. Furthermore, few investigations evaluated antibacterial activity in infected wound models or quantified bacterial load reduction in vivo. Future studies should prioritize chemically standardized extracts, testing against resistant clinical strains and mature biofilm models, and validation of safety and therapeutic efficacy in clinical investigations. These findings reveal a gap between traditional use and clinically validated applications, underscoring the urgent need for standardized research approaches and reinforcing Brazil’s potential as a strategic reservoir of bioactive plant resources for primary health care. Addressing these limitations is essential to strengthening the translational basis for the rational use of medicinal plants in primary health care and public health contexts.
Brazil harbors remarkable biological and cultural diversity, reflected in a rich body of traditional knowledge regarding the medicinal use of plants. This study synthesized ethnobotanical evidence on plants traditionally used for skin and wound healing in Brazil and examined their convergence with available antibacterial data. An integrative literature review identified twenty ethnobotanical studies, mainly involving rural populations and local residents, reporting 51 plant species traditionally used for skin and wound healing across 22 genera, predominantly native and mainly documented in the Northeastern and Northern regions. The most frequently cited species included Aloe vera (L.) Burm.f. and Anacardium occidentale L., followed by Stryphnodendron adstringens (Mart.) Coville. Fabaceae and Anacardiaceae concentrated the highest number of species with confirmed antibacterial activity, followed by Piperaceae and Euphorbiaceae, which also showed a high proportional representation of active species. A meaningful convergence between ethnobotanical use and experimental antibacterial evidence was observed for more than half of the plants, frequently against Staphylococcus aureus, a key pathogen in wound infections. Antibacterial data were predominantly derived from in vitro assays using non-standardized extracts, and only a limited number of studies reported possible mechanisms of action, such as membrane disruption and biofilm inhibition. Furthermore, few investigations evaluated antibacterial activity in infected wound models or quantified bacterial load reduction in vivo. Future studies should prioritize chemically standardized extracts, testing against resistant clinical strains and mature biofilm models, and validation of safety and therapeutic efficacy in clinical investigations. These findings reveal a gap between traditional use and clinically validated applications, underscoring the urgent need for standardized research approaches and reinforcing Brazil’s potential as a strategic reservoir of bioactive plant resources for primary health care. Addressing these limitations is essential to strengthening the translational basis for the rational use of medicinal plants in primary health care and public health contexts.
DOI: https://doi.org/10.37349/eds.2026.1008164
This article belongs to the special issue Discovery and development of new antibacterial compounds
Aim:
This study investigated the anti-inflammatory effect of the extract and fractions of Eucalyptus camaldulensis and also profiled the secondary metabolites of its most active fraction.
Methods:
The leaves of E. camaldulensis were collected, authenticated and extracted with methanol. The extract (100, 200, and 400 mg/kg), normal saline (negative control), and aspirin (positive control) were administered orally to egg-induced paw oedema rats of five groups of five rats each. The extract was partitioned, and each of the solvent fractions was assayed for its anti-inflammatory activity. The results obtained were subjected to one-way analysis of variance (ANOVA) followed by Bonferroni post hoc tests, and p < 0.05 was considered significant. Also, the most active fraction was subjected to gas chromatography-mass spectrometry (GC-MS) analysis.
Results:
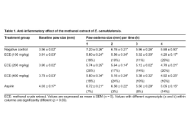
The extract at 100 mg/kg demonstrated the best anti-inflammatory effect at 29%, while the n-hexane (N-HEX) fraction gave the highest inflammatory inhibition at 43%. α-Phellandrene, o-cymene, n-hexadecanoic acid, and beta-sitosterol were identified as the most abundant compounds in the N-HEX fraction.
Conclusions:
The study concluded that the methanol extract of E. camaldulensis possesses good anti-inflammatory properties, and its non-polar fraction was responsible for the observed activity. Bioassay-guided purification of anti-inflammatory constituents of the N-HEX fraction is recommended for future studies.
Aim:
This study investigated the anti-inflammatory effect of the extract and fractions of Eucalyptus camaldulensis and also profiled the secondary metabolites of its most active fraction.
Methods:
The leaves of E. camaldulensis were collected, authenticated and extracted with methanol. The extract (100, 200, and 400 mg/kg), normal saline (negative control), and aspirin (positive control) were administered orally to egg-induced paw oedema rats of five groups of five rats each. The extract was partitioned, and each of the solvent fractions was assayed for its anti-inflammatory activity. The results obtained were subjected to one-way analysis of variance (ANOVA) followed by Bonferroni post hoc tests, and p < 0.05 was considered significant. Also, the most active fraction was subjected to gas chromatography-mass spectrometry (GC-MS) analysis.
Results:
The extract at 100 mg/kg demonstrated the best anti-inflammatory effect at 29%, while the n-hexane (N-HEX) fraction gave the highest inflammatory inhibition at 43%. α-Phellandrene, o-cymene, n-hexadecanoic acid, and beta-sitosterol were identified as the most abundant compounds in the N-HEX fraction.
Conclusions:
The study concluded that the methanol extract of E. camaldulensis possesses good anti-inflammatory properties, and its non-polar fraction was responsible for the observed activity. Bioassay-guided purification of anti-inflammatory constituents of the N-HEX fraction is recommended for future studies.
DOI: https://doi.org/10.37349/eds.2026.1008163
Advanced Therapy Medicinal Products (ATMPs) represent a transformative class of innovative therapies based on genes, cells, or engineered tissues that aim to modify, repair, or replace biological functions at a fundamental level. This review provides a foundational overview of ATMPs, addressing their scientific basis, regulatory classification, clinical translation, and key challenges for future development. The article outlines the four principal categories of ATMPs: gene therapy medicinal products, somatic cell therapy medicinal products, tissue-engineered products, and combined ATMPs, and discusses their distinct mechanisms of action and therapeutic applications. Recent clinical successes, including chimeric antigen receptor T-cell therapies, gene replacement therapies for inherited disorders, and tissue-engineered constructs for regenerative medicine, demonstrate the paradigm shift from symptomatic management towards disease modification or potential cure. However, the clinical implementation of ATMPs presents substantial challenges related to safety monitoring, long-term efficacy, complex manufacturing processes, regulatory evaluation, high treatment costs, and equitable patient access. Ethical considerations, including informed consent, long-term follow-up obligations, and global disparities in availability, further complicate their integration into routine clinical practice. Regulatory agencies have introduced adaptive pathways and accelerated approval mechanisms to facilitate timely patient access while maintaining rigorous standards of quality, safety, and efficacy. Continued technological innovation, coupled with sustainable healthcare policies and international collaboration, will be essential to realise the full therapeutic potential of ATMPs. As evidence accumulates from clinical trials and real-world use, ATMPs are expected to play an increasingly prominent role in modern medicine and the future of personalised healthcare.
Advanced Therapy Medicinal Products (ATMPs) represent a transformative class of innovative therapies based on genes, cells, or engineered tissues that aim to modify, repair, or replace biological functions at a fundamental level. This review provides a foundational overview of ATMPs, addressing their scientific basis, regulatory classification, clinical translation, and key challenges for future development. The article outlines the four principal categories of ATMPs: gene therapy medicinal products, somatic cell therapy medicinal products, tissue-engineered products, and combined ATMPs, and discusses their distinct mechanisms of action and therapeutic applications. Recent clinical successes, including chimeric antigen receptor T-cell therapies, gene replacement therapies for inherited disorders, and tissue-engineered constructs for regenerative medicine, demonstrate the paradigm shift from symptomatic management towards disease modification or potential cure. However, the clinical implementation of ATMPs presents substantial challenges related to safety monitoring, long-term efficacy, complex manufacturing processes, regulatory evaluation, high treatment costs, and equitable patient access. Ethical considerations, including informed consent, long-term follow-up obligations, and global disparities in availability, further complicate their integration into routine clinical practice. Regulatory agencies have introduced adaptive pathways and accelerated approval mechanisms to facilitate timely patient access while maintaining rigorous standards of quality, safety, and efficacy. Continued technological innovation, coupled with sustainable healthcare policies and international collaboration, will be essential to realise the full therapeutic potential of ATMPs. As evidence accumulates from clinical trials and real-world use, ATMPs are expected to play an increasingly prominent role in modern medicine and the future of personalised healthcare.
DOI: https://doi.org/10.37349/eds.2026.1008162
The global rise of antimicrobial resistance (AMR) has emerged as one of the most pressing threats to public health. This crisis calls for the urgent development of alternative therapeutic agents against antibiotic-resistant pathogens. Antimicrobial peptides (AMPs) are widely present in nature, with a broad range of effects and a low risk of causing drug resistance. Therefore, they are an ideal choice for the development of the next generation of antimicrobial drugs. To overcome the inefficiencies of traditional AMP discovery, artificial intelligence (AI) and machine learning (ML) technologies have been increasingly used to predict and design AMPs. Multiple AMP databases were used to train ML models for predicting the activity of AMPs or generating AMP sequences. This review briefly provides a comprehensive overview of AMP databases and computational tools, highlighting their capabilities and challenges. Future work should integrate larger datasets and experimental validation to accelerate clinical translation.
The global rise of antimicrobial resistance (AMR) has emerged as one of the most pressing threats to public health. This crisis calls for the urgent development of alternative therapeutic agents against antibiotic-resistant pathogens. Antimicrobial peptides (AMPs) are widely present in nature, with a broad range of effects and a low risk of causing drug resistance. Therefore, they are an ideal choice for the development of the next generation of antimicrobial drugs. To overcome the inefficiencies of traditional AMP discovery, artificial intelligence (AI) and machine learning (ML) technologies have been increasingly used to predict and design AMPs. Multiple AMP databases were used to train ML models for predicting the activity of AMPs or generating AMP sequences. This review briefly provides a comprehensive overview of AMP databases and computational tools, highlighting their capabilities and challenges. Future work should integrate larger datasets and experimental validation to accelerate clinical translation.
DOI: https://doi.org/10.37349/eds.2026.1008161
This article belongs to the special issue Discovery and development of new antibacterial compounds
Aim:
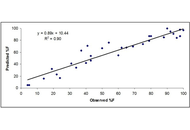
Two empirical methods were used to predict the absolute bioavailability (F) of medicines in adults and children following oral administration in the absence of intravenous (IV) dosing. This study systematically evaluates the predictive performance of Equation 1 to predict F in adults and children.
Methods:
Equation 3 [F = Q/(Q + CLoral)] was used for the prediction of F in adults and children. In Equation 3, clearance is the observed oral clearance following oral administration of a medicine and Q is either liver blood or plasma flow rate. The predictive performance of Equation 3 was evaluated in adults and children for three categories of medicines; medicines which are mainly metabolized in the liver, medicines which are metabolized both in the liver and the gut, and medicines which are mainly renally excreted. From the literature, oral clearance and F values for adults and children were obtained. The predictive performance of these two methods (blood or plasma flow rate) was assessed by comparing the predicted F of the medicines used in this study with the observed F (obtained from clinical studies).
Results:
More than 90% predicted F values were within 0.5–2-fold prediction error in adults and children by both methods for all three categories of medicines. Plasma flow rate provided slightly better results than the blood flow rate.
Conclusions:
The proposed methods indicate that the estimation of F of medicines in adults and children is possible with reasonable accuracy (within 0.5–2-fold prediction error). The method is useful to estimate F, especially in children, because it is not ethical to administer medicines by both IV and oral routes to children just for the sake of estimating F.
Aim:
Two empirical methods were used to predict the absolute bioavailability (F) of medicines in adults and children following oral administration in the absence of intravenous (IV) dosing. This study systematically evaluates the predictive performance of Equation 1 to predict F in adults and children.
Methods:
Equation 3 [F = Q/(Q + CLoral)] was used for the prediction of F in adults and children. In Equation 3, clearance is the observed oral clearance following oral administration of a medicine and Q is either liver blood or plasma flow rate. The predictive performance of Equation 3 was evaluated in adults and children for three categories of medicines; medicines which are mainly metabolized in the liver, medicines which are metabolized both in the liver and the gut, and medicines which are mainly renally excreted. From the literature, oral clearance and F values for adults and children were obtained. The predictive performance of these two methods (blood or plasma flow rate) was assessed by comparing the predicted F of the medicines used in this study with the observed F (obtained from clinical studies).
Results:
More than 90% predicted F values were within 0.5–2-fold prediction error in adults and children by both methods for all three categories of medicines. Plasma flow rate provided slightly better results than the blood flow rate.
Conclusions:
The proposed methods indicate that the estimation of F of medicines in adults and children is possible with reasonable accuracy (within 0.5–2-fold prediction error). The method is useful to estimate F, especially in children, because it is not ethical to administer medicines by both IV and oral routes to children just for the sake of estimating F.
DOI: https://doi.org/10.37349/eds.2026.1008160
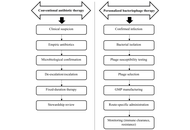
Antimicrobial resistance (AMR) poses a growing global health threat, progressively undermining the clinical effectiveness of conventional antibiotic therapies. Despite their proven efficacy and standardized clinical frameworks, antibiotics exert strong selective pressures that accelerate resistance, disrupt host microbiota, and limit treatment options for chronic, biofilm-associated, and multidrug-resistant infections. Bacteriophage therapy has re-emerged as a potential adjunct or alternative approach, offering pathogen-specific antibacterial activity, preservation of commensal microbiota, and the capacity for co-evolution with bacterial hosts. This focused review critically compares antibiotics and bacteriophage therapy across mechanistic foundations, preclinical and clinical evidence, translational readiness, and real-world implementation challenges. While preclinical models consistently demonstrate robust antibacterial activity of bacteriophages, clinical evidence remains heterogeneous, with few randomized controlled studies available. Key system-level barriers, including regulatory inconsistency, manufacturing complexity, and lack of standardization, currently limit widespread clinical integration. Rather than positioning bacteriophages as replacements for antibiotics, this review emphasizes their potential role as complementary agents, particularly through bacteriophage-antibiotic synergy, to enhance treatment efficacy and mitigate resistance. Addressing methodological gaps, standardizing clinical trial designs, and developing integrated stewardship models will be critical to defining the future role of bacteriophage therapy in modern infectious disease management.
Antimicrobial resistance (AMR) poses a growing global health threat, progressively undermining the clinical effectiveness of conventional antibiotic therapies. Despite their proven efficacy and standardized clinical frameworks, antibiotics exert strong selective pressures that accelerate resistance, disrupt host microbiota, and limit treatment options for chronic, biofilm-associated, and multidrug-resistant infections. Bacteriophage therapy has re-emerged as a potential adjunct or alternative approach, offering pathogen-specific antibacterial activity, preservation of commensal microbiota, and the capacity for co-evolution with bacterial hosts. This focused review critically compares antibiotics and bacteriophage therapy across mechanistic foundations, preclinical and clinical evidence, translational readiness, and real-world implementation challenges. While preclinical models consistently demonstrate robust antibacterial activity of bacteriophages, clinical evidence remains heterogeneous, with few randomized controlled studies available. Key system-level barriers, including regulatory inconsistency, manufacturing complexity, and lack of standardization, currently limit widespread clinical integration. Rather than positioning bacteriophages as replacements for antibiotics, this review emphasizes their potential role as complementary agents, particularly through bacteriophage-antibiotic synergy, to enhance treatment efficacy and mitigate resistance. Addressing methodological gaps, standardizing clinical trial designs, and developing integrated stewardship models will be critical to defining the future role of bacteriophage therapy in modern infectious disease management.
DOI: https://doi.org/10.37349/eds.2026.1008159
Aim:
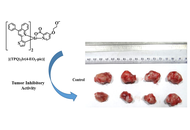
The iridium(III) [Ir(III)] complexes exhibit anticancer properties, and along with their photoluminescence properties, they can be employed as diagnostic agents. Hence, we have tried to evaluate both properties. For this, the complexes of Ir(III) with pyridyl-based heterocyclic ligands, viz., [(TPQ)3Ir] (
Methods:
These complexes were authenticated by NMR (1H and 13C{1H}) spectroscopy and high-resolution mass spectrometry (HRMS). Their photophysical properties were evaluated by UV-Vis spectroscopy and photoluminescence studies. Among them, [(TPQ)3Ir] (
Results:
Among these Ir(III) complexes (
Conclusions:
The [(TPQ)2Ir(4-EO2-pic)] (
Aim:
The iridium(III) [Ir(III)] complexes exhibit anticancer properties, and along with their photoluminescence properties, they can be employed as diagnostic agents. Hence, we have tried to evaluate both properties. For this, the complexes of Ir(III) with pyridyl-based heterocyclic ligands, viz., [(TPQ)3Ir] (
Methods:
These complexes were authenticated by NMR (1H and 13C{1H}) spectroscopy and high-resolution mass spectrometry (HRMS). Their photophysical properties were evaluated by UV-Vis spectroscopy and photoluminescence studies. Among them, [(TPQ)3Ir] (
Results:
Among these Ir(III) complexes (
Conclusions:
The [(TPQ)2Ir(4-EO2-pic)] (
DOI: https://doi.org/10.37349/eds.2026.1008158
Aim:
Chitosan (CHS)-based nanoparticulate systems have gained much interest due to their high drug loading capacity and the simplicity of their fabrication. The physical properties of two types of curcumin-loaded CHS nanoparticles (CHS-NPs) were determined and compared. A new in-vitro release method was developed based on mathematical modeling in which the drug is first released from the NP into the surrounding medium and subsequently diffuses through the membrane.
Methods:
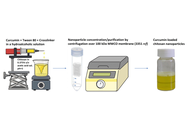
Curcumin-loaded CHS-NPs were fabricated by ionotropic gelation using sodium tripolyphosphate (TPP) and sodium hexametaphosphate (SHMP) crosslinking, and characterized by NP tracking analysis, loading capacity, zeta potential, Fourier transform infrared spectroscopy (FTIR), and in-vitro release rates.
Results:
The data showed that compared to SHMP crosslinked CHS-NPs, TPP crosslinking demonstrated a decrease in entrapment efficiency at a relatively high concentration of the agent, probably by narrowing the space between the polymeric chains. As indicated by zeta potential measurements, TPP crosslinking at all levels was more uniformly distributed inside the NPs, whereas the higher molecular weight SHMP at a low concentration creates NPs mostly by binding onto the surface. It was found that the release rates of curcumin from CHS-NPs crosslinked by SHMP at higher concentrations were about twice as high as the release rates of curcumin from TPP-crosslinked CHS-NPs, accompanied by notable lag times.
Conclusions:
This significant increase in the release rates of curcumin from SHMP-crosslinked CHS-NPs is explained by the large spatial structure of this crosslinker compared to the small TPP molecules. This study advances the literature on drug diffusion by making it possible to accurately determine its release from nanoparticulate systems.
Aim:
Chitosan (CHS)-based nanoparticulate systems have gained much interest due to their high drug loading capacity and the simplicity of their fabrication. The physical properties of two types of curcumin-loaded CHS nanoparticles (CHS-NPs) were determined and compared. A new in-vitro release method was developed based on mathematical modeling in which the drug is first released from the NP into the surrounding medium and subsequently diffuses through the membrane.
Methods:
Curcumin-loaded CHS-NPs were fabricated by ionotropic gelation using sodium tripolyphosphate (TPP) and sodium hexametaphosphate (SHMP) crosslinking, and characterized by NP tracking analysis, loading capacity, zeta potential, Fourier transform infrared spectroscopy (FTIR), and in-vitro release rates.
Results:
The data showed that compared to SHMP crosslinked CHS-NPs, TPP crosslinking demonstrated a decrease in entrapment efficiency at a relatively high concentration of the agent, probably by narrowing the space between the polymeric chains. As indicated by zeta potential measurements, TPP crosslinking at all levels was more uniformly distributed inside the NPs, whereas the higher molecular weight SHMP at a low concentration creates NPs mostly by binding onto the surface. It was found that the release rates of curcumin from CHS-NPs crosslinked by SHMP at higher concentrations were about twice as high as the release rates of curcumin from TPP-crosslinked CHS-NPs, accompanied by notable lag times.
Conclusions:
This significant increase in the release rates of curcumin from SHMP-crosslinked CHS-NPs is explained by the large spatial structure of this crosslinker compared to the small TPP molecules. This study advances the literature on drug diffusion by making it possible to accurately determine its release from nanoparticulate systems.
DOI: https://doi.org/10.37349/eds.2026.1008157
Aim:
Bisoprolol fumarate (BF), commonly prescribed for cardiovascular conditions, is usually split to achieve specific doses. This study evaluated the effects of tablet splitting on the quality parameters of scored BF tablets from three different brands marketed in Saudi Arabia.
Methods:
The products were evaluated for weight variation, content uniformity, and dissolution for intact and split tablets. A UPLC-sensitive assay was used for drug quantification.
Results:
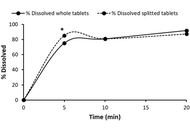
The results showed that all products lost less than 3% of its weight upon splitting, meeting the USP requirements. Content uniformity was between 85% and 115% for all products, complying with pharmacopoeial standards. Dissolution studies showed some variation between intact and split tablets. The f2 similarity factor was calculated to compare the dissolution profiles of BF from both forms. The f2 values showed a similar dissolution profile for the innovator product (f2 was 62.53), but dissimilar profiles for Generic-1 and -2 (f2 values were 48.90 and 34.43, respectively).
Conclusions:
These results should be taken into consideration by healthcare professionals to avoid sub-therapeutic or toxic effects resulting from tablet splitting.
Aim:
Bisoprolol fumarate (BF), commonly prescribed for cardiovascular conditions, is usually split to achieve specific doses. This study evaluated the effects of tablet splitting on the quality parameters of scored BF tablets from three different brands marketed in Saudi Arabia.
Methods:
The products were evaluated for weight variation, content uniformity, and dissolution for intact and split tablets. A UPLC-sensitive assay was used for drug quantification.
Results:
The results showed that all products lost less than 3% of its weight upon splitting, meeting the USP requirements. Content uniformity was between 85% and 115% for all products, complying with pharmacopoeial standards. Dissolution studies showed some variation between intact and split tablets. The f2 similarity factor was calculated to compare the dissolution profiles of BF from both forms. The f2 values showed a similar dissolution profile for the innovator product (f2 was 62.53), but dissimilar profiles for Generic-1 and -2 (f2 values were 48.90 and 34.43, respectively).
Conclusions:
These results should be taken into consideration by healthcare professionals to avoid sub-therapeutic or toxic effects resulting from tablet splitting.
DOI: https://doi.org/10.37349/eds.2026.1008156
Antibodies currently represent a leading segment of the biopharmaceutical market and are expected to maintain a significant presence in the therapeutic landscape. Development of therapeutic antibodies represents one of the most transformative advances in the modern medicine field, with hundreds of successful products on the market that have changed the lives of millions and the history of mankind by revolutionizing medical treatment. In its broadest context, antibody-based products consist of full-length antibodies, antibody fragments, polyvalent and polyspecific antibodies, and their conjugates (for targeted delivery of other therapeutic drugs/agents). High target specificity, tailored mechanisms of action, and broad applicability have made antibodies indispensable in a variety of diseases. With advancements in protein engineering as well as growing integration of computational biology, the field of antibody development continues to push boundaries and is expected to drive the next era of breakthroughs. This article charts the journey of therapeutic antibodies from their conceptual roots to their central role in current medicine, and offers a forward-looking perspective on what lies ahead in this dynamic field.
Antibodies currently represent a leading segment of the biopharmaceutical market and are expected to maintain a significant presence in the therapeutic landscape. Development of therapeutic antibodies represents one of the most transformative advances in the modern medicine field, with hundreds of successful products on the market that have changed the lives of millions and the history of mankind by revolutionizing medical treatment. In its broadest context, antibody-based products consist of full-length antibodies, antibody fragments, polyvalent and polyspecific antibodies, and their conjugates (for targeted delivery of other therapeutic drugs/agents). High target specificity, tailored mechanisms of action, and broad applicability have made antibodies indispensable in a variety of diseases. With advancements in protein engineering as well as growing integration of computational biology, the field of antibody development continues to push boundaries and is expected to drive the next era of breakthroughs. This article charts the journey of therapeutic antibodies from their conceptual roots to their central role in current medicine, and offers a forward-looking perspective on what lies ahead in this dynamic field.
DOI: https://doi.org/10.37349/eds.2026.1008155
Intranasal drug delivery exhibits therapeutic potential for the treatment of central nervous system (CNS) diseases, as it allows pharmaceuticals to bypass the blood-brain barrier (BBB) via the olfactory and trigeminal pathways. The primary advantage of this method lies in its non-invasiveness and low systemic toxicity. Nevertheless, this delivery method faces notable challenges, including limited nasal mucosal absorption and short residence time in the olfactory region. To address these limitations, intranasal nanomedicine has gained research attention. Nanomedicines can improve brain bioavailability by enhancing drug solubility, permeability, and stability. The following discussion summarizes recent advancements in nanotechnology-enabled nose-to-brain delivery systems, with the aim of synthesizing progress in the field and outlining future research directions.
Intranasal drug delivery exhibits therapeutic potential for the treatment of central nervous system (CNS) diseases, as it allows pharmaceuticals to bypass the blood-brain barrier (BBB) via the olfactory and trigeminal pathways. The primary advantage of this method lies in its non-invasiveness and low systemic toxicity. Nevertheless, this delivery method faces notable challenges, including limited nasal mucosal absorption and short residence time in the olfactory region. To address these limitations, intranasal nanomedicine has gained research attention. Nanomedicines can improve brain bioavailability by enhancing drug solubility, permeability, and stability. The following discussion summarizes recent advancements in nanotechnology-enabled nose-to-brain delivery systems, with the aim of synthesizing progress in the field and outlining future research directions.
DOI: https://doi.org/10.37349/eds.2026.1008154
This article belongs to the special issue Nanoformulations for Non-Intravenous Drug Delivery
DOI: https://doi.org/10.37349/eds.2026.1008153
Ageing is a gradual, multifactorial process that leads to the deterioration of physical and mental health, increasing the risk of disease and eventually death. Indicators of ageing manifest at the molecular level, including genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, loss of proteostasis, and dysregulation of key signalling pathways such as the mechanistic target of rapamycin (mTOR) and insulin signalling. These molecular hallmarks of ageing are interconnected, amplifying one another over time. The resulting cellular stress triggers apoptosis or drives cells into a pathological state known as cellular senescence, in which they secrete inflammatory, pro-ageing factors. Consequently, there is a progressive decline in tissue function and regenerative capacity, accompanied by atrophy and stem cell exhaustion under a chronically inflamed microenvironment. Although functional decline with age is irreversible, research indicates it can be delayed. In this review, we discuss the hallmarks of ageing, conventional pharmacological interventions with demonstrated anti-ageing effects in cellular and animal models, and emerging therapeutic strategies being explored as ageing becomes increasingly recognized as a major risk factor for disease development.
Ageing is a gradual, multifactorial process that leads to the deterioration of physical and mental health, increasing the risk of disease and eventually death. Indicators of ageing manifest at the molecular level, including genomic instability, telomere attrition, epigenetic alterations, mitochondrial dysfunction, loss of proteostasis, and dysregulation of key signalling pathways such as the mechanistic target of rapamycin (mTOR) and insulin signalling. These molecular hallmarks of ageing are interconnected, amplifying one another over time. The resulting cellular stress triggers apoptosis or drives cells into a pathological state known as cellular senescence, in which they secrete inflammatory, pro-ageing factors. Consequently, there is a progressive decline in tissue function and regenerative capacity, accompanied by atrophy and stem cell exhaustion under a chronically inflamed microenvironment. Although functional decline with age is irreversible, research indicates it can be delayed. In this review, we discuss the hallmarks of ageing, conventional pharmacological interventions with demonstrated anti-ageing effects in cellular and animal models, and emerging therapeutic strategies being explored as ageing becomes increasingly recognized as a major risk factor for disease development.
DOI: https://doi.org/10.37349/eds.2026.1008152
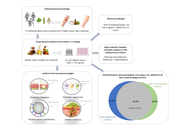
The significant medicinal constituents and pharmacological potential of several botanicals suggest promising therapeutic applications. Scorzonera undulata displayed a diverse phytochemical profile, with 25 volatile and 21 phenolic compounds identified, including quinic and chlorogenic acids, along with flavonoids such as kaempferol, apigenin, luteolin derivatives, quercitrin, and naringin—mostly concentrated in the aerial parts. These extracts exhibited notable antioxidant, antimicrobial, anti-inflammatory, and cytotoxic activities, especially methanolic extracts against MCF-7 breast cancer cells, indicating therapeutic relevance. Andrographis paniculata extracts, rich in andrographolide, showed clinical potential in alleviating mild COVID-19 symptoms. However, the compound’s nonlinear pharmacokinetics highlight the need for optimized delivery strategies. Morinda citrifolia fruit extracts demonstrated considerable in vitro antimicrobial effects and moderate cytotoxicity, supported by UPLC–Orbitrap MS identification of unique bioactives. These findings reinforce the need for further pharmacological and clinical validation. The antiviral efficacy of Houttuynia cordata against dengue virus type 2 was evident, with aqueous extracts showing strong virucidal action and inhibition of viral replication. Hyperoside was identified as the dominant active constituent, supported by a rich phytochemical profile including flavonoids, aristolactams, and triterpenoids. Genotoxicity assessments indicated a favorable safety profile, suggesting potential for phytotherapeutic development. Achillea millefolium (yarrow) contained essential oils enriched in camphor, 1,8-cineole, artemisia ketone, and azulene derivatives, alongside phenolic acids and flavonoids like chlorogenic acid, apigenin, luteolin, and quercetin. These contributed to its antioxidant, anti-inflammatory, antimicrobial, and hemostatic effects, validating traditional medicinal applications and warranting clinical standardization. Flavonoids such as luteolin and apigenin offered anticancer and cardiovascular benefits by inhibiting PD-L1 via STAT3 suppression and promoting autophagy to counter vascular calcification. Bryophyllum pinnatum demonstrated broad pharmacological activity attributed to bufadienolides, flavonoids, and phenolic acids, supporting its ethnomedicinal use while emphasizing the need for clinical safety validation.
The significant medicinal constituents and pharmacological potential of several botanicals suggest promising therapeutic applications. Scorzonera undulata displayed a diverse phytochemical profile, with 25 volatile and 21 phenolic compounds identified, including quinic and chlorogenic acids, along with flavonoids such as kaempferol, apigenin, luteolin derivatives, quercitrin, and naringin—mostly concentrated in the aerial parts. These extracts exhibited notable antioxidant, antimicrobial, anti-inflammatory, and cytotoxic activities, especially methanolic extracts against MCF-7 breast cancer cells, indicating therapeutic relevance. Andrographis paniculata extracts, rich in andrographolide, showed clinical potential in alleviating mild COVID-19 symptoms. However, the compound’s nonlinear pharmacokinetics highlight the need for optimized delivery strategies. Morinda citrifolia fruit extracts demonstrated considerable in vitro antimicrobial effects and moderate cytotoxicity, supported by UPLC–Orbitrap MS identification of unique bioactives. These findings reinforce the need for further pharmacological and clinical validation. The antiviral efficacy of Houttuynia cordata against dengue virus type 2 was evident, with aqueous extracts showing strong virucidal action and inhibition of viral replication. Hyperoside was identified as the dominant active constituent, supported by a rich phytochemical profile including flavonoids, aristolactams, and triterpenoids. Genotoxicity assessments indicated a favorable safety profile, suggesting potential for phytotherapeutic development. Achillea millefolium (yarrow) contained essential oils enriched in camphor, 1,8-cineole, artemisia ketone, and azulene derivatives, alongside phenolic acids and flavonoids like chlorogenic acid, apigenin, luteolin, and quercetin. These contributed to its antioxidant, anti-inflammatory, antimicrobial, and hemostatic effects, validating traditional medicinal applications and warranting clinical standardization. Flavonoids such as luteolin and apigenin offered anticancer and cardiovascular benefits by inhibiting PD-L1 via STAT3 suppression and promoting autophagy to counter vascular calcification. Bryophyllum pinnatum demonstrated broad pharmacological activity attributed to bufadienolides, flavonoids, and phenolic acids, supporting its ethnomedicinal use while emphasizing the need for clinical safety validation.
DOI: https://doi.org/10.37349/eds.2026.1008151
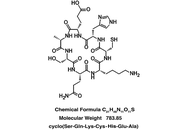
Aim:
A seven amino acid cyclic peptide has been applied to human blood plasma treated with glucose metabolite methylglyoxal (MG) in “proof of concept” experiments to determine the peptide’s ability to counteract pathologies associated with hyperglycemia. Similar pathologies are evident during aging and in age-related disorders. In fact, elevated MG levels in the blood lead directly to diabetic complications and accelerated aging, including cognitive decline. These changes are attributed to oxidant stress and amyloidogenesis, the latter involving toxic accumulations of blood and tissue proteins.
Methods:
cSKE7 was redesigned from cell survival-promoting and anti-inflammatory fragments near the N-terminus of human/primate “orphan” protein DSEP/Dermcidin and incubated at low micromolar concentrations with the MG-stressed human plasma for 24 hours. The modified design of the new compound offers several practical advantages over predecessors including cyclic stability and a marked increase in aqueous solubility.
Results:
The peptide dispersed thioflavin-T-stained amyloid aggregates and reduced oxidant stress as measured by plasma levels of free thiols and of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity. Since these N-terminal fragments of DSEP/Dermcidin have been shown to bind and influence the activity of heat shock protein 70 (HSP70), HSP70 inhibitor pifithrin-μ was added to the plasma prior to peptide treatment. The inhibitor disrupted amyloid dispersion and both peptide-induced and, in some cases, normally occurring antioxidant effects, suggesting these reparative activities are HSP70 dependent.
Conclusions:
The results are discussed in terms of their potential use in new therapies for the complications of metabolic disease and disorders of aging that result from a deterioration of the quality control mechanisms of proteostasis.
Aim:
A seven amino acid cyclic peptide has been applied to human blood plasma treated with glucose metabolite methylglyoxal (MG) in “proof of concept” experiments to determine the peptide’s ability to counteract pathologies associated with hyperglycemia. Similar pathologies are evident during aging and in age-related disorders. In fact, elevated MG levels in the blood lead directly to diabetic complications and accelerated aging, including cognitive decline. These changes are attributed to oxidant stress and amyloidogenesis, the latter involving toxic accumulations of blood and tissue proteins.
Methods:
cSKE7 was redesigned from cell survival-promoting and anti-inflammatory fragments near the N-terminus of human/primate “orphan” protein DSEP/Dermcidin and incubated at low micromolar concentrations with the MG-stressed human plasma for 24 hours. The modified design of the new compound offers several practical advantages over predecessors including cyclic stability and a marked increase in aqueous solubility.
Results:
The peptide dispersed thioflavin-T-stained amyloid aggregates and reduced oxidant stress as measured by plasma levels of free thiols and of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity. Since these N-terminal fragments of DSEP/Dermcidin have been shown to bind and influence the activity of heat shock protein 70 (HSP70), HSP70 inhibitor pifithrin-μ was added to the plasma prior to peptide treatment. The inhibitor disrupted amyloid dispersion and both peptide-induced and, in some cases, normally occurring antioxidant effects, suggesting these reparative activities are HSP70 dependent.
Conclusions:
The results are discussed in terms of their potential use in new therapies for the complications of metabolic disease and disorders of aging that result from a deterioration of the quality control mechanisms of proteostasis.
DOI: https://doi.org/10.37349/eds.2026.1008150
This article belongs to the special issue Peptide Science Without Borders: Novel Insights for Drug Discovery
The s-triazine scaffold has emerged as a privileged heterocyclic nucleus/moiety in pharmaceutical discovery and development, owing to its presence in several natural products and clinically relevant therapeutic agents, including enasidenib, gedatolisib, bimiralisib, atrazine, indaziflam, and triaziflam. s-Triazine derivatives are not only economically accessible and synthetically versatile, but they also exhibit a broad spectrum of noteworthy biological activities, encompassing anticancer, anti-inflammatory, antiviral, antidiabetic, anticonvulsant, antitubercular, and antimicrobial properties. Their widespread utility is further supported by the ease of synthesis from inexpensive precursors such as amidines or the readily available 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride), which enables sequential functionalization and the rapid generation of diverse analogues. The heightened reactivity and modularity of the s-triazine core have facilitated the development of structurally rich heterocyclic hybrids with enhanced potency and improved pharmacological profiles. These multitarget-directed systems offer exciting opportunities for addressing various forms of cancer. Considering the increasing pace of innovation in this field, a comprehensive overview of recent advancements in s-triazine-based hybrid molecules is both timely and necessary. This review highlights current progress, key design strategies, and emerging perspectives to inspire continued efforts toward the identification of promising s-triazine-based lead candidates for future drug development as anticancer agents.
The s-triazine scaffold has emerged as a privileged heterocyclic nucleus/moiety in pharmaceutical discovery and development, owing to its presence in several natural products and clinically relevant therapeutic agents, including enasidenib, gedatolisib, bimiralisib, atrazine, indaziflam, and triaziflam. s-Triazine derivatives are not only economically accessible and synthetically versatile, but they also exhibit a broad spectrum of noteworthy biological activities, encompassing anticancer, anti-inflammatory, antiviral, antidiabetic, anticonvulsant, antitubercular, and antimicrobial properties. Their widespread utility is further supported by the ease of synthesis from inexpensive precursors such as amidines or the readily available 2,4,6-trichloro-1,3,5-triazine (cyanuric chloride), which enables sequential functionalization and the rapid generation of diverse analogues. The heightened reactivity and modularity of the s-triazine core have facilitated the development of structurally rich heterocyclic hybrids with enhanced potency and improved pharmacological profiles. These multitarget-directed systems offer exciting opportunities for addressing various forms of cancer. Considering the increasing pace of innovation in this field, a comprehensive overview of recent advancements in s-triazine-based hybrid molecules is both timely and necessary. This review highlights current progress, key design strategies, and emerging perspectives to inspire continued efforts toward the identification of promising s-triazine-based lead candidates for future drug development as anticancer agents.
DOI: https://doi.org/10.37349/eds.2026.1008149
This article belongs to the special issue The Role of Triazine Scaffolds in Modern Drug Development
Aim:
To evaluate the real-world effectiveness of prophylactic metoclopramide in preventing opioid-induced nausea and vomiting (OINV) during the initial phase of strong opioid therapy in opioid-naïve patients with cancer-related pain.
Methods:
This retrospective, single-center observational cohort study included adult patients with pathologically confirmed malignancies who initiated strong opioid therapy between January 2023 and December 2024. Patients were categorized into a prophylactic metoclopramide group or a no-prophylaxis control group. Complete control (CC) of OINV during the first 7 days was defined as the absence of nausea, vomiting, and rescue antiemetic use. Univariate and multivariate logistic regression analyses were performed to identify factors associated with CC, adjusting for age, sex, body mass index, cancer subtype, cancer stage, comorbidity status, and morphine-equivalent daily dose (MEDD). Subgroup analyses were conducted based on age, sex, and cancer subtype.
Results:
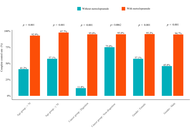
A total of 244 patients were included, of whom 199 received prophylactic metoclopramide, and 45 received no prophylaxis. The prophylactic group achieved significantly higher CC rates than the control group (74.9% vs. 37.8%, p < 0.001). Multivariate logistic regression confirmed that prophylactic metoclopramide was independently associated with higher odds of achieving CC (adjusted OR = 0.20, 95% CI: 0.10–0.40; p < 0.001). Similar improvements were observed for nausea and vomiting control. Subgroup analyses demonstrated consistent benefits across age and sex groups, with particularly notable effects in patients with gastrointestinal cancers.
Conclusions:
Prophylactic metoclopramide significantly improves OINV control in opioid-naïve patients with cancer-related pain during the initiation of strong opioids. These findings support the rational use of early antiemetic prophylaxis in routine clinical practice. Prospective randomized trials are warranted to validate these real-world results and assess long-term safety.
Aim:
To evaluate the real-world effectiveness of prophylactic metoclopramide in preventing opioid-induced nausea and vomiting (OINV) during the initial phase of strong opioid therapy in opioid-naïve patients with cancer-related pain.
Methods:
This retrospective, single-center observational cohort study included adult patients with pathologically confirmed malignancies who initiated strong opioid therapy between January 2023 and December 2024. Patients were categorized into a prophylactic metoclopramide group or a no-prophylaxis control group. Complete control (CC) of OINV during the first 7 days was defined as the absence of nausea, vomiting, and rescue antiemetic use. Univariate and multivariate logistic regression analyses were performed to identify factors associated with CC, adjusting for age, sex, body mass index, cancer subtype, cancer stage, comorbidity status, and morphine-equivalent daily dose (MEDD). Subgroup analyses were conducted based on age, sex, and cancer subtype.
Results:
A total of 244 patients were included, of whom 199 received prophylactic metoclopramide, and 45 received no prophylaxis. The prophylactic group achieved significantly higher CC rates than the control group (74.9% vs. 37.8%, p < 0.001). Multivariate logistic regression confirmed that prophylactic metoclopramide was independently associated with higher odds of achieving CC (adjusted OR = 0.20, 95% CI: 0.10–0.40; p < 0.001). Similar improvements were observed for nausea and vomiting control. Subgroup analyses demonstrated consistent benefits across age and sex groups, with particularly notable effects in patients with gastrointestinal cancers.
Conclusions:
Prophylactic metoclopramide significantly improves OINV control in opioid-naïve patients with cancer-related pain during the initiation of strong opioids. These findings support the rational use of early antiemetic prophylaxis in routine clinical practice. Prospective randomized trials are warranted to validate these real-world results and assess long-term safety.
DOI: https://doi.org/10.37349/eds.2026.1008148
Aim:
To design, synthesize, and test small molecules and fragment-based compounds with putative selective anti-mycobacterial activity.
Methods:
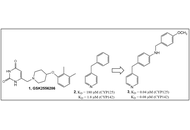
Standard chemosynthetic processes were used to synthesize 42 compounds. A cell-based phenotypic screen for inhibitors of mycobacterial growth was used to identify several fragments and small molecules as representatives of urea-, carbamothioate-, and α,β-unsaturated systems (Michael acceptors) chemotypes.
Results:
All 42 compounds exhibited selective toxicity for mycobacteria as demonstrated by their lack of activity against various Gram-positive and Gram-negative bacteria and acid-fast Corynebacterium glutamicum. A thiadiazole compound, similar to (3-((5-(methylthio)-1,3,4-thiadiazol-2-yl)thio)pyrazine-2-carbonitrile), which activates the human lecitin: cholesterol acyltransferase (LCAT), exhibits growth-inhibitory activity [0.6 μg/mL in bovine serum albumin (BSA)-free media] against drug-susceptible Mycobacterium tuberculosis (Mtb). From the urea class, a 1,2,4-triazole-containing urea demonstrated anti-Mtb activity (4.7 μg/mL in BSA-free media). Several carbamothioate-based fragments demonstrated activity against Mycobacterium marinum [with a best minimum inhibitory concentration (MIC) of 6.25 μg/mL in minimal BSA-free media].
Conclusions:
This foundational study demonstrates the utility of these newly designed and synthesized low molecular-weight compounds and fragments as potential antimycobacterials.
Aim:
To design, synthesize, and test small molecules and fragment-based compounds with putative selective anti-mycobacterial activity.
Methods:
Standard chemosynthetic processes were used to synthesize 42 compounds. A cell-based phenotypic screen for inhibitors of mycobacterial growth was used to identify several fragments and small molecules as representatives of urea-, carbamothioate-, and α,β-unsaturated systems (Michael acceptors) chemotypes.
Results:
All 42 compounds exhibited selective toxicity for mycobacteria as demonstrated by their lack of activity against various Gram-positive and Gram-negative bacteria and acid-fast Corynebacterium glutamicum. A thiadiazole compound, similar to (3-((5-(methylthio)-1,3,4-thiadiazol-2-yl)thio)pyrazine-2-carbonitrile), which activates the human lecitin: cholesterol acyltransferase (LCAT), exhibits growth-inhibitory activity [0.6 μg/mL in bovine serum albumin (BSA)-free media] against drug-susceptible Mycobacterium tuberculosis (Mtb). From the urea class, a 1,2,4-triazole-containing urea demonstrated anti-Mtb activity (4.7 μg/mL in BSA-free media). Several carbamothioate-based fragments demonstrated activity against Mycobacterium marinum [with a best minimum inhibitory concentration (MIC) of 6.25 μg/mL in minimal BSA-free media].
Conclusions:
This foundational study demonstrates the utility of these newly designed and synthesized low molecular-weight compounds and fragments as potential antimycobacterials.
DOI: https://doi.org/10.37349/eds.2026.1008145
This article belongs to the special issue Discovery and development of new antibacterial compounds
