 Original Article
Original Article

Affiliation:
Department of Biotechnology, Vignan’s Foundation for Science, Technology and Research, Vadlamudi 522213, Andhra Pradesh, India
Email: krupanidhi.srirma@gmail.com
ORCID: https://orcid.org/0000-0001-6161-8785
Explor Immunol. 2021;1:155–165 DOI: https://doi.org/10.37349/ei.2021.00012
Received: June 04, 2021 Accepted: August 13, 2021 Published: August 31, 2021
Academic Editor: Wangxue Chen, National Research Council Canada, Canada
The article belongs to the special issue Vaccine-induced Immune Responses Against SARS-CoV-2 Infections
Aim: The envelope protein of novel coronavirus 2 (nCoV2) was reported to be highly conserved compared to its spike (S) protein which was shown to undergo several alterations in their amino acid sequences in the span of one year (2020–2021). Therefore, it is aimed to consider highly conserved structural protein of nCov2 namely envelope (E) protein to design the polytope for the formulation of the vaccine against coronavirus disease 2019 (Covid-19).
Methods: Online in silico tools were employed to decipher the conservancy and antigenicity of E-protein of nCoV2. They are: to evaluate the molecular affinities among the chosen representatives of alpha and beta coronaviruses, the Molecular Evolutionary Genetics Analysis (MEGA) X 10.1.1 was used. Immune Epitope Database (IEDB)-NetMHCpan (ver. 4.1) tool was used to predict the epitopes of E protein binding to the frequently distributed major histocompatibility complex (MHC) I alleles. ProtParam, VaxJen, ToxinPred and AllerTop online tools were used to assess the physicochemical features, antigenicity, non-toxin and non-allergen aspects of constructed polytope. Secondary structure analysis and homology modelling validation of polytope were done using Phyre2 online tool. Discontinuous and linear epitopes of the designed polytope were predicted through IEDB Ellipro tool. Population coverage of epitopes of the polytope was performed using IEDB online tool with the frequent distribution of human leukocyte antigen (HLA) I alleles in the South Indian Asian population.
Results: The phylogeny of envelope proteins of chosen representatives of Coronaviridae confirmed its conservancy and possible origin of nCoV2 from alpha coronaviruses through vampire CoV2. The designed polytope of E-protein was with 53 amino acid residues. The same was developed by linking with cysteine and serine (CS) residues in between epitopes.
Conclusion: The antigenicity, non-allergen, non-toxin, homology modelling, discontinuous and linear epitopes of the designed polytope authenticate to explore the envelope protein for prophylactic measures. The epitopes of polytope were found to restrict to MHC I alleles occurring frequently among South Indian Asians.
The coronavirus disease 2019 (Covid-19) is an unanticipated pandemic and it became uncontrolled for a period of six months in the beginning of the year 2020 [1]. Its severity was witnessed by losing several fellow men, women and children of all ages. People experienced both its first wave and also recurrence of second wave in a short duration with the same intensity of severity. To some extent, the treatment regimens which included supplementation with oxygen, quarantine, plasma therapy, anti-inflammatory, and anti-viral drugs saved more than half of the victims. In the long run, to avoid Covid-19 recurrence, it is advisable to take precautionary measures such as the production of prophylactic vaccines and mass immunization.
Several scientists across the world have come out with the design and formulations of vaccines using the S protein of nCoV2 [2–4]. A few biopharma industries also have made vaccines with the S protein as a main target antigen. However, it is noticed that there is the viral escape due to mutations in amino acid residues of S protein [5–7] and hence the designed vaccines may not be totally appropriate. As this phenomenon of mutation is nurtured by natural selection, an alternative strategy needs to be taken to avoid its recurrence. Hence, in the present article, the envelope (E) protein of nCoV2 which is highly conserved and small structural protein is considered to design a polytope.
In brief, envelope protein of coronaviruses is designated as E protein. This is found as the smallest among the structural proteins of coronaviruses. It plays a significant role in the life cycle of virus which includes the assembly, envelope formation, pathogenesis and budding [8, 9]. By participating as viroporin, E protein allows the flux of ions and enzymes for its replication [8, 9]. Further, it is found to prompt pro-inflammatory pathways. Abdelmageed et al. [8] have designed a multiepitope-based peptide vaccine using the E-protein and suggested as the promising candidate for prophylactic vaccine to cover the population of China, Europe and East Asia. Further, these authors have designed ten peptides from E protein and evaluated molecular docking of ligand epitopes with HLA-A*02.01 (human leukocyte antigen). In yet another instance, Tilocca et al. [9] have shown the major immunogenic domains of E protein. Therefore, considering the importance of E protein to build in the literature, in the present study the E protein of nCoV2 is chosen to explore its conservancy and design of polytope to meet the on-going challenges in the Covid-19 pandemic.
The representative envelope protein sequences, each with ~75 residues, of Coronaviridae were browsed from GenBank database. A total of eight envelope protein sequences comprising of Middle East respiratory syndrome (MERS), severe acute respiratory syndrome coronavirus(SARS CoV) 1 and SARS CoV2 were selected. The aim of choosing the representative sequences was to assess the envelope protein residue conservancy among the chosen sequences, phylogenetic and evolutionary affinities with their possible ancestry. These protein sequences of envelope proteins were retrieved in a notepad in FASTA format. The phylogenetic software suit namely MEGA X 10.1.1 version [10] was used for creating multiple sequence alignment using ‘MUSCLE’ option. The aligned file was exported in Mega format to the desktop.
The multiple aligned sequences of the representative envelope proteins of Coronaviridae in Mega format were retrieved again in MEGA10.1.1 suit and conducted the phylogeny in maximum likelihood method with a bootstrap value of 100. The positions of amino acids in the aligned sequences containing gaps and possible missing data were allowed to eliminate in the chosen method. The resultant phylogenetic tree was retrieved in Pan Gu (PNG) format with the display of bootstrap value at each node along with the branch lengths of each operational taxonomic units (OTU) representing the possible recency of their emergence.
The Immune Epitope Database (IEDB) online tool was employed to derive epitopes of the envelope protein sequence of SARS CoV2 namely gi|1826682072|gb|QIS30437.1| [11]. Upon submission of 75 residues bearing E protein sequence in IEDB analysis resource NetMHCpan (ver. 4.1) tool window with the option of most frequent occurrence of (major histocompatibility complex) MHC I alleles, there resulted an innumerable number of combinations. Of which, the first five epitopes with more than 75% score were chosen in the present study. These five epitopes were linked with a linker comprising of two amino acid residues cysteine and serine (CS) to facilitate in vivo proteasome cleavage and to display as a single unit namely polytope for further physico-chemical analyses, secondary structure determination, homology modelling, validation and population coverage.
The antigenicity of polytope of envelope protein was predicted through http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html. The confirmation of designed polytope free from toxicity and allergenicity were evaluated through online tools https://webs.iiitd.edu.in/raghava/toxinpred/ and http://www.pharmfac.net/allertop respectively.
The on-line tools namely Phyre2 and PEP2D were employed to determine the homology modeling and secondary structure analysis of the polytope of envelope protein of SARS CoV2. The Ramachandran plot, displaying most of the residues of polytope in the favoured region within the phi and psi values namely −40 to −90 and −50 to −10 respectively, indicated the predominance of alpha helix, was retrieved for the polytope through https://saves.mbi.ucla.edu/results?job=715968&p=procheck.
Prediction of epitopes was done using online tool namely IEDB Ellipro. Two sets of discontinuous and three sets of linear epitopes were obtained and the score of each residue of polytope was displayed. The poses showed in silico binding of these predicted epitopes with an antibody.
The IEDB online tool was explored with an input of polytope sequence and the most frequent distribution of HLA class I alleles in South Indian Asian population namely HLA-A*01:01(8.36%), HLA-A*02:01(3.81%), HLA-A*24:01(11.25%), HLA-A*31:01 (6.74%), HLA-B*07:01 (2.29%) and HLA-B*15:01 (3.60%). The resultant values the percent coverage and restriction of each epitope with respective HLAI.
The nCoV2 is having five structural proteins. Of which, S protein in the span of one year (2020–2021) was shown to undergo several alterations in their amino acid sequences [6]. Even then, a few firms are focusing on the preparations of vaccines that are targeting to neutralize the epitopes of S proteins. Hence, these vaccines in the long run may not give an anticipated prophylactic protection to the vaccinated subjects. Therefore, it is aimed to consider highly conserved structural protein of nCov2 namely envelope protein to design the polytope for the formulation of the vaccine against Covid-19.
The eight E protein sequences chosen from Coronaviridae displayed a phylogram (Figure 1) that revealed significant features. There are three primary clusters and two secondary clusters in the phylogram. The two secondary clusters were rooted on the primary cluster 1 (Figure 1) comprising of representatives from alpha coronavirus and Feline coronavirus suggesting that these must have contributed to the evolutionary origin of beta coronaviruses and the branch length of alpha coronavirus is 1.029 indicating that it is an ancient strain. Further, the primary cluster 3 along with its secondary cluster (Figure 1) showed the grouping of the MERS coronaviruses as one cluster, of course due to their similar taxonomic affinities [7]. Interestingly, the off-shoot of SARS CoV1 having a branch length of 1.007 and paired with the cluster 2 which represents SARS CoV2. Both these OTUs have evolved from East Asia. Moreover, the cluster 2 (Figure 1) showed yet another significant observation that the vampire and human SARS CoV2 are paired together with least branch lengths (0.817) showing recency of their emergence with highest affinities.
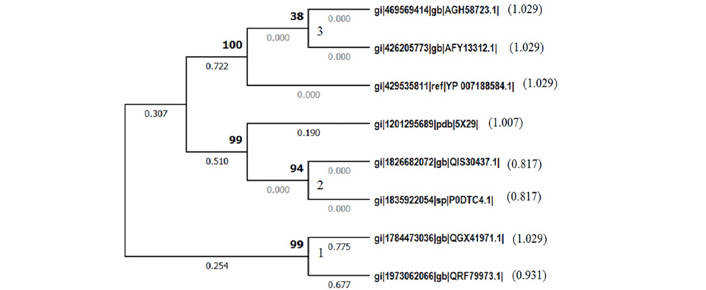
Evolutionary analysis of envelope protein of SARS CoV2 by Maximum Likelihood method conducted in MEGA X 10.1.1. The evolutionary history of representative envelope proteins is inferred by using the Maximum Likelihood method and JTT matrix-based model [1]. The tree with the highest log likelihood (−677.16) is shown. The built-in discrete Gamma distribution is used to model evolutionary rate differences among sites. This analysis involved 8 amino acid sequences. All positions containing gaps and missing data are eliminated.
1. gi|469569414|gb|AGH58723.1| E protein [MERS Jordan-N3/2012] (1.029)
2. gi|426205773|gb|AFY13312.1| E protein [MERS BetacoronavirusEngland 1] (1.029)
3. gi|429535811|ref|YP_007188584.1| [MERS Betacoronavirus England 1] (1.029)
4. gi|1201295689|pdb|5X29| E Chain [SARS CoV1] (1.007)
5. gi|1826682072|gb|QIS30437.1| [SARS CoV2, USA] (0.817)
6. gi|1835922054|sp|P0DTC4.1| Vampire bat_ CoV (0.817)
7. gi|1784473036|gb|QGX41971.1| [Alphacoronavirus sp.] (1.029)
8. gi|1973062066|gb|QRF79973.1| [Feline coronavirus] (0.931) Branch length of each OTU is shown in parentheses
The IEDB online tool was employed for predicting the MHC I binding affinities. The representative envelope protein sequence (gi|1826682072|gb|QIS30437.1|) [11] having affinities with the other two chosen SARS CoVs (Figure 1) was submitted to NetMHCpan (ver 4.1) tool with frequently occurring MHC class I alleles. This yielded innumerable epitopes, of which the first five epitopes with good score in the range of 0.78–0.93 (Table 2) were chosen. They were showing the binding affinities with MHC I. These residues were found located in the range of 51–69 of envelope protein sequence. These epitopes were linked with CS residues to allow the possible in vivo proteolysis and to construct an in silico polytope.
MHC Class I binding predictions of multiepitopes of envelope protein of SARS CoV2 deduced through the IEDB analysis resource NetMHCpan (ver. 4.1) tool with frequently occurring MHC Class I alleles
| Allele | Start | End | Length | Peptide | Score | Rank |
|---|---|---|---|---|---|---|
| HLA-A*31:01 | 61 | 69 | 9 | RVKNLNSSR | 0.939976 | 0.01 |
| HLA-B*08:01 | 57 | 65 | 9 | YVYSRVKNL | 0.894984 | 0.02 |
| HLA-B*15:01 | 51 | 59 | 9 | LVKPSFYVY | 0.890774 | 0.03 |
| HLA-A*02:01 | 50 | 58 | 9 | SLVKPSFYV | 0.846768 | 0.06 |
| HLA-A*02:03 | 57 | 65 | 9 | YVYSRVKNL | 0.783029 | 0.06 |
The physicochemical features of the in silico generated polytope was shown in Table 2. Interestingly, it was shown that its pI was found in the alkaline range and its half-life was shown as one hour. Further, this polytope was found stable with an instability index value of 37.72. The GRAVY that represents hydrophobicity of the polytope was importantly shown within the acceptable range of −2.0 to +2.0 indicating that the designed polytope is more hydrophilic and soluble in aqueous environment. The homology modelling of the polytope shown in Figure 2 was retrieved from the online tool, Phyre2. The predicted model was predominantly with alpha helix, short coils and two turns at the position of proline residues (Figure 3). Majority of residues of the proposed polytope were found placed in the most favourable region in the Ramachandran plot (RP) validating its alpha helical secondary structure (Figure 4). The predicted antigenicity of the polytope was shown to be 0.6456 by VaxiJen tool. The AllerTop tool indicated that the envisaged polytope was found to be non-allergen. The ToxiPred online tool predicted the designed polytope as non-toxin (Table 3). The ToxiPred also indicated the reasonable values of hydrophilicity that would facilitate polytope to interact in an aqueous environment. The discontinuous and linear epitopes of polytope envelope protein were predicted using IEDB-Ellipro online tool (Table 4). There were two sets of discontinuous and three sets of linear epitopes predicted as mimotopes which showed binding poses with an antibody (Figure 5). The MHC I alleles of Indian Asian population was considered to validate the envisaged epitopes to restrict their binding with a reasonable affinity score and percent coverage as shown in Figure 6 and Table 5.
Physicochemical features of polytope are derived using online ProtParam tool (https://web.expasy.org/cgi-bin/protparam/protparam)
| Number of amino acids in the polytope | MW | pI | Total number of negatively charged residues (Asp+Glu) | Total number of positively charged residues (Arg+Lys) | Estimated half-life in mammalian reticulocytes in vitro | II | Aliphatic index | GRAVY |
|---|---|---|---|---|---|---|---|---|
| 53 | 6199.28 | 9.79 | 0 | 9 | 1 hour | 37.72 Stable | 86.04 | –0.004 |
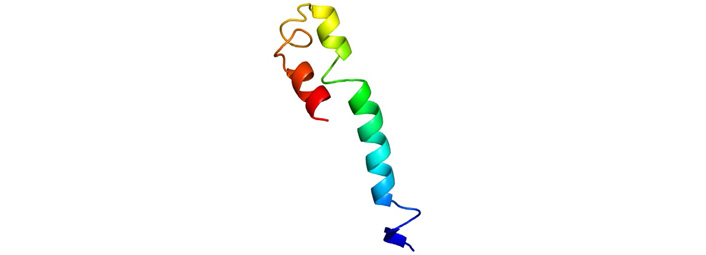
Homology modelling of Polytope of envelope protein derived through Phyre2 online tool
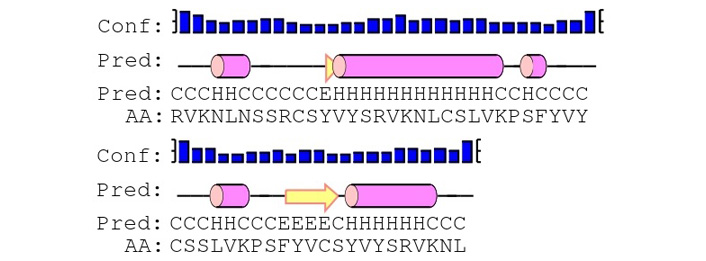
Secondary structure analysis of polytope of envelope protein derived through Phyre2 online tool
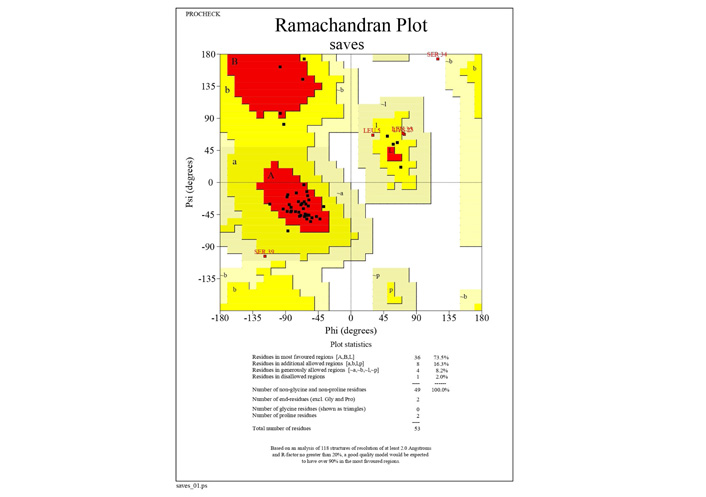
The Ramachandran plot derived through https://saves.mbi.ucla.edu/results?job=715968&p=procheckshowing the distribution of amino acid residues of designed polytope of E protein. 73% of amino acids in the polytope are placed in the most favoured region of the plot. The position of the residues indicates that the polytope is predominantly alpha helix
Prediction of toxicity score of fragmented polytope (< 50 residues) of envelope protein using ToxinPred tool (https://webs.iiitd.edu.in/raghava/toxinpred/multi_submitfreq_S.php?ran=91595)
| Peptide | Peptide Sequence | SVM | Prediction | Hydro-phobicity | Hydro-pathicity | Hydro-philicity | Charge | Molecular Weight |
|---|---|---|---|---|---|---|---|---|
| 1 | RVKNLNSSRCSYVYSRVKN-LCSLVKPSFYVYCSSLV | –0.79 | Non-Toxin | –0.18 | 0.05 | –0.27 | 6.00 | 4263.55 |
| 2 | KPSFYVCSYVYSRVKNL | –1.01 | Non-Toxin | –0.14 | –0.11 | –0.37 | 3.00 | 2166.82 |
Support Vector Machine
Predicted discontinuous and linear epitopes of polytope of envelope protein of SARS CoV2 using http://tools.iedb.org/ellipro/result/predict/
| Predicted Discontinuous Epitope(s) | |||||
|---|---|---|---|---|---|
| No. | Residues | Number of residues | Score | ||
| 1 | _:V2, _:K3, _:N4, _:L5, _:N6, _:S7, _:S8 | 11 | 0.645 | ||
| 2 | _:P26, _:F28, _:Y29, _:V30, _:Y31, _:C32, _:S33, _:S34, _:L35, _:V36, _:K37, _:P38, _:S39, _:Y41, _:V42, _:C43, _:S44 | 5 | 0.577 | ||
| Predicted Linear Epitope(s) | |||||
| No. | Start | End | Peptide | Number of residues | Score |
| 1 | 1 | 9 | RVKNLNSSR | 9 | 0.755 |
| 2 | 28 | 38 | FYVYCSSLVKP | 11 | 0.645 |
| 3 | 40 | 44 | FYVCS | 5 | 0.577 |
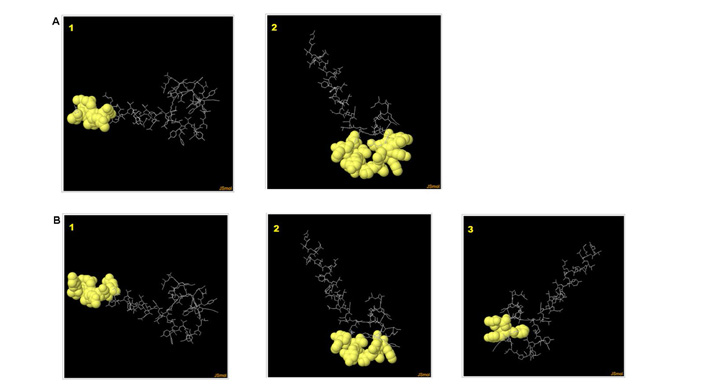
Predicted discontinuous (A) and linear epitopes (B) of polytope shown in poses anchoring to an antibody derived through IEDB-ElliPro software tool
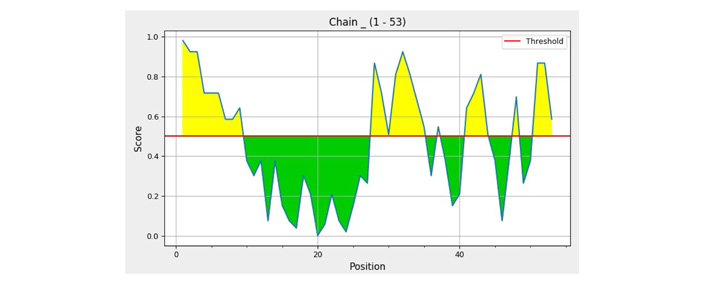
The grid showing the individual residue score of polytope anchoring to an antibody derived through IEDB-ElliPro software tool
Computation of epitopes of polytope restricted to a broad range of MHC I in the Indian Asian population using http://tools.iedb.org/population/result/#India%20Asian
| Epitope | Coverage | HLA allele [genotypic frequency (%)] | Total HLA hits | |||||
|---|---|---|---|---|---|---|---|---|
| Class I | HLA-A*01:01 (8.36) | HLA-A*02:01 (3.81) | HLA-A*24:02 (11.25) | HLA-A*31:01 (6.74) | HLA-B*07:02 (2.29) | HLA-B*15:01 (3.60) | ||
| Epitope #1: RVKNLNSSR | 45.68% | + | + | + | + | + | + | 6 |
| Epitope #2: YVYSRVKNL | 45.68% | + | + | + | + | + | + | 6 |
| Epitope #3: LVKPSFYVY | 45.68% | + | + | + | + | + | + | 6 |
| Epitope #4: SLVKPSFYV | 45.68% | + | + | + | + | + | + | 6 |
| Epitope #5: YVYSRVKNL | 45.68% | + | + | + | + | + | + | 6 |
| Epitope set | 45.68% | 5 | 5 | 5 | 5 | 5 | 5 | 30 |
+: restricted
In the 21st century we witnessed a major devastation due to nCoV2 [1, 12]. Of course, the sophistication in recombinant techniques, bioinformatics tools and social media, have made us awaken and appreciate the intricacies of Covid-19 within a short time of its emergence. The nCoV2 is one among the unique taxonomic groups in the family Coronaviridae within beta coronaviruses with zoonotic origin [6, 7]. It has become highly virulent because of its RBD binding to ACE2 receptors of humans present in the alveolar lung epithelial cells [12–14]. The evolutionary affinities built for nCoV2 based on E protein amino acid sequence showed that it had its origin from East Asia (Wuhan) and again from the animal source [6, 7]. The present study further confirmed through a phylogram (Figure 1) shown with branch lengths reflected that nCoV2 and Vampire CoV2 coupled with SARS CoV1 took the origin from alpha coronaviruses. Importantly, nCoV2 and vampire CoV2 have paired as a single cluster in the phylogram (Figure 1) as closely related members of the family which confirms in compliance with the published reports that human nCoV2 is zoonotic in its origin [7, 12] and it is recent as revealed by the least value of its branch length.
The multi-epitope based design of vaccines considering the spike protein amino acid sequences of nCoV2 have come into vogue since the beginning of the year 2020 from several laboratories [15–19]. Further, the production of vectored vaccines embedded with the gene of S protein and mRNA vaccine with the transcripts from the gene of S protein have been released into the market focussing only on one of the prominent structural proteins of nCoV2 ignoring the fact that the S protein is subjected for rapid mutations as shown by Global Initiative on Sharing All Influenza Data (GISAID) [6]. As a result, the other best alternative to be considered at this juncture is the conserved structural protein namely E protein of nCoV2. Therefore, in the present study the designed polytope of E protein is with 53 amino acids having both discontinuous and linear epitopes, non-toxin, non-allergen with potential for antigenicity and predicted MHC I binding with physico-chemical features for its solubility and stability, all that authenticate well to suit to the destined South Indian Asian population.
In an interesting study, the five structural proteins including E protein of nCoV2 were selected to design a 9 amino acid residue peptide as an epitope [6–9]. Tilocca et al. [9] employed immunoinformatics tools to deduce immunogenic domains in the E protein. Due to extensive mutations appearing in the nCoV2, ten peptides of E protein were projected as a multiepitope-based vaccine [8]. The values obtained for “probable antigen” for the polytope of E protein in our study is 0.6456, whereas similar value reported for the complete E protein was found to be less [8], however, the same authors reported that E protein ranked as the top in the prediction of “probable antigen” among other structural proteins namely M, S and N [17–19]. Most importantly, the SARS-CoV2 of Indian strains are renamed by WHO as Delta and Kappa labels [20] indicating the need for a wide spectrum based prophylaxis. Therefore, the author through this article strongly advocates that the E protein of nCoV2, containing a highly conserved sequence among the members of the family Coronaviridae, for the preparation and formulation of vaccine.
In conclusion, the envelope protein of SARS CoV2 displayed conservancy in its sequence and phylogenetic affinities with SARS CoV1 and vampire CoV2 as evidenced by appearing in one secondary cluster shown in the generated phylogram. The designed polytope of E protein is found as non-allergen, non-toxic and antigenic with the most favoured homology model showing its solubility, stability, MHC I binding and anchoring of the discontinuous and linear epitopes to an antibody. Each predicted epitopes restricted to MHC I alleles, frequently occurring in South Indian Asians, showed its potential as a possible vaccine candidate for formulation.
Covid-19: coronavirus disease 2019
E: envelope
HLA: human leukocyte antigen
IEDB: Immune Epitope Database
MEGA: Molecular Evolutionary Genetics Analysis
MHC: major histocompatibility complex
nCoV2: novel coronavirus
S: spike
SARS CoV: severe acute respiratory syndrome coronavirus
The author profusely acknowledges the management of Vignan’s Foundation for Science, Technology and Research (Deemed to be University) for providing the facility through Centre of Excellence and DST FIST Net-working lab (LSI-576/2013).
The author contributed solely to the work. The concept developed and content projected in the present article are original and prepared by the author.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Available with the author.
No funding.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 4383
Download: 30
Times Cited: 0
Kabeer Haneef ... Zainab Fatima
Eduardo Tosta
Eknath D. Ahire, Sanjay J. Kshirsagar
Kenneth Lundstrom
Sevilay Hintistan, Hatice Demirağ
Mandeep Garg ... Suruchi Garg
Swapan K. Chatterjee ... Maria Nilda M. Munoz
Marileia Chaves Andrade ... Hellen Oliveira Rosa
Devlina Ghosh ... Abhishek Saxena
Yoanna Slabakova ... Tsvetelina Velikova
Balram Ji Omar ... Manju O. Pai
Basista Rabina Sharma, P. Veeranna Ravindra
