 Review
Review

Affiliation:
School of Biological Sciences & Engineering, Yachay Tech University, Urcuquí 100115, Ecuador
Email: sballazg@gmail.com
ORCID: https://orcid.org/0000-0001-5878-8679
Explor Drug Sci. 2025;3:1008125 DOI: https://doi.org/10.37349/eds.2025.1008125
Received: June 02, 2025 Accepted: July 10, 2025 Published: August 18, 2025
Academic Editor: Amedeo Lonardo, University of Modena and Reggio Emilia, Italy
Cholecystokinin (CCK) is the most prevalent neuropeptide in the brain, where it affects satiety, pain modulation, memory, and anxiety. Its effects are mediated by GPCRs known as the “alimentary (gastrointestinal)” CCK1r (CCK 1 receptor) and the brain-specific CCK2r (CCK 2 receptor). While stress causes CCK to be released and full CCK2r agonists are potent panicogenic agents, specific CCK2r antagonists are ineffective at lowering human anxiety. As a result, the therapeutic potential of CCK as a target in psychiatry has been questioned. By compiling relevant new and historical scientific data retrieved from Scopus and PubMed, the aim of this review was to suggest a new function of CCK neurotransmission, the regulation of neuronal homeostasis during stress. Four lines of evidence were discussed that support the hypothesis of a CCK-driven neuronal homoestasis: (1) Homeostatic plasticity including synaptic scaling and intrinsic excitability; (2) its interaction with retrograde endocannabinoid signaling; (3) neuroprotective role; and (4) dynamic neuromodulation of CCK release. CCK functions as a crucial and essential molecular switch of neural circuits and neuroplasticity through its remarkable cell-specific modulation of glutamate and GABA release via CCK2r. CCKergic neurons are downstream of the activation of cannabinoid type-1 (CB1) receptors in order to generate and stabilize rhythmic synchronous network activity in the hippocampus. CCK is also released to modulate other neurotransmitters like dopamine and opioids when neuronal firing is intense during the processing of anxiety/fear, memory, and pain. CCK likely functions to restore baseline neuronal function and protect neurons from harm under these conditions. Anxiety, depression, and schizophrenia could result from compensatory plastic changes of the CCKergic system that go awry during neuronal homeostasis. This review concludes by examining the benefits of putative compounds that exhibit a combination of CCK agonist and antagonist activity at multiple locations within the CCKergic system, as well as off-targets in managing mental conditions.
Cholecystokinin (CCK) comprises a family of intestinal peptide hormones that share the same five C-terminal amino acids as gastrin (Table 1). CCK exists in several forms depending on the number of amino acids it contains and the presence in most of them of a sulfate group attached to a tyrosine located seven residues from the C-terminus (denoted with the letter S) [1]. The secretion of the longest forms [CCK-22S (sulfated 22-aa CCK), CCK-33S, CCK-58S] is linked to the upper gut; meanwhile, sulfated CCK octapeptide (CCK-8S) is expressed in higher quantities than any other neuropeptide in the brain [1–3]. CCK functions through two receptor subtypes: the “alimentary” CCK1r, largely expressed in the gastrointestinal tract, and the “brain” CCK2r, which is predominant in the brain [4, 5] (Table 2).
Family of CCK bioactive peptide hormones present in humans
| CCK forms | Affinity CCK1r | Affinity CCK2r | Release location | CCK C-terminal fragments (sequence of aa residues) |
|---|---|---|---|---|
| CCK-4 | No | Yes | Brain | Trp-Met-Asp-Phe-NH2 |
| CCK-8NS | No | Yes | Digestive tract | Asp-Tyr-Met-Gly-Trp-Met-Asp-Phe-NH2 |
| CCK-8S | Yes | Yes | Brain | Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 |
| CCK-12S | Yes | Yes | Digestive tract | Ile-Ser-Asp-Arg-Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 |
| CCK-22S | Yes | Yes | Digestive tract | Asn-Leu-Gln-Asn-Leu-Asp-Pro-Ser-His-Arg-Ile-Ser-Asp-Arg-Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 |
| CCK-33S | Yes | Yes | Digestive tract | Lys-Ala-Pro-Ser-Gly-Arg-Met-Ser-Ile-Val-Lys-Asn-Leu-Gln-Asn-Leu-Asp-Pro-Ser-His-Arg-Ile-Ser-Asp-Arg-Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 |
| CCK-58S | Yes | Yes | Digestive tract | Val-Ser-Gln-Arg-Thr-Asp-Gly-Glu-Ser-Arg-Ala-His-Leu-Gly-Ala-Leu-Leu-Ala-Arg-Tyr-Ile-Gln-Gln-Ala-Arg-Lys-Ala-Pro-Ser-Gly-Arg-Met-Ser-Ile-Val-Lys-Asn-Leu-Gln-Asn-Leu-Asp-Pro-Ser-His-Arg-Ile-Ser-Asp-Arg-Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2 |
CCK: cholecystokinin; CCK-4: CCK tetrapeptide; CCK-8NS: non-sulfated CCK octapeptide; CCK-8S: sulfated CCK octapeptide; CCK-12S: sulfated 12-aa CCK; CCK-22S: sulfated 22-aa CCK; CCK-33S: sulfated 33-aa CCK; CCK-58S: sulfated 58-aa CCK
Primary anatomical distribution of CCK1r and CCK2r in the nervous system
| Subdivision | Structure | CCK1r | CCK2r |
|---|---|---|---|
| Peripheral nervous system | Vagus nerve | High | High |
| Nodose ganglia | High | Low | |
| Spinal cord | Dorsal root ganglia | Low | Low |
| Myelencephalon | NTS | High | Low |
| Area postrema | High | High | |
| Parabrachial nucleus | High | High | |
| Metencephalon | Cerebellum | High | Absent |
| Mesencephalon | Substantia nigra | Absent | High |
| Ventral tegmental area | Absent | High | |
| Periaqueductal area | High | Low | |
| Dorsal raphe nucleus | High | High | |
| Dielencephalon | Hypothalamic dorsomedial nucleus | High | Low |
| Hypothalamic ventromedial nucleus | Absent | High | |
| Hypothalamic paraventricular nucleus | High | Low | |
| Hypothalamic supraoptical nucleus | High | Absent | |
| Hypothalamic arcuate nucleus | High | Low | |
| Mammillary nuclei | High | Absent | |
| Supramamillary nuclei | High | Absent | |
| Telencephalon | Cortex | High | Low |
| Hyppocampus | High | Low | |
| Striatum | High | High | |
| Nucleus accumbens | High | High | |
| Bed nucleus of the stria terminalis | High | High | |
| Amygdala | High | Low | |
| Olfactory bulbs | High | Low |
CCK is a complex and multifaceted messenger that has undergone over 600 million years of evolutionary history [1]. In the central nervous system, CCK-mediated neurotransmission regulates feeding behavior [6], modulation of opioid-mediated analgesia [7–9], memory, and cognition [10–12]. Interestingly, alterations of the brain CCK system have been linked to the physiopathology of schizophrenia [13–15], major depression [16], suicide [17], addiction [14, 18–20], and particularly anxiety [13, 21, 22]. Despite the high hopes of the preclinical evidence, most of the CCK antagonists have been shown to be unsuccessful in psychiatry clinical trials and as pain medicine [23–26] (see comparative Table 3). These disappointing outcomes sparked contentious debates on the possible therapeutic benefits of drugs that target CCK2r-mediated neurotransmission specifically [27]. Even while interest in CCK’s therapeutic potential subsequently waned, recent discoveries on its importance in the central nervous system have reignited it [3]. In light of this field’s resurgence, and to encourage further clinical research, the goal of this review was to propose a new function of CCK neurotransmission: the regulation of neuronal homeostasis during stress.
Translational gaps on the psychiatry/analgesic potential of CCK2r antagonists
| Preclinical evidence | Clinical trials | ||||
|---|---|---|---|---|---|
| Compound (dosage) length | Outcomes | References | Compound (dosage) length | Outcomes | References |
| CI-988 (0.001–10.0 mg/kg, i.p.)Acute | Anxiolytic-like action in rats elevated the X-maze, rat social interaction test, and mouse light/dark shuttle box | [208] | CI-988 (300 mg/day, thrice daily)Four weeks | No anxiolytic effect in general anxiety disorder | [23] |
| CI-988 (100 mg/day, thrice daily)Six weeks | No anxiolytic effect in panic disorder | [25] | |||
| L-365,260 (3.2, 10, and 32 mg/kg, i.p.)Acute | Antipanic-like effects in rats receiving brain stimulation in the dorsal PAG | [209] | L-365,260 (30 mg/day, four times daily)Six weeks | No anxiolytic effect in panic disorder | [24] |
| CI-988 and L-365,260 (8.9, 0.16, and 0.25 μmol/kg, i.p.)Acute | Anxiolytic-like action in rats elevated the X-maze | [210] | L-365,260 (10–50 mg)Acute | CCK-4 panicogenic effects are antagonized by L-365,260 in panic disorder patients | [211] |
| L-365,260 (0.1 and 0.5 mg/kg, s.c.)Acute | Enhancement of the analgesia induced by a submaximal dose of morphine | [212] | L-365,260 (10 mg and 40 mg thrice daily)Two weeks | L-365,260 fails to augment morphine-induced analgesia in chronic neuropathic pain | [26] |
i.p.: intraperitoneal; s.c.: subcutaneous. PAG: periaqueductal grey; CCK-4: cholecystokinin tetrapeptide
Bioactive peptides of varying lengths with different N-terminal extensions [CCK tetrapeptide (CCK-4), CCK-8, CCK-12, CCK-22, CCK-33, and CCK-58] are synthesized by enzymatic processing of the human CCK preproprotein (115 aa residues) [1]. CCK possesses a sulfated tyrosine at the 7th amino acid residue from the C-terminus. Notice that the C-terminal phenylalanine residue is amidated. The C-terminal sequence (Trp-Met-Asp-Phe-NH2) is highly conserved across different CCK peptides and gastrin, and it’s important for its biological activity.
This review was based on documents retrieved from the Scopus and PubMed databases as of April 1, 2025. All research publications addressing CCK in the nervous systems that were written in English and published in peer-reviewed journals between 1975—the year when CCK was first discovered in the brain—and 2025 met the inclusion criteria. For advanced research, the following keywords were adopted to sight documents: TITLE-ABS-KEY [(“Cholecystokinin”) AND (“CCK-A receptor” OR “CCK-B receptor” OR “central nervous system” OR “neurotransmission” OR “brain” OR “neurons” OR “amygdala” OR “hippocampus” OR “cortex” OR “hypothalamus” OR “anxiety” OR “learning” OR “memory” OR “satiety” OR “pain” OR “peripheral nervous system” OR “gastrointestinal”)] AND PUBYEAR > 1975 AND PUBYEAR < 2025 AND [LIMIT-TO (DOCTYPE, “ar”) OR LIMIT-TO (DOCTYPE, “re”)]. Eligible articles were based on the author’s own experience with the topic.
Several explanations have been proposed to explain the disheartening clinical trials [23–26]. The most plausible one is “dynamic neuromodulation” [10], which means that CCK release is triggered in response to high-frequency neuronal firing [28] to regulate the activity of other neurotransmitters. Another possibility is that CCK could interact with both CCK1r and with CCK2r in the brain [29], producing opposite effects in most cases [30, 31]. The mesolimbic system is the most well-known case of this [32]. CCK colocalized with dopamine in neurons of the tegmental ventral area projecting to the medial nucleus accumbens [33]. In the anterior part of the nucleus accumbens, CCK inhibits dopamine release via CCK2r, whereas CCK via CCK1r promotes DA in the posterior nucleus accumbens [34]. CCK controls dopamine neurotransmission in the limbic system, affecting motivation [35, 36], reward, and anxiety [28]. In the conditioned place preference (CPP) test in the rat, CCK2r and CCK1r antagonists enhance and decrease respectively the rewarding effects of morphine [37]. The close neuroanatomical distribution of CCK with opioids in the limbic system raises the possibility of an opioid-CCK functional link [38, 39]. Remarkably, the activation of CCK1r and CCK2r by endogenous CCK may also have opposite effects in the regulation of antidepressant effects induced by endogenous enkephalins [31]. Herein, the reciprocal relationships between the two CCK receptor subtypes further underscore the neuromodulatory relevance of CCK by fine-tuning the impact of opioid- and dopamine-mediated neurotransmission.
CCK influences brain-wide structural-functional networks across the isocortex [40]. CCK’s function in memory relies on the hippocampal neuronal circuitry [41–44]. For instance, in the developing dentate gyrus, cortical activity guides the formation of the CCK+ basket cell network, which preserves the inhibitory to excitatory balance in the hippocampus, a crucial aspect of learning and memory [45]. More importantly, the evidence shows that CCK release interacts with CCK2r to promote high-frequency stimulation-induced long-term potentiation caused by NMDA receptors [46–48]. CCK is heavily present in neurons of the hippocampus and subiculum, sending fibers to the septum and hypothalamus [49]. In the dorsomedial nucleus of the hypothalamus, CCK shifts the plasticity of GABA synapses from long-term depression to long-term potentiation [50]. There is proof that CCK-containing interneurons play a crucial role in the regulation of place-cell temporal coding and the development of contextual memories [51]. Given the evidence that CCK2r antagonists have carry-over effects in the baseline for anxiety after the drug is cleared [52], it is conceivable that CCK may generate plastic changes in the brain [3, 53–56].
CCKergic pathways appear to be interwoven with key components of the anxiety/fear, memory, and pain networks such as the amygdala, cortex, hippocampus, periaqueductal grey (PAG), and hypothalamus [57] (Figure 1). Intravenous administration of full CCK2r agonists such as CCK-4 [58] and the sinthetic analogue pentagastrin [59] is panicogenic in healthy volunteers and worsens symptoms in panic attack patients. Functional magnetic resonance imaging in humans points to a cerebral activation in anxiety-related brain regions following CCK-4-induced panic challenge [60, 61]. In rodents, CCK-induced anxiety is linked to CCK2r activation at the basolateral amygdala (BLA) [28, 62, 63–65] and cerebral cortex [29, 66].
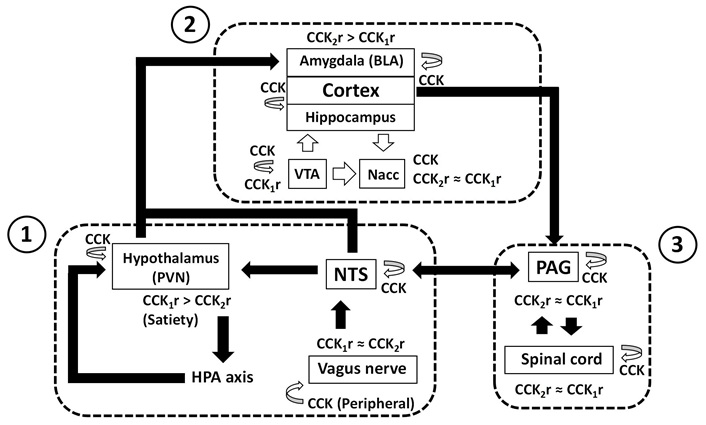
The CCKergic system across anxiety, pain, and memory networks. The diagram illustrates the primary brain networks (framed with dashed lines) that regulate: (1) anxiety and memory at the body-brain interface; (2) cortical processing of anxiety, pain, and memory; and (3) anxiety-pain interactions between the mesencephalon and spinal cord. Solid arrows represent main network paths, while empty arrows imply CCK release. With the exemption of the cortical CCKergic projections to the nucleus accumbens and the mesolimbic and mesocortical dopaminergic and CCKergic projections from the tegmental ventral area, CCK release often happens in local circuits responsible for specific neuronal processing. Additionally, the diagram displays the coexpression of both subtypes of CCK receptors (≈), as well as the prevalence of a certain subtype (>). References [14, 29, 47, 85, 88, 93–95, 98, 100, 103, 107–109, 114, 162, 163, 165, 166, 170–179, 181–184, 187] were used in the diagram construction. CCK: cholecystokinin; BLA: basolateral amygdala; VTA: ventral tegmental area; Nacc: nucleus accumbens; PVN: paraventricular nucleus of the hypothalamus; NTS: nucleus of the solitary tract; HPA: hypothalamic-pituitary-adrenal; PAG: periaqueductal grey
Glutamate-GABA harmony plays a critical role in anxiety [67, 68], pain [69], and memory [70–72]. CCK is interspersed in the excitatory-inhibitory neural circuits of limbic cortices [73, 74]. Enhanced neuronal excitability may be one of the mechanisms by which the selective CCK2r activation causes anxiety and panic attacks in humans [75]. GABA release is crucial for regulating anxiety and fear processing in BLA [62, 76], hippocampus [77, 78], and cerebral cortex [48]. GABAergic inhibition is modulated by CCK2r [62, 79, 80] and to a lesser degree by CCK1r [29, 62]. CCK controls glutamate [81, 82] and GABA [83] release in the hippocampus. When anxiety is expressed, CCK controls electrical activity in the cortex [29]. Similar CCK-mediated mechanisms play an important role in cognition [11, 84]. By facilitating glutamate release and gating GABAergic basket cell activity in the hippocampus, CCK regulates memory rather than encoding it [82].
In inflammatory pain, the CCK/CCK2 system of the central amygdala switches from an anxiogenic to analgesic role that implicates descending control to the spinal cord [85]. CCK takes part in the descending pain facilitation system, particularly in the rostral ventromedial medulla and spinal cord [86, 87]. CCK contributes to pain hypersensitivity and is implicated in the nocebo effect [8]. This effect is likely to be mediated by the CCK input from the anterior cingulate cortex to the lateral PAG [88]. Interestingly, the CCK2r is thought to be responsible for anxiety-induced hyperalgesia states in this structure [89], since it mediates the anxiogenic effect of CCK [90–92]. Integrating aversive memories and mediating defensive and emotional states, including fear, anxiety, and pain, may be important functions for CCK in the PAG [93].
The hypothalamic CCK plays a role in mediating stress responses, particularly the hypothalamic-pituitary-adrenal (HPA) axis [94–96] and stress-induced suppression of appetite [97]. Corticotropin-releasing hormone (CRH) and CCK are strongly related in the human CNS [98]. There is also evidence that the hypothalamus acts as the primary coordinator of memory updating [99]. HPA alterations impact on memory [100]. The paraventricular nucleus of the hypothalamus (PVN) is a major site of CCK concentration in the hypothalamus and where CRH neurons express CCK [101]. The CRH type 1 receptor or CRHR1 interacts with CCK to trigger anxiety [102]. Therefore, it is not extrange that PVN, a node for the CCK-regulated stress responses [103], is also one of the significant sites of glucocorticoid negative feedback regulation of the HPA axis [104]. Besides glucocorticoid feedback via bloodstream, PVN receives projections from the hindbrain neurons in the nucleus of the solitary tract (NTS) [105], the port of entry of vagal afferents.
The vagus nerve, which presents both CCK1r and CCK2r [106], is the route that uses intraperitoneal CCK to enhance memory retention [107, 108]. CCK activating vagal afferent C fibers enhances memory consolidation and retention involved in long-term visceral negative affective state like in irritable bowel syndrome [109]. Peripheral CCK may work partially through centrally projecting neurons from the nucleus tractus solitarius [110], since it has been connected to the activation of brain stem neurons, amygdala, and hypothalamus [111]. In contrast to the central CCK, which uses CCK2r found in the hippocampus to produce its mnemonic effects [47, 48, 56, 112, 113], peripheral CCK may aid in memory formation via CCK1r [114, 115]. Brain-derived neurotrophic factor is likely to be an intermediate of vagus nerve/CCK-1R-mediated memory [116].
Growing preclinical evidence points to an impact of CCK on memory [41, 45, 51, 53, 55, 56, 81, 112, 117–119]. NMDA receptors promote CCK release in the cerebral cortex [120] and the hippocampus, where it switches long-term potentiation [121]. Through neuroplasticity, memory offers tools to rewire anxious brain patterns, lowering hypervigilance, and encouraging more balanced responses [122–124]. Similarly, rather than merely being a moment-to-moment appraisal of a nociceptive input, perception of pain is a dynamic process that is influenced by prior experience and basal anxiety levels [8, 125]. Trace fear memory development is facilitated by neuroplasticity processes through CCKergic projections terminals of the anterior cingulate cortex into the lateral amygdala [126]. Anticipatory stress may be impacted by CCK’s role in fostering associative memory [47, 127, 128]. This implies that, via modulating memory and neural plasticity, CCK may have a great impact on anxiety/fear [126] and nocebo pain effect [8].
Neuronal homeostasis refers to the nervous system’s ability to maintain a stable internal environment and regulate neuronal firing, ensuring proper function and adaptation to changing conditions. CCK would carry out its neuronal homeostatic function in four ways: (1) Homeostatic plasticity mechanisms; (2) interaction with retrograde endocannabinoid signaling; (3) neuroprotection; and (4) dynamic neuromodulation of CCK release occurring after high-frequency neuronal firing (for a summary, see Table 4).
Summary of the key research on CCK-driven neuronal homeostasis
| Process | Main findings | Brain region | References |
|---|---|---|---|
| Homeostatic plasticity (I): synaptic scaling | CCK colocalizes with glutamate neurons and controls glutamatergic excitatory projections and local GABAergic basket cells that gate signal flow and modulate network dynamics | Cortices, hippocampus, amígdala, ventral tegmental area | [62, 73, 74, 79, 80, 82, 83, 153] |
| CCK stimulates glutamate release and promotes long-term potentiation | Cortices, hippocampus, amygdala | [46–48, 81, 82, 120, 121] | |
| CCK shifts the plasticity of GABA synapses from long-term depression to long-term potentiation | Hipothalamus | [50] | |
| Homeostatic plasticity (II): intrinsic excitability | CCK-8 enhances acid-sensing ion channel currents in primary sensory neurons | Spinal cord | [130] |
| Endocannabinoid interactions | Coupling of CCKergic interneurons co-expressing CB1 receptors is involved in the generation and stability of rhythmic synchronous network activity of the hippocampal CA1 subfield | Hippocampus | [136] |
| CB1 and CCK2 receptors work together to modulate cortical GABAergic release in opposite ways | Cortex, periaqueductal grey | [80, 137] | |
| Neuroprotection | CCK triggers anti-oxidative stress pathway | Striatum, substantia nigra | [146] |
| CCK inactivates pro-inflammatory microglia response | Medial prefrontal cortex, caudate-putamen, hippocampus | [147] | |
| Dynamic neuromodulation of CCK release | Serotonin induces CCK release via 5-HT3R | Cortex, nucleus accumbens | [153] |
| GABA regulates CCK release | Cortex | [156] | |
| NMDA receptors promote CCK release | Cortex, hippocampus | [120, 121] | |
| Dopamine controls CCK release | Striatum | [157] | |
| Endogenous opioids mediate CCK release | Spinal cord, frontal cortex | [158, 159] |
CCK: cholecystokinin; CB1: cannabinoid type-1
Synaptic scaling and intrinsic excitability changes are crucial components of neuronal homeostatic plasticity, since they stabilize firing and overall network function around a set-point value in the face of activity fluctuations [129]. CCK can influence synaptic transmission, potentially through synaptic mechanisms (affecting glutamate and GABA release) or postsynaptically altering receptor sensitivity. CCK can alter the properties of ion channels, impacting intrinsic excitability, that is, the neurons’ firing threshold and firing rate. In the context of chronic pain, CCK-8 enhances acid-sensing ion channel currents in rat primary sensory neurons [130]. Through its remarkable cell-specific modulation of excitatory and inhibitory signals and synaptic transmission, the CCK system can influence these mechanisms, contributing as an essential molecular switch to regulate the functional output of neural circuits [74, 131]. Synaptic plasticity is an interesting aspect of neuronal homeostasis in which endogenous CCK release could hypothetically operate to ameliorate the impairment induced by stress to synaptic plasticity [132].
The endocannabinoid system plays a significant homeostatic role in brain functions [133]. Cortical CCK+-GABA basket cells, which exert perisomatic inhibition of pyramidal cells [73, 74, 134], are downstream of the activation of cannabinoid type-1 (CB1) receptors in the forebrain [135]. The endocannabinoid system is involved in the generation and stability of rhythmic synchronous network activity of the CA1 region of the hippocampus that impacts cognitive processes, which is mediated by the chemical and electrical coupling of CCK interneurons co-expressing CB1 receptors [136]. Additionally, CB1 and CCK2 receptors work together to modulate cortical GABAergic release in opposite ways in the cortex, making them relevant to anxiety [80]. The similar thing occurs with CCK1r in the PAG that can both oppose and reinforce opioid and cannabinoid modulation of pain and anxiety within this brain structure [137]. Lastly, the amygdala projection CCK+-glutamatergic neurons to the nucleus accumbens, which regulates mood stability, have CB1 receptors [138]. Thus, CB1 receptors widely mediate endocannabinoid effects on glutamatergic and GABAergic transmission to modulate cortical networks and the expression of anxiety and fear [139]. It is likely that fear-related psychiatric diseases may be the result of the dysfunctional CCK-CB1 homeostatic interactions [140–142].
Homeostatic mechanisms protect neurons from harm, particularly during times of stressful and chronic pain situations, as well as prolonged and intense cognitive efforts accompanied by stress (i.e., cognitive overload). Chronic stress and pain have detrimental effects on the limbic system [143]. Oxidative stress may be a major component of anxiety pathology [144], while chronic pain leads to the weakening or loss of these synaptic connections, leading to maladaptive changes in the brain [145]. Neuroprotection by CCK can occur through an anti-oxidative stress mechanism [146] or by the anti-inflammatory inactivation of microglia through CCK2r [147]. Preclinical evidence suggests that CCK could even help with depression, Parkinson’s and Alzheimer’s diseases through CCK2r [76, 82, 148–150] and cerebral ataxia via CCK1r [151].
The fact that CCK release mostly monitors neuronal activity and that no single neurotransmitter directly causes it reinforces CCK’s role in modulating certain aspects of neuronal homeostasis. It is known that the interaction of CCK with serotonin in the cortex under aversive conditions [152], and that serotonin functions as a strong CCK release factor in the cerebral cortex and nucleus accumbens by activating 5-HT3 receptors on the CCK-releasing terminals [153]. In the ventral tegmental area, CCK is released from the somato-dendrites of dopamine neurons, triggering long-term potentiation of GABAergic synapses onto those same dopamine neurons [154] and dopamine release in the nucleus accumbens and the amygdala via CCK1r [155]. GABA regulates CCK release in the cortex [156], while dopamine controls CCK release in the neostriatum [157], and opioids mediate CCK release in the spinal cord [158] and frontal cortex [159]. Through its ability to colocalize and interact with these neurotransmitters, CCK is actually in the position to modulate a broad range of behaviors and functions.
The most illustrative example is the homeostasis of the opioid system by CCK [11, 39]. CCK2r activity accounts for neuropathic pain [160] and the development of opioid tolerance and/or dependence after chronic administration of opioids [158]. CCK1r also contributes to the anti-opioid action of CCK [161, 162] and visceral pain at the level of dorsal root ganglia [163], but the cooperative stimulation of both CCK1r and CCK2r produces modest opioid-like effects [164, 165]. CCK-opioid interactions are in the onset and manifestation of stress-induced hyperalgesia [89], morphine withdrawal-induced stress [166], and addiction [37].
The dynamic neuromodulatory action of CCK through two receptors [10], its neuroplasticity role [53], and the involvement in overlapping brain processes (anxiety, pain, and memory) suggests that the CCKergic system is a network of three main components (CCK, CCK1r, and CCK2r) interacting together to restore baseline neuronal function. The brain produces C-terminal sulfated octapeptide fragments of CCK-8 or CCK-8S, one of the major neuropeptides in the brain, followed to a lesser degree by CCK-4 [167], which is released in distinct limbic regions under anxiety [168]. The distribution of CCK1r and CCK2r across the peripheral and central nervous system differs, as do their affinities for these fragments. Though sparsely distributed in the brain, CCK1r is highly selective for the CCK-8S, whereas CCK2r is more common in the brain but less selective due to its interchangeable binding with CCK-8S and CCK-4 [169]. The components of the CCKergic system would be positioned conveniently over several network subdivisions responsible for different brain processes, resulting in a particular pattern of CCK1r and CCK2r activation across subdivisions depending on where and how CCK-8S and CCK-4 are released (Figure 1). Any imbalance in the CCKergic system could change how the brain processes memory, pain, and anxiety.
Several neural pathways were connected in this model to bolster the CCKergic system hypothesis. One of them is the HPA axis, which is regulated by CCK in the human brain [98], and whose disruption can lead to alteration of neuronal homeostasis [170]. The HPA axis plays a crucial role in regulating body stress response, including its impact on anxiety and memory [100]. CCK function on the HPA axis might be accomplished through CCK2r [94, 95] (Figure 1). The vagus nerve, which contains both CCK receptors [103], and the NTS, which expresses CCK [171], form the brain-gut axis, the main brain-body communication [172]. Since the NTS is an essential autonomic integration center with reciprocal connections with the PAG, the HPA axis, and the amygdala [173], its participation in CCK-mediated anxiety and memory cannot be excluded [107–109, 174] (Figure 1). It should come as no surprise that endogenous peripheral and brain CCK, each activating unique neural circuits, have anxiogenic and anxiolytic effects respectively through the CCK2r [174, 175], whereas the inhibition of peripheral CCK impairs memory [174].
In the cerebral cortex, NMDA receptors trigger CCK release [47]. Although CCK2r is regarded as the brain-specific receptor, the electrical activity of local neuronal networks in the fronto-parietal neocortex [176] and hippocampus [177] is under the control of diffuse populations of CCK1r. The intercalation of CCK-expressing neurons with excitatory and inhibitory neuronal circuits of the limbic system controls dopamine-mediated neurotransmission [14] (Figure 1), which in turn modulates anxiety-like behaviors [178] and memory [179]. Rat anxiety-like behaviors are undeniably mediated by CCK2r- [180], while CCK1r antagonists also have anxiolytic effects [181, 182]. Both of these effects seem to depend on cortical CCK receptors [29]. The enhancing effects of CCK in memory [183] also require the participation of both receptors, albeit through different pathways [114]. Thus, CCK1r agonists and CCK2r antagonists both enhance memory in an olfactory recognition test in the rat [184]. This could be the reason why the injection of the selective CCK2r agonist BC264 into the nucleus accumbens impairs memory in the rat [185]. Short-term memory is affected in healthy volunteers by the panicogenic agent CCK-4, a full CCK2r agonist [186]. While CCK-4 is raised during stress [168], high levels of CCK-8S during the induction of stress can mitigate the detrimental effects of stress on hippocampal synaptic plasticity and memory [147]. This demonstrates how CCK1r and CCK2r work in conjunction to support CCK function at the cortical intersection of anxiety and memory [29].
The cortical CCKergic system may contribute to pain modulation [187] through the CCK/CCK2r system within the amygdala [85] and likely by the connections of the anterior cingulate cortex to lateral PAG [88]. In the rat, CCK microinjection into the ventrolateral and dorsolateral PAG produces anxiolytic-like and anxiogenic-like effects, respectively [93]. Additionally, through the activation of CCK2r, CCK exerts its pronociceptive and anxiety-induced hyperalgesia effects in the PAG [89, 188]. Spinal CCK1r also contributes to the anti-opioid action of CCK [161, 162]. Because CCK1r and CCK2r produce modest opioid-like effects [164, 165], it is anticipated that coordinated activation of both receptors across the cortex-PAG-spinal cord axis will play a role in the nexus of anxiety and pain (Figure 1).
Neuronal homeostasis over a wide range of temporal and spatial scales requires dynamic plastic changes of neuronal and circuit activity [189]. Because of their active neuromodulatory and neuroplasticity roles, the elements of the CCKergic systems work together to support cognition and affective regulation [10, 48, 53, 64, 76]. During neuronal homeostasis, compensatory plastic changes of the CCKergic system could go awry. Therefore, it should not come as a surprise that certain cortical areas of the schizophrenic brain exhibit abnormal CCK mRNA expression [190, 191], and that the cerebral cortex of suicide victims shows abnormally elevated CCK2r binding [192]. In the rat, interindividual differences in “novelty-seeking”—a behavioral trait associated with anxiety and addiction—are influenced by varying expression of CCK elements across the limbic system. [193, 194]. Restoring the proper function of the entire CCK system, rather than just a single component, may be necessary for the correction of certain mental conditions.
The link between anxiety and CCK2r expression is not as straightforward as it was first thought [195]. Even if intravenous administration of full CCK2r agonists [58] is panicogenic in healthy volunteers and worsens symptoms in panic attack patients, CCK2r antagonists have not been proven to alleviate panic attacks [24, 25]. They are also ineffective in generalized anxiety disorder [23]. Even worse, patients with panic disorder receiving long-term treatment with the drug had an unforeseen higher baseline incidence of panic attacks than those receiving a placebo [24]. The findings are also controversial when it comes to pain management. The potent CCK agonist ceruletide is characterized as a robust analgesic [196], whereas the CCK antagonist proglumide ameliorates neuropathic pain [197], potentiates opioid analgesia [198, 199], and inhibits the nocebo effect [8]. In contrast to preclinical predictions, morphine-induced analgesia is not increased by a full CCK2r antagonist [26]. It follows that the intended therapeutic effect might not be achieved by specifically targeting the brain-specific CCK2r. It might be more appropriate to take into account additional components of the CCKergic system, such as CCK1r [200, 201]. The scientific evidence gathered in this review regarding the intricate neuromodulatory and neuroplasticity functions of the CCK neuropeptide supports the idea that certain affective, pain, and cognitive disorders, and even neurological conditions like epilepsy [202, 203] may be associated with dysregulations of the homeostatic CCKergic system, rather than an overactive CCK2r-mediated neurotransmission.
Novel compounds that have a combination of CCK agonist and antagonist activities could be a good addition to existing mental health drugs. These compounds have the ability to simultaneously affect multiple pathways within the CCKergic system. Combining the two activities may be able to get around the drawbacks of single-target strategies [204] and provide more extensive therapeutic advantages. Given that CCK1r can control imbalances in dopaminergic neurotransmission [14, 155], a compound with CCK1r agonism and CCK2r antagonism may potentially lessen anxiety while influencing other aspects of mood regulation [192]. In a similar vein, these compounds could offer a fresh strategy for managing psychotic symptoms [13–15]. Furthermore, different individuals may exhibit varying degrees of CCK-ergic activity [193, 194] and receptor subtype activity. Compounds with combined agonist/antagonist activity could potentially be tailored to address the individual differences.
Finally, a fascinating but little-known pharmacological feature that merits additional investigation is the discovery of positive allosteric modulators that target the physiologic spatial and temporal engagement of CCK1r by CCK [205]. The synthesis of a single CCK-based ligand with several off-targets is another intriguing research avenue. For example, bifunctional peptides that act as agonists on δ and μ opioid receptors as agonists and with CCK receptors as antagonists provide a good alternative for the treatment of chronic neuropathic pain [206].
In conclusion, the pharmacological development of such putative CCKergic agents is associated with abnormal dopamine and opioid neurotransmitters like schizophrenia [14, 191], depression [16, 192], and addiction [207].
BLA: basolateral amygdala
CB1: cannabinoid type-1
CCK: cholecystokinin
CCK-4: cholecystokinin tetrapeptide
CCK-8S: sulfated cholecystokinin octapeptide
CRH: corticotropin-releasing hormone
HPA: hypothalamic-pituitary-adrenal
NTS: nucleus of the solitary tract
PAG: periaqueductal grey
PVN: paraventricular nucleus of the hypothalamus
SJB: Conceptualization, Investigation, Methodology, Validation, Writing—original draft, Writing—review & editing.
Not applicable.
Santiago J. Ballaz, who is the Guest Editor of Exploration of Drug Science, had no involvement in the decision-making or the review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

