 Review
Review

Affiliation:
1Tumor Immunology and Immunotherapy Group, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC-TMC), Navi Mumbai 410210, India
2Homi Bhabha National Institute, Anushaktinagar, Mumbai 400094, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0002-6485-0691
Affiliation:
1Tumor Immunology and Immunotherapy Group, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC-TMC), Navi Mumbai 410210, India
2Homi Bhabha National Institute, Anushaktinagar, Mumbai 400094, India
†These authors contributed equally to this work.
Affiliation:
1Tumor Immunology and Immunotherapy Group, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC-TMC), Navi Mumbai 410210, India
2Homi Bhabha National Institute, Anushaktinagar, Mumbai 400094, India
†These authors contributed equally to this work.
Affiliation:
1Tumor Immunology and Immunotherapy Group, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC-TMC), Navi Mumbai 410210, India
2Homi Bhabha National Institute, Anushaktinagar, Mumbai 400094, India
Email: shubhachiplunkar@gmail.com
ORCID: https://orcid.org/0000-0003-3421-0076
Explor Immunol. 2021;1:166–198 DOI: https://doi.org/10.37349/ei.2021.00013
Received: May 02, 2021 Accepted: August 12, 2021 Published: August 31, 2021
Academic Editor: Federico Garrido, University of Granada Medical School, Hospital Universitario Virgen de las Nieves, Spain
Head and neck squamous cell carcinoma (HNSCC) is a relatively widespread cancer with high mortality rates. Many patients with locally advanced disease are treated with combinations of surgery, radiation, and chemotherapy, while others are considered incurable and develop recurrent/metastatic (R/M) disease. Despite these treatment modalities, the 5-year survival rate of HNSCC has remained at 50% due to limited treatment options in patients with recurrent disease. Immunotherapy has been shown to induce durable responses in R/M patients, but only a minority of patients currently respond. A major hurdle in tumor immunotherapy is identifying the non-responders and markers to predict resistance in patients who at first responded to the therapy. In HNSCC patients, the tumor microenvironment (TME) assumes a vital role to either diminish or augment immune responses. There is an urgent need for extensive studies to be undertaken to better understand how tumor cells escape immune surveillance and resist immune attack. In this review, the impact of TME on the efficiency of immunotherapy, addressing the factors that mediate therapy resistance are highlighted. The composition of the TME encompassing the immunosuppressive cells including myeloid-derived suppressor cell (MDSC), regulatory T cells (Treg), mesenchymal stem cell (MSC), cancer-associated fibroblast (CAF), and tumor-associated macrophages (TAMs) and intrinsic factors like hypoxia, reactive oxygen species (ROS), extracellular matrix (ECM), angiogenesis, and epithelial-mesenchymal transition (EMT), how this debilitates immunosurveillance, and also discuss existing and potential strategies aimed at targeting these cellular and molecular TME components are reviewed. Understanding the interactions between the TME and immunotherapy is not only important in dissevering the mechanisms of action of immunosuppression but also offers scope for developing newer strategies to improve the competence of current immunotherapies.
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide. HNSCC constitutes a diverse mixture of tumors usually originating in the squamous cells lining the mucosal surfaces of the upper aerodigestive tract which include the oropharynx, oral cavity, hypopharynx, larynx, and paranasal sinuses. Alcohol consumption, tobacco smoking, genetic susceptibility, and human papilloma virus (HPV) infection are risk factors for head and neck cancer. The use of smokeless tobacco in multiple forms including smoked, chewed, applied topically on the gums, or inhaled are major risk factors for HNSCC in Asian countries, particularly in India. Immunotherapy is emerging as an important new route for the treatment of HNSCC in addition to surgery, chemotherapy, and radiation aimed at improving the prognosis of patients [1]. Surgery, chemotherapy, and radiotherapy or a combination are used to treat patients with non-metastatic progressive cancer, however, some patients with recurrent/metastatic (R/M) disease do not respond well to these treatments. The immune system of the HNSCC patients has a major impact on the progression of the disease as well as the response to treatment [2]. A successful immunotherapy regimen involves the activation of the immune system, expansion, and infiltration of the effector cells into the tumor tissue eventually destroying the cancer cells [3]. Cancer immunotherapy involves various strategies to activate the immune system which includes the application of immune agents such as cytokines, vaccines, cell therapies, and use of checkpoint inhibitors (CPI), however, the two treatment modalities that include CPIs and cell-based immunotherapy [4] have received much attention in the treatment of HNSCC and are detailed in this review.
The tumor microenvironment (TME) represents a complex, dynamic network of cells that contribute to tumor progression and metastasis. The TME can also shape the response to therapies and resistance to treatment. Stromal cells, endothelial cells, immune cells, and fibroblasts make up the TME and play a major role in oncogenesis as well as driving intratumoral immune response. The immune cells have tumor promoting as well as inhibitory functions. Pro-tumor immune cells include regulatory T cells (Treg), M2 macrophages, CD4 T helper type (Th) 2 (Th2), and myeloid-derived suppressor cells (MDSCs), on the other hand, natural killer (NK) cells, CD8+ T cells, M1 macrophages, CD4 type 1 helper T cells (Th1), and dendritic cells (DCs) have tumor inhibitory role. Chemokines and cell adhesion molecules regulate the equilibrium between these protumor and antitumor immune cells, which further maintains the tumor milieu. The understanding of “immune contexture” is essential in immunotherapy, the terminology that refers to the immune cell type, location, distribution, and presence of tertiary lymphoid structures (TLS) in the tumor and its microenvironment [5]. TME of HNSCC requires further exploration and understanding using a multiparametric approach.
HNSCC has been characterized into 6 groups: immunoreactive, inflammatory, HPV-like, classical, hypoxemic and mesenchymal [6]. There is a higher infiltration of immune cells into the inflammatory and immunoreactive profiles classified as “hot tumors”, making them more sensitive to immunotherapies. The important tumoral infiltration by the CD8 lymphocytes could further characterize this inflammatory type [7].
HNSCC is one of the few types of cancers categorized as hot tumors. The juxtaposition of these tumors to lymphoid tissue in the Waldeyer’s ring and the exposure to microbiota [especially viruses like HPV, Epstein-Barr virus (EBV)] results in dense infiltrates of immune cells to form an integral component of the tumor categorizing them as “hot” tumors [8]. A number of immune cells and factors secreted by these cells are abundantly found in the HNSCC TME. Both cutaneous skin and aerodigestive tract mucosa are highly immunoreactive organs. In this environment, only the tumor cells which are impervious to the body’s adaptive and innate immune system prevail, following the elimination of other tumor cells.
HPV infection plays an important role in the immunomodulation of HNSCC, influencing the treatment response as well as the immune landscape. HNSCC patients can be categorized as two genetically different groups, HPV infection related (HPV+) and the other alcohol and/or tobacco and oral injury related (HPV–), which display both clinical and biological opposite features. In contrast to the HPV– HNSCC, HPV+ HNSCC frequently occurs in younger patients with negligible or no tobacco exposure [9, 10]. Both HPV– and HPV+ HNSCC have a high prevalence in males showing three to five times higher occurrence in men than women [11]. The 3-year survival rate of HPV+ oropharyngeal HNSCC was 84% compared to the 57%, of HPV– HNSCC [12]. p16 overexpression, cyclin D1, and retinoblastoma (pRB) down-regulation, a low epidermal growth factor (EGF) receptor (EGFR) expression with a high proliferating index [Antigen KI-67 (Ki-67)] are among a few characteristic features of HPV+ tumors compared to HPV– tumors [13, 14]. The variability in the genetic and immune characteristics of HNSCC is associated with the carcinogenesis process that is initiated by tobacco/alcohol and HPV infection. HPV+ HNSCC demonstrates a relatively inflamed immune microenvironment comparedto HPV– HNSCC[15]. Although the treatment approaches (combinationofsurgery, radiotherapy, and chemotherapy) remain the same, HPV+ HNSCC shows a better prognosis and treatment response than HPV– HNSCC. HPV+ HNSCC has a less immunosuppressive TME, as shown by higher infiltration of CD4, CD8, lower Tregs, lower CD4/CD8 ratio compared to their respective levels in HPV– HNSCC. HPV+ HNSCC expressed higher densities of CD20+ B cells with reduced immunosuppressive regulatory B cells [16]. TLS with a higher number of B cells have been observed in HPV+ HNSCC compared to HPV– HNSCC [16].
Expression of immune checkpoint molecules like programmed cell death protein 1 (PD-1) and programmed cell death ligand (PD-L) 1 are also upregulated in HPV+ HNSCC compared to HPV– HNSCC [17]. Tumor mutational burden (TMB) has emerged as a potential biomarker of response to immune CPI in several clinical trials carried out across various cancer types [18–23]. In HPV– HNSCC increased TMB and neoantigen load correlated with response to CPI, however predictive value of these markers in HPV+ HNSCC were contested by other reports [24].
A higher incidence of chromosomal changes is seen in HPV– HNSCC, while HPV+ HNSCC gets its distinct genetic phenotype due to integration of the early (E6 and E7) viral oncoproteins and subsequent genetic alterations [25]. Tobacco and alcohol incite several DNA mutations, additionally modifying the TME. The efficiency of immunotherapy is affected by these alterations in TME and may diminish the response rate to immunotherapy observed in many HNSCC patients [26, 27]. HPV+ HNSCC patients showed better response to PD-1 inhibitor pembrolizumab, as seen in the KEYNOTE-012 trial [overall response rate (ORR) was 32% for patients with HPV+ and 14% for HPV– patients] [28]. Another phase II trial using durvalumab (PD-L1 inhibitor) observed better survival and ORR of 29.4% in HPV+ HNSCC compared to 10.9% in HPV– patients [29]. However, phase III KEYNOTE-040 trial and other clinical trials were not able to reproduce these observations. The inconsistencies could be attributed to other factors like smoking, mutational signatures, and TMB which may have influenced response to CPI.
Resistance to CPI treatment is observed in about 85% of patients with HNSCC, consequently calling attention to understanding the mechanisms underlying HNSCC immune escape [30]. Careful selection of patients based on their “immune contexture” is the key to the success rate of immunotherapy [31]. Histopathological analyses of human tumors have provided evidence that variable numbers of infiltrating immune cells are found in different tumors of the same type, and are found in different locations within and around a tumor [5]. The tumor consists of various immune cell types, including B cells, macrophages, DCs, mast cells, NK cells, naïve and memory lymphocytes, and effector T cells [including various subsets of T cell: T helper cells (Th1 cells, Th2 cells, Th17 cells), Treg cells, T follicular helper (Tfh) cells, and cytotoxic T lymphocytes (CTLs)]. These immune cells can be located in the core of the tumor, at the invasive margin, or in the adjacent TLS [5].
HNSCC can escape immune surveillance by adopting different mechanisms, either the presence of the immunosuppressive cells, release of immunosuppressive factors in the TME, or using other strategies [32]. Impairment in major histocompatibility complex (MHC) class I antigen presentation can hamper tumor-associated antigen (TAA) specific immune response in HNSCC [33, 34]. Tumor antigen presentation is impeded by 1) Dysregulation of proteins involved in antigen processing, like latent membrane protein [LMP; proteasome 20S subunit beta (PSMB) 9)] 2, LMP7 (PSMB8), transporter associated with antigen processing 1 (TAP1). 2) Human leukocyte antigen (HLA) expression is lost or down-regulated. 3) Disruption of the differentiation and function of antigen presenting cells (APCs) [35, 36]. CTLs depend on the HLA class I and class II proteins for effective recognition and binding of tumor peptides, making the HLA system an important target in immune escape. Genetic alterations, including point mutations, loss of heterozygosity (LOH), and loss of function mutations in the antigen processing machinery (APM) are irreversible. Besides these, epigenetic dysregulation, post-translational modifications, post-transcriptional modifications, and oncogene activation can cause reversible alterations in MHC I pathway [37]. Mutations in MHC class I heavy chain and β2 subunits especially can irreversibly affect tumor-specific antigen presentation and influence CPI resistance [38, 39]. There have been investigations to find an association between HLA loss, MHC class I impairment, and reduced response to CPI immunotherapy [40]. Tumor cell mutations [interferon (IFN) γ pathway alterations, phosphatase and tensin homolog (PTEN) mutations, β2 mutation] may not allow immunotherapy to work leading to primary or acquired resistance [39, 41]. NK or γδ T cell-based adoptive cell transfer (ACT; MHC unrestricted) and HLA upregulation by cytokines may be a therapeutic strategy to overcome these obstacles. Genetic instability in antigen presentation and processing machinery has a significant impact on the immunotherapeutic strategies employed and should be considered when designing treatment regimes. Different immune regulatory molecules like PD-1, PD-L1, tumor necrosis receptor superfamily member 4 receptor (CD134/OX40), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) are significantly expressed in tumor-infiltrating lymphocytes (TILs) [42]. The presence of tumor-infiltrating T cells has been correlated with progression free survival (PFS) and/or overall survival (OS) in HNSCC [43]. The mechanism of how these immunosuppressive mediators bring about immunosuppression in HNSCC is discussed in detail in the following sections.
The pathogenesis of HNSCC is associated with inflammation and T cell tolerance. Chronic inflammation is described as the overproduction of an array of immunosuppressive and inflammatory mediators. The TME is a complex network of cellular non-malignant host cells [MDSCs, TAMs, Tregs, and IL (interleukin)-17 producing CD4+ T cells (Th17)] and non-cellular components [hypoxia, reactive oxygen species (ROS) and epithelial-mesenchymal transition (EMT)]. Crosstalk between the cellular and non-cellular components in the TME contributes to the induction of chronic inflammation and immune suppression (Figure 1).
Treg cells represent a minor subset of CD4+ T cells (5%) in peripheral blood, marked by CD25+CD127– forkhead box P3 (FOXP3)+ expression. Treg levels increase with tumor staging and high levels of Treg cells correlate with the advanced stage of the disease [44]. In TME immunosuppressive factors like transforming growth factor (TGF)-β, PD-L1 can endorse the conversion of naïve CD4+ T cells to Tregs, which in turn supports their suppressive capabilities [45, 46]. A meta-analysis of 76 studies showed that FOXP3+ Treg infiltration in the tumor is related to patient survival [47]. In Tregs infiltrating the tumor, inhibitory receptors like PD-1, CTLA4, and T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) were significantly elevated [48, 49]. Immunotherapy against Tregs involves targeting the specific surface markers expressed, like CD25, CTLA4, glucocorticoid-induced tumor necrosis factor-related (GITR) receptor, and C-C chemokine receptor-4 (CCR4). The anti-CD25 treatment has been developed to inhibit the suppressive action of Tregs, but the results have been ambiguous in different cancers. A study in which anti-CD25 was used in combination with signal transducer and activator of transcription (STAT) 3 inhibition in HNSCC showed a decrease in Tregs and improved the anti-tumor functions of T cells [50]. Jie et al. [51] showed that treatment of HNSCC patients with cetuximab increased the intratumoral Tregs expressing CTLA4, Tim-3, CD39, and TGF-β. Ex vivo experiments demonstrated that inhibition of Tregs with ipilimumab (anti-CTLA4) reversed the suppressive activity of Tregs and enhanced NK cell-mediated antibody-dependent cell cytotoxicity (ADCC). The CCR4 was predominantly expressed by FOXP3hiCD45RA–CD4+ activated Tregs (aTregs), a subset of Tregs in the peripheral blood of HNSCC patients. Monocyte chemoattractant protein-1 (MCP-1), an endogenous ligand of CCR4 was upregulated in the HNSCC microenvironment [52]. In vivo studies in mice models showed that CCR4 antagonist and anti-MCP-1 antibodies blocked aTreg cell trafficking to the tumors, resulting in inhibition of tumor growth and prolonged survival. GITR and OX40 (CD134) are co-stimulatory signals involved in the activation of effector T cells and Tregs. In different solid tumors including HNSCC agonist antibodies for OX40 and GITR have shown abrogation of immune suppression and inhibition of Tregs [53, 54]. In general, inhibition of the function of Tregs in HNSCC elevates the NK and T cell-mediated anti-tumor effector functions.
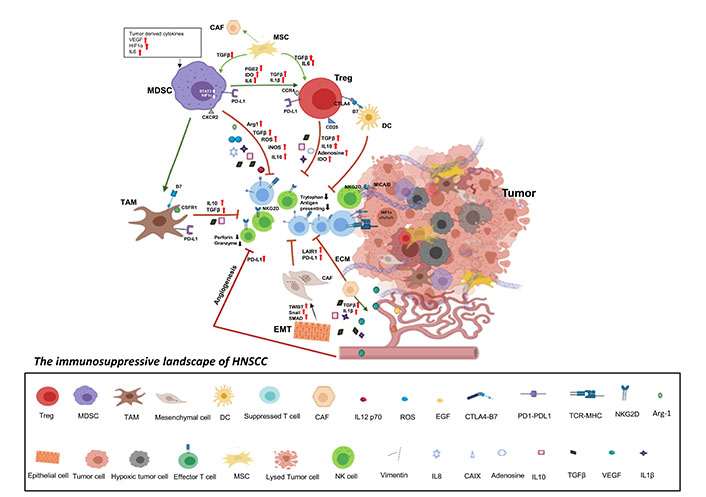
The immunosuppressive landscape of HNSCC. Various tumor-derived factors and hypoxia recruit immune suppressive cells including MDSCs, Tregs, CAFs, MSCs, and TAMs to the tumors. These immune suppressive cells secrete several pro-tumor factors like TGF-β, PGE2, Arg-1, IL-6, iNOS, IL-10, and IDO. In addition, high expression of adenosine, PD-L1, and CTLA4 on the immune suppressive cells results in dampening of the anti-tumor effector functions of the immune cells in the TME. Hypoxia, tumor intrinsic factors, like ROS, ECM, and EMT form a physical barricade, hindering the infiltration of immune cells. These factors also favor the proliferation of immune suppressive cells which decrease the proliferation and anti-tumor effector functions of tumor-infiltrating immune cells including NK cells and CTLs. Induction of neoangiogenesis leads to increased migration of tumor cells resulting in metastasis. In addition, VEGF induces PD-L1 expression on MDSCs reducing T cell activation Increased mesenchymal cells in the tumor, as a result of EMT, also express the immune check-points along with the secretion of immunosuppressive factors like TGF-β. Thus, either directly or in a combination, the tumor intrinsic factors and the recruited non-malignant host cells aid tumor immune evasion in HNSCC. HIF: hypoxia-inducible factors; CAF: cancer-associated fibroblast; CXCR: C-X-C motif chemokine receptor; MSC: mesenchymal stem cell; PGE2: prostaglandin E2; IDO: indoleamine 2,3-dioxygenase; B7: B7 family proteins; iNOS: inducible nitric oxide (NO) synthase; Arg 1: arginase 1; CSFR1: colony-stimulating factor receptor 1; NKG2D: Natural Killer Group 2D receptor; TWIST: twist transcription factor; SMAD signal transducers for receptors of the TGF-β superfamily; an acronym from the fusion of Caenorhabditis elegans Sma genes and the Drosophila Mad, (Mothers against decapentaplegic); TCR: T cell receptor; CAIX: carbonic anhydrase IX; VEGF: vascular endothelial growth factor; ECM: extracellular matrix
MDSC are a subset of immature myeloid cells (IMCs) which are abundantly present in the peripheral circulation and TME of cancer patients. Hyper inflammatory or chronic inflammatory conditions in TME lead to the generation of IMCs which are normally produced by the bone marrow. Hematopoietic stem cells (HSCs) in the bone marrow produce common myeloid progenitor cells (MPCs) and IMCs which then differentiate into mature macrophages, DCs, and granulocytes in peripheral organs. The conversion of HSCs into MDSCs is governed by two different signals. The first signal supports the expansion of impaired differentiation of myeloid cells through granulocyte and macrophage colony-stimulating factor, β-fibroblast growth factor (FGF; β-FGF) and VEGF [55]. The second signal is the “activation signal” which promotes suppressive functionality, mediated by tumor-stromal factors like IL-1β, PGE2, and toll-like receptor (TLR) ligands [56]. Cyclooxygenases are key players of inflammation in TME. The levels of cyclooxygenase-2 (COX-2) were markedly up-regulated in HNSCC, which contributes to elevated levels of prostaglandins [57].
Acute inflammatory stimuli induce activation of mature cell populations which mediates phagocytosis and release of pro-inflammatory signals, and also supports tissue remodeling once the inflammatory state is resolved [58]. MDSCs can be grouped into two major subtypes based on their cell surface markers and morphology: polymorphonuclear MDSCs (PMN-MDSC) and monocytic MDSCs (M-MDSC). PMN-MDSC can be characterized by expression of HLA-DR–CD14–CD11b+CD15+and M-MDSCs by surface expression of HLA-DR–CD14+CD11b+CD15– [59]. MDSCs are recruited by tumor-derived factors which bring about suppression of host immunity and favors tumor progression.
In oral squamous cell carcinoma (OSCC), immune evasion is channelized by MDSC released immunosuppressive factors and surface-expressed molecules like PD-L1 and PD-L2 or via the formation of vasculature by modulating the expression of VEGF and β-FGF isoforms [60]. MDSC contributes to the immunosuppressive microenvironment by hampering T cell proliferation, cytokine production, and effector functions by sequestering the amino acids cysteine and arginine or by converting arginine to ROS and NO that supports the suppression of T cell effector functions. Different strategies to inhibit MDSCs have been discovered which can be used for immunotherapy. These curbs either the function or homing of MDSCs in HNSCC TME. MDSC impairs the differentiation of CD8+ T cells into effector populations, without hindering their activation status, production of IL-2, or signaling through the TCR [61].
In TME, inflammatory cytokines like IL-1β and IL-6 are potent inducers of MDSCs expansion in addition to myeloid-specific growth factors. IL-6 accumulation is associated with the enrichment of MDSCs in TME, IL-6, and its downstream transcription factor (TF) STAT3 aids in the accumulation of MDSCs and in acquiring immunosuppressive phenotype [62]. In pre-clinical models, targeting IL-6/IL-6 receptor (IL-6R) was found to be effective due to the direct effect on tumor cells, including the inhibition of their survival, growth, invasiveness, and metastasis. A study based on different murine cancer models suggested that IL-6 blockade could exert anti-tumor effects via the suppression of MDSCs [63]. STAT3 is a key player in regulating both expansion and activation of MDSCs [64], cytokines like IL-6, IL-10, IL-1β, and tumor necrosis factor-α (TNF-α) are known to alter the expression of STAT3. Elevated levels of IL-6, IL-10, TNF-α, IL-1β, IL-8, TGF-β, PGE2, and VEGF present in sera of OSCC patients helps in regulating immunosuppressive functions of MDSC. The mechanism used by MDSCs to suppress T cell proliferation and reduce levels of key signaling molecule CD3-ζ chain is through the expression of STAT3 regulated Arg 1 and IDO [65]. The therapeutic approach based on inhibiting STAT3 in MDSC is of clinical relevance since inhibition of STAT3 signaling in OSCC patients will not only stall the EGFR mediated tumor progression but also aid in revitalizing the antitumor immunity by inhibiting the production and function of MDSCs. Studies based on targeting STAT3 signaling showed a reduction of MDSCs in HNSCC [66]. Silencing of STAT3 gene using small interfering RNA (siRNA) in MDSC effectively decreased the expression of Arg 1 [67]. Chemokines play a key role in the trafficking of MDSCs to the TME among them, CXCR 2 and CXCR1 are involved in the mobilization of MDSCs in HNSCC. Preventing MDSCs trafficking and localization by inhibiting CXCR2 is of clinical interest to develop new treatment modalities. A pre-clinical study (NCT02499328) assessed the efficacy of durvalumab [STAT3 inhibitor (AZD9150)] and danvatirsen [CXCR2 inhibitor (AZD5069)] on HNSCC patients in two different cohorts. The first cohort consisted of patients treated with durvalumab and danvatirsen in combination or as a monotherapy. The second cohort included anti-PD-L1 pre-treated/naïve patients followed by administration of durvalumab or danvatirsen. The outcome of the study showed a 26% overall ORR as the primary endpoint with four complete and six partial responses in the second cohort [68, 69]. Duvelisib (IPI-145), an inhibitor of phosphoinositide 3-kinase (PI3K, PI3Kδ and PI3Kγ isoforms) was found to inhibit the function of MDSCs in a pre-clinical tumor model of HNSCC. This study showed that the effector functions of CD8+ T cells were elevated on the treatment of IPI-145 combined with PD-L1 inhibitor [70].
Induced differentiation of MDSCs into a mature and less immune-suppressive phenotype is one treatment modality. MDSCs in the peripheral blood of HNSCC patients were differentiated using α,25-dihydroxyvitamin D3. This led to a decrease in circulating MDSCs and increased levels of IFN, IL-2. It was also observed that the levels of CD4, CD8, and activated T cells expressing CD69 increased along with the increase in mature DCs [71]. CXCR2, which is expressed by MDSCs can be inhibited using small molecules or antibodies. Disruption of CXCR2 reduced the recruitment and subsequent immune-suppressive action of MDSCs [58]. HNSCC mice models showed enhanced recruitment of CXCR2+ MDSCs. Treatment with SX-682, a CXCR 1/2 inhibitor impeded the MDSC accumulation into a tumor, increased immune infiltration, and reverted anti-tumor effector functions of NK cells. The study concluded that hindering the MDSC trafficking into the TME can foster the efficacy of NK cell-based immunotherapy [72]. Phosphodiesterase 5 (PDE5) inhibitors can functionally inhibit MDSCs by blocking iNOS. Tadalafil, a PDE5 inhibitor has been used for immunotherapy in several HNSCC clinical trials. It was found to reverse the immune suppression and increase levels of CD4, CD8 T cells in HNSCC patients [73, 74]. TLR8 agonist, motolimod in conjugation with cetuximab was used in a clinical trial. Infiltration of MDSC in HNSCC TME decreased on treatment with motolimod along with the increase in tumor antigen-specific CD8 T cells [75]. Studies show that outcome of immunotherapy for HNSCC can be enhanced by targeting MDSCs.
MSCs are adult non-hematopoietic, multipotent stem cells isolated from different tissues [76] and are known to home into HNSCC. The presence of intratumoral MSC correlates with the tumor size and inhibits T cell proliferation [77]. MSC homing is dependent on changes such as hyperoxia/hypoxia/anoxia, low/high pH, low/high ion gradients which can result in either increased heterogeneity or clonogenic convergence in the TME [78].
The presence of TNF-α, IL-1β, and IFNγ or hypoxic conditions all stimulate MSCs to release proangiogenic and immunosuppressive factors including EGF, platelet-derived growth factor (PDGF), FGF, VEGF, and IL-6 and IL-8 [79, 80]. TLRs are involved in the activation of MSCs and determining anti- or pro-inflammatory phenotype of MSCs. TLR induced activation of MSCs promotes tumor development and immune suppression [81]. Immune responses (adaptive as well as innate) against various tumors are modulated by the presence of MSCs. MSCs mediate immune suppression indirectly by inducing the differentiation of immune suppressive cells Tregs and MDSC [82–86]. Targeting the MSC-derived factors or suppressive functions of MSCs can be an attractive treatment modality in solid tumors like HNSCC. Fibroblast activating protein (FAP) is a major promoter of CAFs, and its secretion is supported by MSCs [87]. MSCs secrete VEGF, EGF, PDGF and support the formation of the tumor milieu. Targeting the various molecules like IDO, hemeoxygenase (HO), Arg I and II, NO synthase-2 (NOS-2), PGE2, and TGF-β, using specific inhibitors used in therapeutic regimens, can inhibit MSC induced tumor growth [80]. The dismal prognosis of locally advanced and metastatic HNSCC could be due to the development of resistance to therapy attributing to the MSC-induced immune escape leading to EMT and metastasis [76, 88].
Macrophages are the miscellaneous population of immune cells arising from a common myeloid progenitor and exhibit dynamic physiological plasticity, which depends upon environmental signals, giving rise to different subpopulations of macrophages [89]. Macrophages are known professional phagocytes of the innate immune system. Depending on the exposure to danger signals and endogenous molecules (cytokines like IFNγ and IL-4, microbial products, nucleotide derivatives, glucocorticoids) these macrophages serve specialized functions [90]. Conventionally macrophages are categorized into two different subtypes: the classical M1 and the alternative M2 macrophages. M1 phenotype is able to orchestrate cytotoxic anti-pathogen and antitumor responses, whereas M2 macrophages support tumor progression, angiogenesis, and suppression of adaptive immunity [91].
In the adult, the bone marrow produces circulating monocytes which, give rise to tissue-resident and inflammatory macrophages. Likewise, TAMs are derived from circulating monocytes and tissue-resident macrophages [92]. Various factors like chemokines, ECM, cytokines, hypoxia, growth factors secreted from tumor and stromal cells in TME are involved in the recruitment of TAMs into the tumor. Mobilization of monocytes and macrophages toward inflamed tissue is influenced by C-C chemokine ligand (CCL) 2 (CCL2; MCP-1) [93]. TAMs associated with M2 macrophage-like state possess a poor antigen-presenting capability and secrete immunosuppressive factors like IL-10 and TGF-β which curbs the immune response of T cells [92].
Macrophage mobilization is guided by chemo-attractants released from stromal cells in TME. Inhibiting these chemo-attractants may support better clinical outcomes. Upon impeding CCL2, the TAM recruitment into the tumor was hampered along with a reduction in cancer progression [94]. Re-polarization of tumor-promoting M2 to M1 macrophages, which support antitumor activity, appears to be a better therapeutic target for cancer therapy. Some studies have reported that TLR activation modulates tumor-supporting macrophages to become tumoricidal effectors. A study by Shime and colleagues on a tumor-bearing mice model showed that activation of TLR3 by poly(I:C) rapidly stimulated the production of pro-inflammatory cytokines followed by an acceleration of M1 macrophage polarization [95]. Activation of immune cells and a decrease in immune suppression is brought about by TLR agonists [96]. In syngeneic mouse models of HNSCC, intratumoral delivery of IV270 (TLR7 agonist) or SD-101 (TLR9 agonist) in combination with anti-PD1 antibody suppressed tumor growth effectively not only at the injected site but also at uninjected distant sites (abscopal effect). The mechanism of action was by activating TAMs, increasing their antigen presentation, M1/M2 TAM ratio, and recruitment of CD8 T cells in the TME. Although the combination of TLR agonist and CPIs have shown promising results in preclinical models of HNSCC, human clinical trials are warranted to establish their therapeutic benefit [97].
CAF are heterogeneous cells and represent the main components of TME. CAFs are the most abundant stromal cell type present in epithelial tumors, including HNSCC. CAFs play an important role in facilitating tumor progression in HNSCC and are associated with poor outcomes [98]. Tumor-associated CAFs express α-smooth muscle actin (α-SMA), integrin α6, and FAP. CAFs are involved in the remodeling of the ECM and facilitate metastasis by secreting matrix metalloproteinases (MMPs), VEGF, and EGF. Although CAFs express PD-L1 in 40% of HNSCC, this has not been associated with clinical outcomes [99]. In HPV+ HNSCC, increased expression of PD-L1, PD-L2 on CAFs was mediated through TLR9 [100]. Interactive reciprocity among the tumor and stromal cells increases the overall PD-L1 level, leading to the development of an immune-suppressive environment. The mechanism behind PD-L1+ CAFs participating in remodeling the inflammatory TME and modulating the T cell responses can be mediated either through direct contact or secreted cytokines and needs to be further investigated. CAFs are known to express high levels of CD73, an ecto-5’-nucleotidase and a rate-limiting enzyme that converts adenosine tri-phosphate (ATP) to adenosine which inhibits activation of T cells and induces apoptosis [101]. In an in vitro 3D culture model consisting of HNSCC and CAFs it was demonstrated that CAFs affect proliferation, EGFR expression, induce EMT, and stem cell phenotype on HNSCC, which may influence the response to cetuximab or cisplatin treatment [102].
Solid tumors like HNSCC form many layers of immunosuppressive stroma which helps in immune evasion and also creates a physical barricade for immune cells. TME plays a major role in supporting tumor growth and resisting therapeutic interventions. Hypoxia is an established characteristic of head and neck cancer [103]. Only some tumor cells are exposed to the hypoxic environment while others exist in normoxic conditions. It is well known that hypoxia is associated with decreased survival and poor prognosis in patients. This may be because hypoxic tumors are usually more aggressive and resistant to treatments than non-hypoxic tumors. Hypoxia promotes tumorigenic milieu by increasing expression of angiogenesis genes, drug resistance, inducing metastasis, and promoting the proliferation of apoptosis-resistant tumor cells. Although the exact role of hypoxia in the interaction between the immune system and tumor cells is not completely understood, it is studied that hypoxia impedes the functions of the immune system by dysregulating the homing of immune cells to the tumor [103].
HIFs are fundamental mediators of hypoxic signaling and are a family of TFs. They are comprised of 3 subunits: HIF1α, HIF2α, and HIF3α that form a heterodimer with HIF1β [104]. The oxygen content determines the transcriptional activity of HIF, which is upregulated under hypoxia. The proline residues on HIF1α are hydroxylated by proly-4-hydroxylase under normoxic conditions and this hydroxylated HIF1α can be downregulated by proteasomal degradation or ubiquitination. Stabilization of HIF1α and/or HIF2α under hypoxia leads to upregulation of transcription of various downstream genes, causing metabolic and immune modulation of tumor cells. Hypoxia upregulates IL-8, VEGF, and osteopontin causing an increase in angiogenesis in the tumor [105]. Enhanced expression of genes involved in EMT like Vimentin (VIM), SNAI1 and SNAI2 (Snail Family Transcriptional factors), TWIST has been observed in hypoxic TME [106]. Hypoxia also affects cell-cell adhesion by downregulating E-cadherin on tumor cells. There have been studies suggesting that the hypoxia-HIF-EMT niche is important in maintaining cancer stem cells [107]. Under certain hypoxic conditions, HIF signaling can act as an oncogenic stimulus leading to immune suppression, evasion and tumor development, metastasis [108].
Hypoxic regions in HNSCC also show infiltration of immunosuppressive cells like MDSCs, Tregs, and TAMs along with tumor cells. This infiltration is facilitated by stromal cell-derived factor 1α (SDF1α), which regulates the differentiation of MDSCs into the monocytic phenotype (Mϕ MDSC) in TME. SDF1α is produced by the tumor cells and binds to IL-6, IL-8, and CXCR causing monocytic MDSC recruitment to hypoxic HNSCC [109]. It is also noted that MDSCs derived from tumors are more strongly immunosuppressive than splenic MDSCs. This may be due to nitric acid production in tumor MDSCs and arginase activity impelled by HIF1α. Tregs in TME suppress anti-tumor functions of immune cells and also promote neoangiogenesis [110]. There is a strong correlation between the accumulation of Tregs in TME and hypoxia, proven by the increase in expression of FOXP3 on account of HIF1α [111]. FOXP3 is a key regulator in the development of Tregs and also a marker for the presence of Tregs. Under hypoxia, tumor cells attract CXCR10 positive Tregs because of the high production of CCL28, instigating angiogenesis and immune evasion. Along with the regulation of immunosuppressive cells like Tregs and MDSCs, hypoxia influences checkpoint molecules like PD-L1, HLA-G, and the cluster of differentiation 47 (CD47) [112]. This occurs via HIF1α dependent upregulation of these molecules in hypoxic TME [113]. Our group has recently shown that the presence of hypoxia abrogated the anti-tumor effector functions of γδ T cells and upregulated the differentiation of γδ T cells into pro-tumorigenic γδT17 cells in HNSCC [114].
Various metabolites are activated in response to hypoxia to bring about immune suppression, and one of them is adenosine. HIF1α prevents re-phosphorylation of adenosine to adenosine monophosphate (AMP) causing adenosine accumulation. Hypoxia can enhance the expression of the A2A adenosine receptor (A2AR) which is a G-protein-coupled receptor (GPCR) involved in adenosine signaling. Activation, as well as anti-tumor effector functions of T cells, are hindered by the adenosine accumulated in TME [115]. Furthermore, the binding of adenosine to A2AR on T cells can lead to T cell apoptosis. This high level of adenosine in the hypoxic environment of HNSCC can lead to immune suppression [116]. Hypoxia acts in a myriad of ways to cause immune suppression in HNSCC, including direct disruption of the immune cell functions and acting in tandem with other factors like ROS, immune checkpoints, ECM, etc.
ROS is a common name given for free oxygen radicals like superoxide (O2-), hydroxyl radical (OH-), singlet oxygen, and hydrogen peroxide (H2O2) formed as a result of various activities in the cell. ROS are produced in various sites in the cell-like mitochondria during aerobic metabolism, endoplasmic reticulum, redox reactions in the cytoplasm, non-mitochondrial electron transport chain, etc. In a normal cell, free radical scavengers like superoxide dismutase regulate and neutralize ROS, but the imbalance between the levels of these antioxidants and ROS leads to oxidative stress. Tumorigenesis, tumor immune evasion, and TME all have been associated with increased levels of ROS in cancer [117]. Hypoxia, which is a distinct characteristic of head and neck cancer, needs mitochondrial ROS for stabilizing HIF. ROS is also produced by various cells in TME like tumor and stromal cells. As the tumor progresses, tumor cells become tolerant to ROS, much unlike normal cells wherein elevated ROS induces autophagy. This is called “ROS addiction” [118]. These elevated levels of ROS can influence and support tumorigenicity.
Oxidative stress due to uncontrolled ROS affects the regulation of TAMs, MDSCs, and T cells. Tregs showed an increase in their immunosuppressive function in response to increased ROS [119]. MDSCs, which are known to alter TCR signaling, express high levels of ROS. It was found that inhibition of ROS in MDSCs could revert the damaged immune system [120]. Inflammatory pathways like nuclear factor-kappa B (NF-κB) and Janus kinase (JAK)/STAT may be activated due to enhanced expression of ROS and reduce the anti-tumor immune function. In a study by Kesarvani et al. [121], it was found that lower ROS levels were necessary for T cell survival, and enhanced ROS lead to apoptosis of T cells. In fact, decreased levels of ROS corresponded with better T cell survival and anti-tumor effector functions. Increased resistance to PD-1/PD-L1 immunotherapy was reported in tumors with enhanced ROS. ROS also influences the immunometabolic response to chemotherapy and this has been studied in HNSCC. High ROS and increased glucose-dependent metabolism correlated with a low CD4/CD8 ratio in TILs [122]. This was also enumerated in HPV+ HNSCC where high levels of ROS corresponded to chemotherapy resistance and a mitochondrial-rich cancer metabolism [123]. Besides hypoxia and pH change, ROS may play a major role in the metabolic pathways used by tumor cells for survival and immune evasion [124]. Elevated ROS levels also affect the efficacy of therapies used in HNSCC [125]. Upon inhibition of nuclear factor erythroid 2-related factor 2 (NRF2), an important TF involved in the regulation of oxidative stress, the resistance of cancer cells to ferroptotic death was reversed [126]. Thus normalizing ROS activity in TME may help in improving the anti-tumor immune responses and other treatment strategies.
ECM is composed of a complex network of various fibrous proteins [elastin, collagen (Col)], chondroitin sulfate, hyaluronic acid, fibronectin, laminins, and fibrils that form a scaffold of organs and tissues. In solid tumors like HNSCC, ECM makes up the majority of the tumor mass. In cancers, ECM proteins can be produced by the tumor as well as stromal cells, but CAFs play a major role in the assembly, secretion, and reformation of ECM [127]. Along with the proteins and covalent cross-linkages, factors like structure, molecular density, rigor/rigidity, and tension are also major characteristics that influence ECM. Subsequently, the ECM is adaptable and prone to remodeling, which is brought about by tumor and stromal cells. Various changes within the TME, as well as chemokines, cytokines, circulating tumor cells, growth factors, monitor and regulate the ECM. In HNSCC, crosstalk between the malignant epithelial cells and stromal cells prompts the upregulation of a set of ECM components that play a role in inducing carcinoma cell migration, modulate the cytokine environment and promote immune evasion in these tumors [128]. ECM can promote metastasis by regulating both the tissue stiffness and the entry of a drug into the tumor. These factors can also alter the ECM of other organs to become more susceptible to alteration and promote metastasis [129].
Col, one of the proteins that form the ECM network, is especially important for cancer progression as it can be regulated by various TFs, pathways, mutated genes, and even cancer cells [130]. Col also interacts with other ECM components like fibronectin, hyaluronic acid to support tumor cell functions [131]. Higher Col percent in ECM composition correlates to hypoxia which leads to immune suppression and tumor growth. In addition to Col, ECM also enhances the expression of immune suppressive molecules like TGF-β, PGE2, and IL-10. ECM increases VEGF expression, encouraging angiogenesis and EMT [132]. ECM inhibits infiltration of cytolytic immune cells and diffusion of drugs into the tumor by modifying the structural integrity. Certain components like transmembrane Col XVIII in ECM act via leukocyte-associated immunoglobulin-like receptor 1 (LAIR1) to induce inhibitory signals in NK and B cells [133]. Hyaluronic acid has been hypothesized to increase Treg levels and suppress the immune system. Hyaluronic acid is also involved in increasing PD-1/ PD-L1 levels in ECM [134]. A recent study correlated EMT gene signature with response to immunotherapy in HNSCC patients. The mesenchymal subgroup of HNSCC patients expressed elevated levels of immune checkpoint genes PDCD1, PD-L1 and CTLA4 and showed a worse prognosis compared to the epithelial subgroup [135]. TAMs are the most abundant immune cells in ECM and have both anti-inflammatory and pro-inflammatory functions depending on their polarization. Various components of ECM, such as fibronectin, hyaluronic acid, and Col directly affect the polarization of TAMs and thus may be involved in regulating the immune response in cancer [136]. ECM stiffness influences the resistance to cancer therapies (especially chemotherapeutic agents like cisplatin), however, the exact molecular mechanisms need to be studied [137].
Angiogenesis is the process of the formation of new blood vessels. In HNSCC, the “angiogenic switch” is a crucial step that ensures the survival and growth of the tumor. This process of forming new vasculature is a limiting step in tumor formation and can be targeted in the therapy of HNSCC. It is mainly induced by VEGF-A, along with VEGF-B, VEGF-C VEGF-D, VEGF-E, and placental growth factor (PIGF), which belongs to the PDGF superfamily. VEGF can be induced by the hypoxia-mediated production of HIF1α. VEGF ligands function through their surface receptors VEGF receptor (VEGFR)-1, VEGFR-2, VEGFR-3 which are tyrosine kinases [138]. On endothelial cells, the receptors facilitate VEGF to exert their vascular permeabilizing, mitogenic, and chemotactic effects. Neurolipin (NRP; NRP1 and NRP2) are co-receptors for VEGF and improve its biological activity by bolstering the VEGF-VEGFR interactions. Other factors like COX2, EGF, prostaglandins, PDGF, and IL-6 are also involved in angiogenesis. In HNSCC, overexpression of VEGF has been correlated to tumor resistance, poor prognosis, and even metastasis [139].
Elevated levels of IL-8/C-X-C motif ligand (CXCL) 8 along with FGF and VEGF promote angiogenesis in HNSCC [140]. These cytokines work in tandem and have different modes of action, but both result in angiogenesis in HNSCC [141]. Along with angiogenesis, VEGF is also involved in immune suppression, further enabling tumor survival [142]. VEGF interferes with hematopoiesis which curbs the differentiation of immune cells from their progenitors. It can deregulate T cell development and function, affecting their anti-tumor immune response. VEGF binds to VEGFR on MDSCs and induces PD-L1 expression, prevents maturation, and reduces T cell activation [143]. VEGF/VEGFR reduces T cell extravasation and adhesion in TME, supporting immune escape [144]. It also enhances Tregs differentiation, causing a decrease in proliferation of T cells, suppressing DCs, and increasing CTLA4 [139]. Anti-angiogenic agents, which block VEGF, VEGFR, and EGFR can be used in combination with immune CPI for immunotherapy.
EMT is the process when epithelial cells lose their characteristic features and obtain mesenchymal-like properties. Although EMT is a fundamental process in wound healing and during development, tumor cells initiate EMT for obtaining immune resistance and mobility [77]. The key features of EMT are loss of cell-cell adhesions and basal-apical polarity, gene reprogramming, alteration in signaling cascade to change cell shape, and rearrangement of cytoskeleton. Tumor cells undergo epithelial to mesenchymal transition when migrating from the primary tumor [145]. This can be the first step towards metastasis, though only EMT is not enough for the tumor to metastasize. In HNSCC, the expression of key regulators and hallmarks of EMT has been widely studied. Keck et al. [123] correlated the mesenchymal phenotype with a high influx of CTLs. In addition, the epithelial subgroup showed a better prognosis than the mesenchymal subgroup. The mesenchymal subgroup displayed significantly elevated PD-1, PD-L1, and CTLA4 expression along with enhanced levels of IFNγ, cytolytic T cell homing, better overall immune infiltration, and immune signature scores [135]. Tumor-associated inflammation results in the release of various cytokines and chemokines, causing a “cytokine storm”. This supports tumorigenesis and the tumor cells undergo EMT as a means to adapt to the changing TME [145]. EMT-like changes in tumor cells have been associated with treatment resistance, tumor survival, immunosuppression, and poor prognosis. EMT in HNSCC has been associated with high levels of IL-1β and IL-6 [146]. This results in loss of e-cadherin, increase in VIM expression, and activation of the Snail pathway.
Recent work has shown the ability of HNSCC cells to achieve an intermediate state of EMT, called partial EMT which prompts a high metastasis compared to complete EMT. Among the genes related to partial EMT, serine protease inhibitor clade E member 1 (SERPINE1), integrin subunit alpha 5 (ITGA5), TGF-beta-induced gene (TGFBI), prolyl-4-hydroxylase alpha subunit 2 (P4HA2), tumor suppressor gene H-cadherin (CDH13), and laminin gamma 2 (LAMC2) were highly expressed in HNSCC cases and correlated with poor survival of HNSCC patients [147]. The EMT gene signature was reported to be associated with poor prognosis in HNSCC and could be used as a prognostic marker [135]. It was reported that miRNAs participate in regulating recurrence, EMT, and metastasis in HNSCC. Under an inflammatory stimulation, the endogenous miRNAs (miR-18a-5p, miR-18b-5p, miR-19a-3p, miR-19b-3p, miR-21-5p, miR-27a-3p, miR-30a-5p) were identified as miRNAs promoting survival/EMT in HNSCC cells [148].
TFs like twist-related protein (TWIST), Snail (Zinc finger protein SNAI1), and Zinc finger E-box binding homeobox (ZEB) are involved in EMT regulation, tumor progression, and immune evasion. Snail induces EMT, enhancing the expression of suppressive cells and promoting metastasis. Snail also supports the secretion of CCL2 and thereby induces infiltration of Tregs, MDSCs, and TAMs into TME [149]. CCL2 also inhibits CD8 T cells and promotes immunosuppressive DCs [150]. TWIST, another important TF is upregulated by PD-L1 and enhances Tregs while inhibiting CD8 T cells and NK cells [151]. In addition, TWIST reduces TNF-α and NF-κB expression resulting in a decrease in inflammation [152]. Tumor cells that undergo EMT or those that have mesenchymal-like properties are more resistant to the anti-tumor functions of NK and CD8 T cells. In HNSCC, EMT has a strong correlation with immune checkpoints, especially PD-L1 [153]. High expression of PD-L1 was associated with the high probability of an EMT signature. Moreover, survival analysis showed the PD-L1+/EMT+ patients had a poorer prognosis compared to PD-L1+/EMT– HNSCC patients [154]. Increased expression of VIM and low expression of E-cadherin resulted in higher PD-L1 expression. TGF-β is secreted during EMT due to decreased E-cadherin and causes immune suppression in the later stages of tumor progression. Recent studies have suggested EMT as a predictive marker to be explored for immunotherapy outcomes.
Immune checkpoint molecules are crucial to prevent autoimmune diseases and also inhibit cell-mediated immune damage. Although they exist to modulate self-tolerance by the immune system, tumor cells hijack these immune checkpoints and cause immune evasion [155]. The essential checkpoint molecules targeted by tumor cells that have gained the most clinical interest in HNSCC include CTLA4 and PD-1/PD-L1 [156]. CPI PD-1 and PD-L1 were the first to be approved as second-line treatments for HNSCC [157]. The CPIs that have been tested on HNSCC incorporate PD-1 (nivolumab and pembrolizumab), PD-L1 (atezolizumab, durvalumab, and avelumab), and CTLA4 (ipilimumab and tremelimumab) [158–161]. Besides these, lymphocyte activated gene (LAG)-3, Tim-3, and T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) are also checkpoint molecules associated with the tumor immune evasion but their role in HNSCC is not well understood.
CTLA4 and CD28 are homologous receptors expressed on both CD4 and CD8 T cells and mediate opposing functions on T cell activation. These receptors bind to CD80/CD86 ligands expressed on antigen-presenting cells mediating T cell co-stimulation in conjunction with TCR signaling. On the contrary, the interaction of CTLA4 with the ligands, inhibit T cell responses. Tregs express CTLA4 constitutively due to the high levels of TF FOXP3 known to regulate CTLA4 expression [51]. Interaction of CTLA4 with CD80 expressed on antigen-presenting cells, thwarts the T cell effector functions and proliferation [162]. Also, CTLA4 is expressed on Tregs which drive further impairment of immune cell function in tumors [51]. There are currently two human anti-CTLA4 antibodies utilized in phase III clinical trials, ipilimumab, and tremelimumab. The safety and toxicity profile from phase I/II studies have shown the efficacy of both the antibodies as monotherapy or when combined with IL-2 conventional chemotherapy [32].
PD-1 (CD279) is a cell surface receptor expressed on T cells, B cells, and NK cells. PD-1 is expressed on Tregs, NK cells, activated monocytes, and myeloid DCs. PD-L1 [B7 homolog 1 (B7-H1)] and PD-L2 [B7 family member, whose expression is highly restricted to DCs (B7-DC)] act as ligands for PD-1and are expressed on T cells, B cells, and a variety of cell types that include vascular endothelial cells, epithelial cells, astrocytes, neurons, trophoblasts in the placenta, etc. [42, 163, 164]. Tumor cells are known to express PD-L1 in solid cancers including HNSCC and hematological malignancies [163]. High expression of PD-L1 in melanoma and non-small-cell lung carcinoma has been correlated with poor response to immunotherapy. Immune suppression is triggered when dysregulated PD-1 pathway in tumor cells leads to higher production of PD-L1. Inflammatory cytokines like VEGF, TNF-α, and IFNs can induce PD-L1 and influence T cell activity. This pathway is also activated in a chronic inflammatory environment, which is seen in tumors [29]. The activation of the PD-1/PD-L1 pathway leads to the treatment resistance observed in HPV+ HNSCC [29, 165]. The levels of PD-1, PD-L1 were higher in HPV+ HNSCC compared to HPV– HNSCC. The interaction between PD-1/PD-L1 can modulate the function of T cells, Tregs, MDSCs and directly influence the TME. Inhibition of PD-1 on Tregs decreases their functional ability. On effector T cells, PD-1/PD-L1 can stimulate the apoptotic pathway and hinder the anti-tumor functions [166]. This occurs via phosphorylation of tyrosine in the immunoreceptor tyrosine-based switch motif (ITSM) domain of PD-1. Downstream inhibition of the PI3K/AKT pathway following this phosphorylation leads to transcription inhibition of the genes required for T cell activation and function. The binding of PD-1/PD-L1 hinders T cell proliferation and cytokine secretion. PD-1/PD-L1 also affects the interactions between different immune cells and can be considered to be an exhaustion marker for T cells [151]. Subsequently, blocking PD-1/PD-L1 increased the release of IFNγ [167]. Chen et al. [168] reported that IFNγ induces PD-L1 expression by upregulating protein kinase D isoform 2 (PKD2), a downstream target of PI3K, in a time and dose-dependent manner in OSCC cells suggesting a negative feedback mechanism of CTLs effector response. Although the inhibitors of PD-1/PD-L1 are effective in the therapy of several malignancies, it has been recently revealed that hindering the PD-1/PD-L1 axis enhances the expression of other immune checkpoints like Tim-3, which may be implicated in adaptive resistance [32].
Along with T cells, PD-1 also affects NK cells and a high expression of PD-1+ NK population was found in HNSCC [163]. Inhibition of PD-1 on these cells increased the cytolytic function of NK cells [169]. PD-1 blockade increased cytotoxicity of IFNβ-activated NK cells toward nasopharyngeal carcinoma cells [170]. These checkpoint molecules are attractive targets for immunotherapy in HNSCC [32].
Nivolumab, an anti-PD-1 monoclonal antibody (mAb) was the foremost drug to be permitted by the Food and Drug Administration (FDA). The Checkmate 141 trial showed an improvement in OS of 7.7 months versus 5.1 months contrasted and monotherapy of docetaxel, methotrexate, or cetuximab in HNSCC [171]. A single institution, single-arm, phase II clinical trial of neoadjuvant presurgical nivolumab for histologically demonstrated, locoregionally advanced HNSCC was conducted. Phenotypic analyses of peripheral blood T-cells before and after nivolumab treatment showed a decline in the frequency of circulatory CD4+ cells however did not affect the expression of IFNγ. Nivolumab treatment also showed an increase in the proportion of CD8+cells, as well as the expression of granzyme B. Treatment with nivolumab, did not affect the levels of the NK containing CD8dimCD3− or the secretion of IFNγ or granzyme B [172]. This study demonstrates that treatment of nivolumab results in stimulating mainly CD8+ cell subpopulations, instead of inducing the whole immune system which was an expected outcome [172]. Significant infiltration of CD8+ and a high PD-1/Tim3 expression ratio (regardless of HPV status and smoking status) showed an increase in the OS (84 months vs. 13 months) with anti-PD-1 treatment, compared with non-inflammatory tumors [173].
Another PD-1 inhibitor, pembrolizumab was studied in the phase III study, and it was found that the survival significantly increased from 7.9 to 11.6 months in favor of pembrolizumab in patients with PD-L1 > 50% [174]. A phase III clinical investigation utilizing pembrolizumab (KEYNOTE 048) in the treatment of R/M HNSCC reported superior treatment results from pembrolizumab. Pembrolizumab in combination with chemotherapy improved the OS compared with cetuximab combined with chemotherapy (13.0 months vs. 10.7 months). In light of the observed efficacy and safety results, pembrolizumab combined with chemotherapy is now the first-line treatment for patients with metastatic or intermittent HNSCC, whereas pembrolizumab monotherapy is the preferred treatment for patients with relapsed or metastatic PD-L1-positive HNSCC [175].
Durvalumab is another fully human mAb that hinders the interaction of PD-L1 with its of these receptors PD-1 and CD80. The EAGLE trial compared an anti-PD-L1 (durvalumab) alone or with an anti-CTLA4 (tremelimumab) to standard chemotherapy. This investigation showed a reduction in OS, but using durvalumab as monotherapy was concurrent with the data of nivolumab and pembrolizumab showing a median OS at 7.6 months and a response rate of 17.9% [176]. Durvalumab treatment in patients is successful in R/M HNSCC in whom PD-L1 expression was found in more than 25% of tumor cells after the failure of platinum-based chemotherapy [177].
A meta-analysis reported that the PD-L1 expression accounted for 42% of HNSCC patients and was related to the HPV status. HPV+ HNSCC tissues had more lymphocytes and elevated levels of PD-L1 than HPV– HNSCC tissues, and the infiltrated CTLs expressed more PD-1. Interactions between PD-L1 and PD-1 can directly regulate the TME and have different functional significance in modulating the effects of T cells, DCs, MDSCs, and Tregs [32]. Reports show that the HPV+ tumors respond better to anti-PD-1 [7]. HPV+ tumors show enhanced immune infiltrates and more markers of activated T cells resulting in better efficacy of anti-PD-1 therapy. A retrospective analysis of HNSCC patients who did not respond to platinum therapy was treated with pembrolizumab or nivolumab and significantly increased the OS for HPV+ patients: 17 months vs. 4.5 months was observed [7]. A meta-analysis of PD-L1 expression was performed to predict the survival of HNSCC patients, no significant variance in OS between PD-L1+ and PD-L1-HNSCC patients were reported [178].
PD-L1 expression is observed at the interface between the tumor nests and the surrounding inflammatory stroma [165]. Epigenomic deconvolution on HNSCC samples from The Cancer Genome Atlas (TCGA) datasets revealed the distinct molecular and epigenetic profile of HPV+ and HPV–HNSCC. HPV+ HNSCC had a higher level of infiltrated CD8+ T and B cells in the tumors and also exhibited higher PD-1 expression in these infiltrated immune cells, which may result in a better response rate to PD-1 targeted therapy [179]. It is important to note that the crosstalk of PD-L1 high stromal cells with the PD-1 high CD8+ T and B cells in the tumors is important in deciphering the patient outcome. These outcomes propose that the PD-1/PD-L1 pathway has an essential function in the pathogenesis and progression of HPV+ HNSCC [42].
Recovery of NK cell exhaustion was observed in HNSCC when Tim3, which is over-expressed on tumor cells, was inhibited [180]. In an HNSCC mouse model, Tim3 inhibition disrupted tumor growth [181]. TILs in HNSCC show enhanced expression of other checkpoint molecules, LAG-3, particularly in metastatic or recurrent disease, and are associated with poor survival. Similar to Tim-3, LAG-3 inhibition also restricted tumor growth in the mouse model [182]. The anti-cancer agents thus help in accelerating the CTL effector response in inducing tumor cell death and predict the survival outcome of the patient.
CPI-based treatment modalities have reported improvement in the OS in patients with HNSCC and better quality of life compared with concurrent chemotherapy and radiation therapy [156, 183]. Although the use of CPIs has been approved by the FDA as the second-line treatment of R/M HNSCC, the relatively high rate of adverse events and the low response rate call for new immunotherapy strategies, either as monotherapy or in combination with existing CPIs.
Targeted therapies have made a major impact on the management of HNSCC patients. Several promising therapeutic targets are being explored for HNSCC, but it is beyond the scope of the present article to include all of these, and therefore, recently published articles on this topic are cited [184–186]. The most promising targeted therapy is the inhibition of EGF-based cellular signaling mediated by EGFR binding mAb, cetuximab. Tumor aggressiveness in HNSCC patients correlates with the increase in EGFR expression observed [187]. Different EGFR inhibitors have been recognized that work either by binding to the extracellular domain of the EGFR (e.g., mAbs such as cetuximab, zalutumumab, panitumumab, nimotuzumab) or by inhibiting the intracellular tyrosine kinase activity (TIKs) of the receptor [188]. Gefitinib and erlotinib (mAbs targeting EGFR) and a small molecule osimertinib are FDA-approved drugs against HNSCC used either alone or in combination with chemotherapy and radiotherapy [188, 189]. NK cell activation in response to cetuximab can up-regulate the PD-1 expression on NK cells. This cetuximab-regulated ADCC is elevated by combination with PD-1 inhibitors, prompting tumor cytolysis [190]. Cetuximab induces a tumor antigen-specific adaptive immune response through DC maturation, further causing CTL activation and major histocompatbility complex class I chain-related molecule A (MICA) recognition by NKG2D expressing NK cells in HNSCC patients [191]. NK cells activated by cetuximab induced the upregulation of HLA-C on tumor cells. NK-cell induced DC maturation in response to cetuximab aided cross-presentation to CTLs which were specific for EGFR and Melanoma-associated antigen 3 (MAGE-A3; another tumor antigen) [191]. Monalizumab is a humanized IgG4 against natural killer cell receptor group 2A (NKG2A) mAb currently under investigation for R/M HNSCC. An ORR of 27.5% was observed in HNSCC patients treated with a combination of cetuximab and monalizumab [192]. Monalizumab gives the additional advantage of enhancing cetuximab-induced NK-mediated ADCC [193]. A de-escalate randomized phase III clinical trial showed a better prognosis in low-risk HPV+ OSCC when treated with cisplatin or cetuximab in combination with radiotherapy [194]. Low EGFR expression in HPV+ phenotype compared to HPV– may also influence the response to cetuximab [195].
The various immunotherapeutic drugs currently in clinical development for the treatment of HNSCC are enumerated in Table 1 and Figure 2.
| Trial ID | Target | Intervention | Study type | Patient population |
|---|---|---|---|---|
| NCT04375384 | EGFR inhibitor | Cetuximab | Phase II treatment, non-randomized, open-label clinical trial | HNSCC patients with R/M after the failure or intolerance of the immunotherapeutic drug |
| NCT04326257 | LAG-3 CTLA4 PD-1 | Nivolumab + Relatlimab Nivolumab + Ipilimumab | Phase II, open-label, parallel arms trial | R/M HNSCC patients with preceding failure of immunotherapy with anti-PD-1 or PD-L1, having a high expression of LAG-3 and CTLA4 genes |
| NCT04428151 | RTK and VEGFR1 inhibitor PD-1 | Lenvatinib (E7080/MK-7902) Pembrolizumab (MK-3475) | Phase II, randomized, open-label, three-arm clinical trial | R/M HNSCC patients with the progressed disease after platinum therapy and immunotherapy (PD-1/ PD-L1 inhibitors) |
| NCT03283605 | PD-1 CTLA4 | Durvalumab (MEDI4736) + Tremelimumab + stereotactic body radiotherapy (SBRT) | Phase II, open-label, single group | HNSCC metastatic patients following prior treatment failures |
| NCT04555837 | Serine/threonine-protein kinase Aurora-A inhibitor PD-1 | Alisertib Pembrolizumab | A Phase I-II, open-label study | Retinoblastoma (Rb)-deficient, HPV+ HNSCC patients. |
| NCT03022409 | ATR inhibitor PARP inhibitor | Ceralasertib Olaparib | Phase I, randomized, open-label | Treatment naïve HNSCC patients |
| NCT04634825 | B7-H3 PD-1 LAG-3 | Enoblituzumab Enoblituzumab + Tebotelimab | Phase II, open-label, non-randomized | Patients with R/M HNSCC |
| NCT04193293 | PI3K inhibitor PD-1 | Duvelsib Pembrolizumab | Phase Ia/IIb multicentre, non-randomized, open-label study | Patients with R/M HNSCC |
| NCT03565783 | PD-1 | Cemiplimab | Phase II, open-label | Patients with stage III-IV R/M HNSCC before surgery |
| NCT03854032 | PD-1 IDO1 | Nivolumab BMS-986205 | Phase 2 Randomized, open-label | Stage II-IV HNSCC patients |
| NCT04080804 | PD-1 LAG-3 CTLA4 | Nivolumab + Relatlimab Nivolumab + Ipilimumab | Phase II Neoadjuvant study, randomized, open-label | Locally advanced resectable HNSCC patients prior to surgical resection |
| NCT03795610 | PI3Kγ inhibitor | IPI-549 | Phase II, open-label | Locally advanced HPV+ and HPV– HNSCC patients |
| NCT04144517 | IL-2 PD-1 | ALKS 4230 Pembrolizumab | Phase II, open-label | Patients with HNSCC prior treated with anti-PD-L1 therapy but not achieved a CR |
| NCT03336606 | OX40 agonist | MEDI0562 | Phase1, randomized, open-label | HNSCC patients prior to surgical resection |
| NCT04602013 | PD-1 | Sintilimab | Phase II, open-label, multicentre | Locally advanced OSCC patients |
| NCT03818061 | PD-L1 VEGF | Atezolizumab Bevacizumab | Phase II, multi-center, non-randomized | Advanced stage patients with recurrence of HNSCC |
| NCT04393506 | VEGFR2 inhibitor PD-1 | Apatinib Camrelizumab | Phase I, open-label | Patients with locally advanced and resectable OSCC |
| CTRI/2020/ 11/028953 | PD-1 | Nivolumab | Phase I and II, randomized group | HNSCC patients under palliative systemic therapy |
RTK: receptor tyrosine kinase; ATR: ataxia telangiectasia and Rad3-related; PARP: Poly (ADP-ribose) polymerase; B7-H3: CD276; DART: dual-affinity re-targeting; ALKS 4230: nemvaleukin Alfa; CR: complete response
ACT is a form of passive immunotherapy that entails ex vivo development and alteration of the patient’s immune cells, followed by their reinfusion. TILs or peripheral blood mononuclear cells (PBMCs) from the patient are collected, expanded, and reintroduced in patients for anti-tumor immunity. ACT can be effectively used to target tumor cells and prevent recurrence [198, 199]. Activated undifferentiated CD44lowCD62L+CD8+ T cells [referred to as stem cell memory T cells (TSCM)] transferred into mice models showed higher tumor killing than the mice treated with TEM cells suggesting that the abrogation of CD8+ T cell differentiation increased their efficacy after ACT [200]. Jiang et al. [201] studied two groups of patients, the ACT group (ACT was repeatedly given after chemotherapy) and the control group (patients without ACT). The ACT group showed a pronounced increase in PFS and OS (56 vs. 40; 58 vs. 45 months) compared to the control group [201]. In this study, ACT, which included the expansion and characterizing of cytokine-induced killer cells (CIKs), reversed the immune suppression following chemotherapy and also enhanced the effector functions of CIKs [201]. Patients with metastatic EBV+ nasopharyngeal cancer were treated with EBV-directed autologous T cells expanded ex vivo in a phase I study. EBV-directed T cell expansion was successful in 16 patients who demonstrated a median OS of 523 days. On the other hand, patients without successful T cell expansion had a median OS of 220 days [202]. Patients having platinum resistance or high cisplatin sensitivity may be considered for the combination of ACT and chemotherapy. Combining cytomegalovirus (CMV) p65 antigen-specific CTLs with radiotherapy or chemotherapy was found to impart cytotoxicity against OSCC cell lines overexpressing the CMVpp65 antigen [203]. Several teams have launched clinical trials of γδ T cell-based cancer therapies targeting hematological malignancies (follicular lymphoma, multiple myeloma, acute and chronic myeloid leukemia), as well as solid tumors (renal cell, breast, and prostate carcinomas) [204]. Immunotherapies based on Vγ9Vδ2 T cells have been examined for head and neck cancer [205]. The effectiveness of γδ T cell-based immunotherapy is limited due to its suppressive function in the TME [206]. Two main therapeutic strategies based on Vγ9Vδ2 T cells have been proposed for tumor immunotherapy: the in vivo expansion of Vγ9Vδ2 T cells by aminobisphosphonates and the adoptive transfer of ex vivo-expanded Vγ9Vδ2 T cells combination of γδ T cell adoptive transfer with CPI is a useful strategy to enhance the antitumor activity of these cells [207]. For ACT, the major limitation is the difficulty to expand Vγ9Vδ2 T cells ex vivo from the advanced cancer patients especially after radiotherapy and chemotherapy [208].
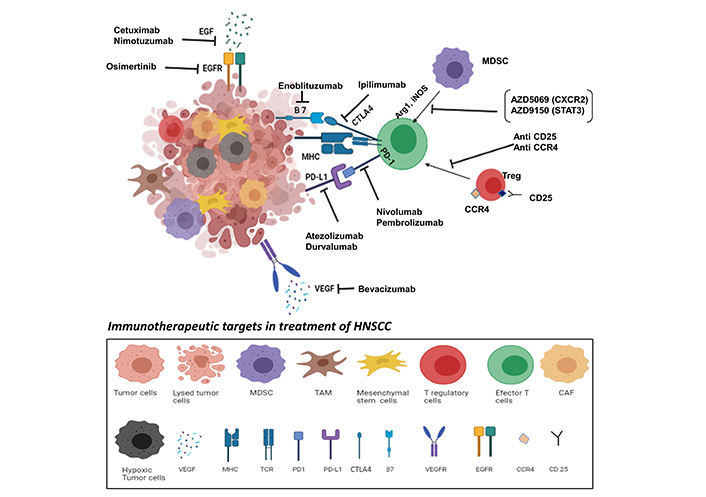
Immunotherapeutic targets in the treatment of HNSCC. Modification of immunosuppressive TME to favor anti-tumor immunity can be achieved by enhancing the infiltration of effector cells like CTLs and NK cells, APCs, or by blocking the activity of immunosuppressive cells like MDSCs, TAMs, and Tregs. Immune CPI based monoclonal antibodies: nivolumab (anti-PD-1), pembrolizumab (anti-PD-1), atezolizumab (anti-PD-L1), ipilimumab (anti-CTLA4), and durvalumab (anti-PD-L1) are being used as targeted treatment modalities that help in restoring functions of effector T cells. Danvatirsen (AZD9150) and AZD5069 which are STAT3 and CXCR2 inhibitors respectively are used to block the trafficking of MDSCs in TME. Cetuximab and nimotuzumab bind specifically to the EGFR on tumor cells to competitively inhibit the binding of EGF. In addition, osimertinib an EGFR tyrosine kinase inhibitor, directly blocks EGFR. Enoblituzumab recognizes B7-H3, a member of the B7 family of immune regulators, evoking an anti-tumor immune response. Bevacizumab, an anti-angiogenic drug, selectively binds to circulating VEGF and inhibits its binding to VEGFR on endothelial cells, limiting blood supply to tumor tissues. Antibodies against CCR4 and CD25 selectively deplete FOXP3+ Tregs which suppress the CTLs. Overpowering the suppressive effects of TME through immunotherapeutic interventions is an attractive treatment modality for HNSCC
A tumor antigen-specific TCR is introduced to genetically modify T cells that have sensitive recognition of tumor antigens, resulting in a specific and effective immune response against tumor cells. A chimeric antigen receptor (CAR) on the other hand is synthesized by linking signaling domains from T cells signaling machinery, a transmembrane unit, and a tumor-specific antibody fragment, resulting in a hybrid receptor. CAR-T cells have been developed for immunotherapy in HNSCC. CAR-mucin 1 (MUC1)-IL-22 T cells were found to have a robust and more effective cytotoxic function against MUC1+ HNSCC cells [209]. A retroviral human CD70 CAR construct, using truncated CD27 conjugated with 4-1BB (CD137/TNFRSF9) and CD3-zeta costimulatory molecules, was used to transduce activated human T cells to generate CD70 CAR-T cells which efficiently eliminated CD70-positive HNSCC cells [210]. A phase I clinical trial to evaluate intratumoral delivery of erythroblastic leukemia viral oncogene homologue (ErbB) targeted CAR T cells in locally advanced or recurrent HNSCC patients is initiated (NCT 01818323) [211].
Adoptive immunotherapy has shown promising outcomes in early-stage trials for many cancers. Lack of cancer-specific antigens, poor trafficking and penetration of CAR-expressing T cells into malignant deposits, and a hostile anti-inflammatory, hypoxic, and metabolically compromised TME all contribute to this challenge [212]. The successful approaches in HNSCC will require addressing all these challenges that are inherent in many solid tumors, such as identifying the right homing pathway, attaining adequate T-cell infiltration into the tumor, the discovery of an antigen that will not result in off-tumor effects, and most importantly persistence in the immunosuppressive TME.
Vaccination strategies employed for HNSCC are influenced by HPV infection. HPV vaccines are basically virus-like particles (VLPs) and can be effective against HPV-related diseases like head and neck cancer [213, 214]. These VLPs are derived in vitro using recombinant proteins and can stimulate naïve B cells. Several vaccines like Cervarix and Gardasil have been approved by United States-FDA against HPV [215, 216]. However, these vaccines are only preventative approaches against HPV and HPV-associated disorders [217]. HPV vaccines in relation to HNSCC specifically have not been used as a treatment strategy yet but they have shown to be promising in clinical trials [218].
Therapeutic cancer vaccines in HNSCC involve various vaccine delivery platforms that include cell-based vaccines, DNA vaccines, mRNA vaccines, peptide vaccines, and viral/bacterial vector-based vaccines. TLRs are important receptors involved in the systemic immune response against DNA and mRNA vaccines [219, 220]. Cell-based vaccines involve the use of modified tumor cells or immune cells with specific antigens for immunotherapy. Autologous modified tumor cells primed with co-stimulatory molecules may be used to enhance the immune response. DCs altered with specific epitopes is a well-investigated model for use as vaccines [221, 222]. One such example is AlloVax, a personalized vaccine for recurrent HNSCC undergoing a phase II study (NCT01998542). The immune activity induced in response to vaccines includes activation of CTLs, presentation of the various antigens by DCs, and maintenance of CTL activity by Th1 cells. Tumor-specific antigens (TSA), as well as TAAs, are used as targets for vaccines.
HNSCC has a relatively high mutational burden and the expressed “neoantigens” are likely targets for vaccine development [223]. Neoantigen recognizing T cells are key mediators of antitumoral immunity, especially in CPI treatment strategies [18, 224]. A recent study characterized the genomic and neoantigen changes between paired primary and recurrent HNSCC and identified increased T cell infiltration in tumors that shared these neoantigens [225].
TSAs include mutated neoepitopes and cancer-specific antigens that can elicit a potent immunogenic response but are less frequently available than TAAs like MUC1 and carcinoembryonic antigen (CEA). Trials targeting these TAAs are underway (NCT02544880, NCT00924092, NCT00027534). Vaccines against p53 and rat sarcoma (RAS) are also being investigated as immunotherapy in HNSCC [30, 226]. Cancer testis antigens (CTAs) are a type of TAAs that can generate a strong immune response owing to their expression on the tumor periphery. CTAs are considered good candidate targets for immunotherapy as they are characterized by a restricted expression in normal somatic tissues concomitant with a re-expression in solid epithelial cancers [227]. Various CTAs like melanoma antigen gene [melanoma-associated antigen family (MAGEA) 1, MAGEA3/6], lymphocyte antigen 6 complex locus K (LY6K), and cell division cycle associated 1 (CDCA1) have been identified as potential vaccine targets [228–232]. Combining stimulatory molecules like cytokines and vaccines targeting specific antigens is a better immunotherapy strategy than monotherapy. Depletion of immune suppressive cells like Tregs and MDSCs can aid in enhancing the efficacy of vaccines [233]. Vaccines in HNSCC are an attractive treatment modality because they can be personalized and provide a tumor-specific adaptive immune response.
HNSCC encompasses a heterogeneous group of diseases and exhibits varying degrees of immune dysregulation. Although currently approved immunotherapies have shown promising results, only a fraction of patients benefits from the therapy. It is therefore important to have an in-depth understanding of the complex TME in HNSCC that contributes to immune evasion (Figure 1). The immune landscape of HPV+ and HPV– tumors clearly indicates that these are to be treated as two independent entities. Future research should focus on understanding how the unique vulnerabilities of the HPV+ tumors can be exploited for improved immunotherapeutic interventions in these subsets of patients. Moreover, the high mutation burden in HNSCC may drive the neoantigen load in these tumors and help identify novel targets for immune intervention (Figure 2). Several challenges need to be addressed to harness the potential of immunotherapy for better treatment and management of HNSCC. The selection of the right combination of checkpoint molecules that can be targeted, biomarker identification to predict response or resistance to immunotherapy, and use of appropriate pharmacological compounds to inhibit the accumulation, trafficking, and functions of immunosuppressive cells in the TME is required to improve the outcome of immunotherapy in HNSCC.
ACT: adoptive cell transfer
ADCC: antibody-dependent cell cytotoxicity
Arg 1: arginase 1
CAF: cancer-associated fibroblast
CAR: chimeric antigen receptor
CCL: C-C chemokine ligand
CCR4: C-C chemokine receptor-4
Col: collagen
CPIs: checkpoint inhibitors
CTAs: cancer testis antigens
CTLA4: cytotoxic T-lymphocyte-associated antigen 4
CTLs: cytotoxic T lymphocytes
CXCR: C-X-C motif chemokine receptor
DCs: dendritic cells
EBV: Epstein-Barr virus
ECM: extracellular matrix
EGF: epidermal growth factor
EGFR: epidermal growth factor receptor
EMT: epithelial-mesenchymal transition
FDA: Food and Drug Administration
FGF: fibroblast growth factor
FOXP3: forkhead box P3
GITR: glucocorticoid-induced tumor necrosis factor-related
HIF: hypoxia-inducible factors
HLA: human leukocyte antigen
HNSCC: head and neck squamous cell carcinoma
HPV: human papilloma virus
IDO: indoleamine 2,3-dioxygenase
IFN: interferon
IL: interleukin
IMCs: immature myeloid cells
iNOS: inducible nitric oxide synthase
LAG: lymphocyte activated gene
mAb: monoclonal antibody
MAGE: melanoma-associated antigen family
MCP-1: monocyte chemoattractant protein-1
MDSC: myeloid-derived suppressor cell
MHC: major histocompatibility complex
MSC: mesenchymal stem cell
MUC1: mucin 1
NK: natural killer
NO: nitric oxide
NRP: neurolipin
ORR: overall response rate
OS: overall survival
OSCC: oral squamous cell carcinoma
PD-1: programmed cell death protein 1
PDGF: platelet-derived growth factor
PD-L: programmed cell death ligand
PGE2: prostaglandin E2
PI3K: phosphoinositide 3-kinase
R/M: recurrent/metastatic
ROS: reactive oxygen species
STAT: signal transducer and activator of transcription
TAA: tumor-associated antigen
TCR: T cell receptor
TF: transcription factor
TGF: transforming growth factor
Th: T helper type
TILs: tumor-infiltrating lymphocytes
Tim-3: mucin-domain containing-3
TLR: toll-like receptor
TLS: tertiary lymphoid structures
TMB: tumor mutational burden
TME: tumor microenvironment
TNF-α: tumor necrosis factor-α
Treg: regulatory T cells
TWIST: twist transcription factor
VEGF: vascular endothelial growth factor
VEGFR: vascular endothelial growth factor receptor
VIM: vimentin
We thank the Department of Atomic Energy, Government of India for providing fellowship to SKS, Integrated Cancer Treatment and Research Centre (ICTRC), Pune, India for providing fellowship to JHG and Central Council for Research in Ayurvedic Sciences (CCRAS), Government of India for providing fellowship to AV.
SVC conceptualized the content of the manuscript and helped in drafting the manuscript. SKS, JHG and AV contributed equally in drafting the manuscript. SKS, JHG and SVC reviewed and edited the final manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

