 Review
Review

Affiliation:
Independent Researcher, 13505 Berlin, Germany
Email: w.w.krause@web.de
ORCID: https://orcid.org/0000-0002-5397-9430
Explor Med. 2025;6:1001364 DOI: https://doi.org/10.37349/emed.2025.1001364
Received: July 10, 2025 Accepted: August 14, 2025 Published: October 15, 2025
Academic Editor: Hongzhou Lu, Shenzhen Third People’s Hospital, National Clinical Research Center for Infectious Diseases, China
The article belongs to the special issue Global Perspectives on the Clinical Diagnosis, Treatment, and Functional Cure of HIV Infection in the Post-ART Era
HIV/AIDS has changed from a deadly disease in the early 1990s to a chronic treatment following huge research efforts. HIV had a great impact due to the long period until its fatal consequences in the form of AIDS appeared. As a consequence, the spread of the disease was global. However, even now, after many years of extensive research, there is still no functional cure or eradication possible. This review provides an overview on HIV/AIDS covering a description of the disease, the mechanism of infection, HIV/AIDS symptoms, the current treatment options, the formation of latent reservoirs, and the efforts to provide a cure of HIV including CCR5Δ32/Δ32 donor stem cell transplantation, gene therapy, broadly neutralizing antibodies, HIV vaccination, chimeric antigen receptor cells, latency-addressing agents, and combination approaches.
The human immunodeficiency virus (HIV) is a retrovirus with a size of approximately 120 nm [1] and belongs to the group of lentiviruses. It contains a genome with two RNA strands, the enzymes integrase, reverse transcriptase, and protease, and the TAT protein. The viral genomic RNA comprises nine genes (gag, pol, env, tat, rev, nef, vif, vpr, and vpu) with a total size of less than 10 kb. It is capable of initiating the production of 15 proteins needed for replication in the host cell.
There are two species of virus, HIV-1 and HIV-2. Both have their origin in monkeys in Africa, HIV-1 in chimpanzees, and HIV-2 in the sooty mangabey. Probably at the beginning of the 20th century, the virus jumped species from animal to man via at least three stations, leading to the three groups of the virus, M, N, and O [2]. HIV-2 is less aggressive in terms of virulence and infectivity and is therefore more or less confined to West Africa, whereas HIV-1 has spread globally. In the following, the term HIV is used—if not explicitly otherwise stated—for HIV-1 for which the majority of research is applied. The reason for the worldwide spread of HIV lies in the fact that the virus only leads to clear symptoms years after infection, in some cases, early on to flu-like symptoms, and thereby is able to spread to quite a number of new hosts before treatment is initiated, resulting in exponential distribution. This behavior makes the difference to other virus infections, such as Ebola, which kill their hosts within days after infection and thereby prevent global spread.
HIV is sexually transmitted via semen or by the transfer of blood or breast milk of feeding mothers. Other body fluids, such as tears, saliva, or sweat, are not involved. Untreated, HIV infection finally leads to acquired immunodeficiency syndrome (AIDS). Life expectancy is approximately 11 years for untreated patients [3]. AIDS was first identified in 1981, and its connection to HIV was established in 1983 [4, 5].
HIV infection/AIDS proceeds in three phases. Following infection, flu-like symptoms, fever, or swelling of lymph nodes can occur and might last for one or two weeks. The next phase is symptomless and might last on average 11 years [6] but can reach up to 20 years [7]. The last phase is the AIDS phase and is characterized by opportunistic infections of any of the organ systems. The impact of HIV/AIDS has been tremendous regarding both society as a whole and individuals [8, 9].
Upon entry of the host’s body, the virus starts infecting immune cells, in particular T cells (CD4+ T cells), macrophages, and dendritic cells (DCs), and begins to replicate at an extremely high speed, leading to plasma levels of viruses in the millions per mL [10]. In parallel, the number of CD4+ T cells declines, and CD8+ T cells are activated and start killing viruses and producing antibodies directed against the virus. This leads to a decline in viral plasma levels after an initial peak. In the long run, the levels of CD4+ T cells become too low to be able to control opportunistic infections, and the patient inevitably develops AIDS, which finally leads to death. The AIDS phase begins when the levels of immune cells have decreased to approximately 200 lymphocytes per mm3. At that time point, the levels of virus had increased again to approximately 500 copies per mL.
HIV is diagnosed via blood tests, which are based on antibody or antibody + p24 antigen or PCR testing. They are possible starting 23–90 days after infection (antibody), 18–45 days after infection (antibody + p24), or 10–33 days after infection (PCR) [11].
HIV disseminates throughout the human body within 1–2 weeks [12]. Attachment of the virus to the host cell proceeds via the HIV envelope (Env) glycoprotein complex, which is built from three gp41 and three gp120 proteins that are formed following post-translational cleavage of the gp160 polypeptide [13]. The proteins are heavily protected using glycosylation against attacks by the host’s immune system [14] (Figure 1).
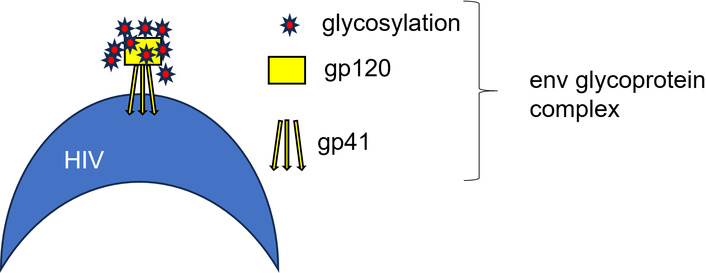
Composition of the Env glycoprotein complex of HIV. The Env ectodomain has a median of 30 N-linked glycosylation sites per protomer [15], which strongly limits accessibility to the protein surface by host defense systems. This and the enormous genetic variability due to the mutation of HIV make it nearly impossible for the host to generate antibodies that can neutralize HIV [16]. The same issue holds for the production of vaccines for HIV treatment, which will be discussed later.
The attachment procedure runs via HIV-mediated CD4 receptor and C-X-C chemokine receptor type 4 (CXCR4) or C-C chemokine receptor type 5 (CCR5) coreceptor interaction, leading to a gp120 conformation change, which is necessary for coreceptor coupling [17]. For the fusion of the virus and host cell membranes, gp41 is responsible [15]. CXCR4 primarily facilitates infection in CD4+ T cells, while CCR5 is mainly associated with macrophage infection [18]. Following attachment and fusion, the cell content of the virus (genome, reverse transcriptase, integrase, protease) is released into the host cell’s cytoplasm. The viral RNA is then converted into viral DNA with the help of reverse transcriptase, transported into the nucleus of the host cell through the nuclear pore, and embedded in chromatin, followed by integration into the host genome using the enzyme integrase. The integrated viral DNA is called provirus. It remains dormant, forming latent reservoirs unless the host cell activates it. Multiple copies of viral RNA are then created by the transcription of the provirus by the host cell RNA polymerase II enzyme [19] and transported into the cytoplasm. The other building blocks of the virus are created by the host’s ribosomes according to the plan laid down in the HIV RNA and inserted into the host’s genome. At the host cell membrane, the viral RNA genome and proteins are assembled to form multiple immature virions. After formation of an Env, the immature virions emerge from the host cell membrane [20, 21] and, after maturation, move on to infect further host cells. A summary of the procedure is provided in Figure 2. Productive infection is initially established in a pool of phenotypically and clonotypically distinct T cells, and latently infected cells are generated simultaneously [12].
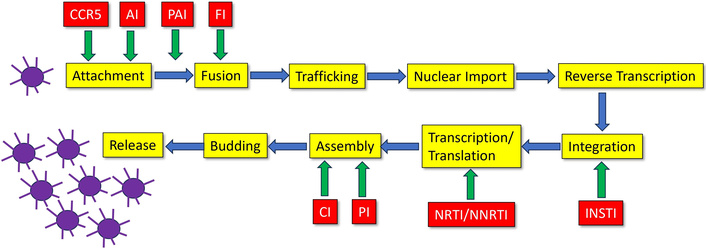
The HIV replication cycle of a single virus leads to multiple copies. NRTI: nucleoside reverse transcriptase inhibitors; NNRTI: non-nucleoside reverse transcriptase inhibitors; INSTI: integrase strand transfer inhibitors; PI: protease inhibitors; FI: fusion inhibitors; CCR5: C-C chemokine receptor type 5; AI: attachment inhibitors: bind to the gp120 protein of the virus; PAI: post-attachment inhibitors (block CD4 receptors on immune cells); CI: capsid inhibitors.
The HIV-1 genome contains 9,173 nucleotides and is primarily a coding RNA with nine open reading frames, which is able to produce 15 proteins [22]. The matrix of the virus, capsid and nucleocapsid, and p6 proteins are produced by the proteolytically processed Gag polyprotein precursor. Protease, reverse transcriptase, and integrase are compiled by the Gag-Pol polyprotein. The proteins gp120 and gp41 are encoded in the env gene. Virions contain genomic RNA as a non-covalent dimer, which is 5' capped and 3' polyadenylated.
According to WHO [23], HIV infection is diagnosed by antibody testing based on two tests with different antibodies and/or by two positive virological tests for HIV or its components (HIV-RNA or HIV-DNA or ultrasensitive HIV p24 antigen). The immune status of a patient is assessed by measuring the absolute number (per mm3) or percentage of CD4+ T cells. Increasing depletion of CD4+ T cells is a clear sign of progression coupled with an increased likelihood of opportunistic infections and other clinical events, leading, finally, if untreated, to death.
Clinical criteria for diagnosis of advanced HIV in adults and children with confirmed HIV infection [23]:
Presumptive or definitive diagnosis of any stage 3 or stage 4 condition and/or;
Immunological criteria for diagnosing advanced HIV in adults and children five years or older with confirmed HIV infection:
CD4 count less than 350 per mm3 of blood in an HIV-infected adult or child and/or;
Immunological criteria for diagnosing advanced HIV in a child younger than five years of age with confirmed HIV infection:
%CD4+ < 30 among those younger than 12 months;
%CD4+ < 25 among those aged 12–35 months;
%CD4+ < 20 among those aged 36–59 months;
HIV/AIDS develops in three stages: acute infection, clinical latency, and AIDS [23]. Primary infection usually presents as an acute febrile illness 2–4 weeks post-exposure, often with lymphadenopathy, pharyngitis, maculopapular rash, orogenital ulcers, and meningoencephalitis. Clinical staging of established HIV infection comprises four stages: asymptomatic (1), mild (2, e.g., moderate weight loss), advanced (3, severe weight loss), and severe symptoms (4, substantial weight loss). More details are available in [23].
HIV infection is still an uncurable disease necessitating lifelong treatment. Start of ART (antiretroviral therapy, formerly called highly active antiretroviral therapy, HAART) is recommended as soon as the diagnosis has been made [14, 24] and irrespective of baseline CD4 count. It may even be provided the same day [17]. Based on the different parts of the mechanism of HIV replication, there are currently nine classes and more than 30 drugs described on the National Institute of Health (NIH) website [25], ranging from single-drug to four-drug tablets, as outlined in Tables 1 and 2.
FDA-approved HIV drugs.
| Drug | Class | FDA approval | Brandname | Remarks |
|---|---|---|---|---|
| Zidovudine | NRTI | 1987 | Retrovir | |
| Lamivudine | NRTI | 1995 | Epivir | |
| Abacavir | NRTI | 1998 | Ziagen | |
| Tenofovir disoproxil fumarate | NRTI | 2001 | Viread | |
| Emtricitabine | NRTI | 2003 | Emtriva | |
| Tenofovir alafenamide | NRTI | 2016 | Vemlidy | |
| Nevirapine | NNRTI | 1996 | Viramune | Discontinued |
| Efavirenz | NNRTI | 1998 | Sustiva | Discontinued |
| Etravirine | NNRTI | 2008 | Intelence | |
| Nevirapine extended release | NNRTI | 2011 | Viramune XR | Discontinued |
| Rilpivirine | NNRTI | 2011 | Edurant | Discontinued |
| 2024 | Edurant PED | |||
| Doravirine | NNRTI | 2018 | Pifeltro | |
| Raltegravir | INSTI | 2007 | Isentress | |
| 2017 | Isentress HD | |||
| Dolutegravir | INSTI | 2013 | Tivicay | |
| 2020 | Tivicay PD | |||
| Cabotegravir | INSTI | 2021 | Apretude | Injection bimonthlyTablet |
| 2021 | Vocabria | |||
| Ritonavir | PI | 1996 | Norvir | |
| Atazanvir | PI | 2003 | Reyataz | |
| Fosamprenavir | PI | 2003 | Lexiva | Discontinued |
| Tipranvir | PI | 2005 | Aptivus | |
| Darunavir | PI | 2006 | Prezista | |
| Enfuvirtide | FI | 2003 | Fuzeon | Discontinued |
| Maraviroc | CCR5 | 2007 | Selzentry | |
| Cobicistat | PE | 2014 | Tybost | |
| Fostemsavir | AI | 2020 | Rukobia | |
| Ibalizumab | PAI | 2018 | Trogarzo | |
| Lenacapavir | CI | 2022 | Sunlenca | Prophylaxis injection, 2 per year |
| 2025 | Yeztugo |
NRTI: nucleoside reverse transcriptase inhibitors; NNRTI: non-nucleoside reverse transcriptase inhibitors; INSTI: integrase strand transfer inhibitors; PI: protease inhibitors; FI: fusion inhibitors; CCR5: C-C chemokine receptor type 5; PE: pharmacokinetic enhancer; AI: attachment inhibitors: bind to the gp120 protein of the virus; PAI: post-attachment inhibitors (block CD4 receptors on immune cells); CI: capsid inhibitors.
FDA-approved combination drugs.
| Drugs | Classes | FDA approval | Brandname | Remarks |
|---|---|---|---|---|
| Abacavir, Lamivudine | NRTI, NRTI | 2004 | Epzicom | Discontinued |
| Emtricitabine, Tenofovir disoproxil fumarate | NRTI, NRTI | 2004 | Truvada | |
| Atazanavir, Cobicistat | PI, PE | 2015 | Evotaz | |
| Darunavir, Cobicistat | PI, PE | 2015 | Prezcobix | |
| Emtricitabine, Tenofovir alafenamide | NRTI, NRTI | 2016 | Descovy | |
| Dolutegravir, Rilpivirine | INSTI, NNRTI | 2017 | Juluca | |
| Lamivudine, Tenofovir disoproxil fumarate | NRTI, NRTI | 2018 | Cimduo | |
| Dolutegravir, Lamivudine | INSTI, NRTI | 2019 | Dovato | |
| Cabotegravir, Rilpivirine | INSTI, NNRTI | 2021 | Cabenuva | |
| Abacavir, Lamivudine, Zidovudine | NRTI, NRTI, NRTI | 2000 | Trizivir | Discontinued |
| Efavirenz, Emtricitabine, Tenofovir disoproxil fumarate | NNRTI, NRTI, NRTI | 2006 | Atripla | Discontinued |
| Emtricitabine, Rilpivirine, Tenofovir disoproxil fumarate | NRTI, NNRTI, NRTI | 2011 | Complera | |
| Abacavir, Dolutegravir, Lamivudine | NRTI, INSTI, NRTI, | 2014 | Triumeq | |
| 2022 | Triumeq PD | |||
| Bictegravir, Emtricitabine, Tenofovir alafenamide | INSTI, NRTI, NRTI | 2018 | Biktarvy | |
| Doravirine, Lamivudine, Tenofovir disoproxil fumarate | NNRTI, NRTI, NRTI | 2018 | Delstrigo | |
| Efavirenz, Lamivudine, Tenofovir disoproxil fumarate | NNRTI, NRTI, NRTI | 2018 | Symfi | Discontinued |
| 2018 | Symfi Lo | |||
| Elvitegravir, Cobicistat, Emtricitabine, Tenofovir disoproxil fumarate | INSTI, PE, NRTI, NRTI | 2012 | Stribild | |
| Elvitegravir, Cobicistat, Emtricitabine, Tenofovir alafenamide | INSTI, PE, NRTI, NRTI | 2015 | Gemvoya | |
| Emtricitabine, Rilpivirine, Tenofovir alafenamide | NRTI, NNRTI, NRTI | 2016 | Odefsey | |
| Darunavir, Cobicistat, Emtricitabine, Tenofovir alafenamide | PI, PE, NRTI, NRTI | 2018 | Symtuza |
NRTI: nucleoside reverse transcriptase inhibitors; PI: protease inhibitors; PE: pharmacokinetic enhancer; NNRTI: non-nucleoside reverse transcriptase inhibitors; INSTI: integrase strand transfer inhibitors.
Due to rapid viral reproduction, peak levels of RNA can reach up to 107 copies per mL of plasma [26]. T cells infected before ART decline rapidly, with an estimated half-life of 1–2 days [27, 28]. ART leads to a decay of viral RNA in the plasma of patients down to 0.6 copies/mL. Adherence to continuous drug intake according to prescription is the most important issue of HIV medication. It allows a nearly normal life of the patient with viral plasma levels below the detection limit. As a further result, the patient is no longer transmittable regarding the virus. According to Matsui et al. [29], “undetectable = untransmittable” (U = U). Based on the same U = U principle, lenacapavir was developed for the prophylaxis of HIV infection [30, 31]. Additionally, it is active against highly resistant treatment of HIV [32]. Lenacapavir is a capsid inhibitor that is prophylactically injected every 6 months.
Cessation of drug intake on ART inevitably results in recurrence of the virus in the blood or viral rebound, which occurs within several weeks, with a mean of two weeks, in the majority of patients [33, 34]. The cause of viral rebound is that a small fraction of infected circulating CD4+ T cells with intact viral genomes (below 5–10%) transition into a state of latency and only regain activity as quickly as 14 days after ART is discontinued [35]. That is the main reason why ART is a lifelong treatment.
However, approx. 0.2–0.5% of HIV patients (“elite controllers”) are able to sustain viral load suppression and sufficiently high CD4+ T cell counts after discontinuing ART [17, 36]. Elite controllers are characterized by broad, specific, and superior T-cell, innate cell, and natural killer (NK) cell effector functions. Detailed analyses for previously untreated elite controllers have been reported by Turk et al. [37] and by Jiang et al. [38]. They conclude that these elite controllers seem to exemplify attributes of a “block and lock” mechanism of viral control, defined by silencing of pro-viral gene expression through chromosomal integration into repressive chromatin locations caused by cell-mediated immune selection forces that preferentially eliminate pro-viral sequences more permissive to viral transcription, in a process that they suggest referring to as the “autologous shock and kill” mechanism.
Latent HIV reservoirs are the major hurdle to HIV cure. The HIV reservoir is shaped by viral transcriptional suppression and by clonal expansion [39]. Genetic diversity of the virus increases rapidly during infection, due to errors in reverse transcription [40] and high rates of viral recombination and turnover [21, 27]. Latency is established very early in the course of infection, typically within the first days or weeks following exposure [41] and cannot be blocked by early ART [42]. However, early treatment does have an effect on the time of viral rebound [43]. Currently, a clinical trial (NCT02140255) is recruiting infants with HIV using early intensive treatment to achieve HIV remission. The ART regimen consists of three to four components out of nucleoside reverse transcriptase inhibitors (NRTIs), nevirapine, raltegravir, dolutegravir, VRC07-523LS, and VRC01 administered within 48 h of birth. In a simian immunodeficiency virus (SIV) monkey model, latency was observed as early as three days after infection [44]. HIV integrates into the host genome as a translationally inactive provirus, resulting in a long-lived (latent) reservoir of infected cells. The potential cell targets range from T cells to macrophages, myeloid cells, mast cells, NK cells, microglia, and DCs [45–48]. Most commonly, resting memory CD4+ T cells are involved [38]. Intact HIV proviruses are predominantly integrated in heterochromatin [49]. Transcriptional silence might only occur in a subset of proviruses integrated in repressive heterochromatin locations, such as repetitive satellite DNA or zinc finger (ZNF) genes that are loaded with inhibitory histone modifications deposited by the human silencing hub (HUSH) complex [50].
The affected body parts include lymphoid tissues, which cover most of the HIV reservoir [49, 51, 52], in particular the gut mucosa and gut-associated lymphoid tissue (GALT) [53], the central nervous system [36, 54] (where astrocytes [55], pericytes [56] and myeloid-resident cells, such as microglia and perivascular macrophages [57] can be infected), reproductive organs, lung, heart, liver, kidneys, thymus, and bone marrow [58]. These are body regions, some of which cannot easily be reached by ART. But even if they could be reached, it would not help, since the proviruses are protected by their translational inactivity.
HIV latency in T cells is maintained by blocking transcriptional elongation, completion, and splicing [59, 60], keeping the phenotype of the cells unchanged [61–64]. More than 50% of all latently infected cells after ART treatment result from some type of clonal expansion [41]. Latency can be induced by two mechanisms: the loss or inhibition of host post-transcriptional regulatory systems and the scarcity of several RNA exporters, such as polypyrimidine tract-binding protein (PTB) or matrin3 (MATR3), which causes nucleus retention of HIV RNAs in resting CD4+ T cells [65, 66]. Various studies have shown that the half-life of the HIV reservoir can range somewhere between 44 months and 13 years, and in some cohorts, no decay was observed at all [39].
Before going into detail, we need to define what “HIV cure” means. It should comprise the following characteristics:
No further treatment
Permanent suppression of HIV transcription
No disease progression
Undetectable virus replication
No viral transmission
In a consensus meeting, a Target Product Profile for a procedure or drug that achieves HIV cure has been agreed upon, including the steps to come up with a single drug that cures HIV in one administration [67, 68].
The currently approved drugs used in ART are characterized by the fact that they are not able to kill the virus but, instead, inhibit viral replication. However, any drug able to actually kill the virus, additionally, would need the ability to reach it, wherever it is hiding, which is a second, and hardly achievable goal. In principle, the human immune system should be the first line of defense. Unfortunately, this is actually the major point of attack of the virus, shutting down the immune system, not completely, but long enough to allow for widespread distribution of the virus within the population. Shutting the immune system down, however, is not enough for the virus; disabling any attack by the immune system comes on top. Furthermore, constant change of the viral surface makes it nearly impossible to use antibodies for the treatment directed against the virus. Any vaccination efforts have therefore so far been unsuccessful.
Attempts to cure HIV can be characterized into the following categories, which—except for the last one—need combinations of more than one type:
Kill the virus
Unlock the latent reservoirs
Permanently close down the reservoirs
Modify the surface of the T cells (CCR5Δ32 mutation)
We will start with the last category, which has been performed by stem cell transplantation and which has provided a cure for HIV to a small number of patients.
As already mentioned above, CCR5, a β-chemokine receptor gene, is needed as a co-receptor in addition to the CD4 receptor for the virus to infect CD4+ T-cells. A mutation with deletion of 32 base pairs (CCR5Δ32/Δ32) in the coding region prevents CCR5 expression on the surface of the cells and makes the potential host immune to infection, in case of homozygosity [69, 70]. The “Berlin Patient”, who suffered from acute myeloid leukemia (AML) on top of HIV infection and who was on ART treatment (4 years of efavirenz, emtricitabine, and tenofovir), needed a stem cell transplantation to treat AML. He received two CCR5Δ32/Δ32 allogeneic stem cell transplantations and was cured of HIV. He died 12 years later from a relapse of AML [71]. More examples are the London patient (tenofovir disoproxil fumarate, emtricitabine, and efavirenz), who suffered from Hodgkin lymphoma [72], the Düsseldorf patient (tenofovir disoproxil fumarate, emtricitabine, and darunavir) with AML [73], and the New York patient (tenofovir, emtricitabine, and raltegravir) with leukemia [74]. Further details are summarized in Table 3.
HIV patients cured by stem cell transplantation.
| Patient | HIV (D)ART (T) | 2nd Disease | Conditioning | Transplantation | Remarks | Reference |
|---|---|---|---|---|---|---|
| Berlin | 1995 (D), 2003 (T) | AML, 2007 | Chemotherapy, total body irradiation (200 cGy) | 2× CCR5Δ32/Δ32, allo-HSCT, blood stem cells | Died in 2020 from AML | [71] |
| London | 2003 (D), 2012 (T) | Hodgkin lymphoma, 2012 | Alemtuzumab, cyclosporine A, methotrexate | CCR5Δ32/Δ32, allo-HSCT, bone marrow | ART post-transplantation for 510 days | [72] |
| Düsseldorf | 2008 (D), 2010 (T) | AML, 2011 | Reduced-intensity conditioning | CCR5Δ32/Δ32, allo-HSCT, blood stem cells | ART post-transplantation for 69 months | [73] |
| New York | 2013 (D), 2017 (T) | AML, 2017 | Fludarabine, melphalan, total body irradiation, anti-thymocyte globulin | CCR5Δ32/Δ32, haplocord and blood stem cells | ART post-transplantation for 37 months | [69] |
| City of Hope | 1988 (D), 1997 (T) | AML, 2019 | Fludarabine, melphalan | CCR5Δ32/Δ32, allo-HSCT, blood stem cells | ART post-transplantation for 25 months | [75] |
| Geneva | 1990 (D), 1990 (T) | Myeloid sarcoma, 2018 | High intensity, irradiation (8 Gray) | CCR5WT/WT, allo-HSCT, blood stem cells, 2018 | ART + ruxolitinib until 11.2021 | [76] |
| Berlin #2 | 2009 (D) | AML, 2015 | Total body irradiation, intensive chemotherapy | CCR5Δ32/WT, allo-HSCT, bone marrow, 2015 | ART post-transplantation until 2018 | [77] |
| NN | 1999 (D) | AML, 2020 | Cyclophosphamide, irradiation (200 cGy) | CCR5Δ32/Δ32 allo-HSCT, 2020 | Analytic treatment interruption in 10.2023 | [78] |
HIV(D): year of HIV diagnosis; ART(T): start of antiretroviral therapy; AML: acute myeloid leukemia; allo-HSCT: allogeneic hematopoietic stem cell transplantation; WT: wild type.
The reduction in the size of the HIV reservoir in these patients seems to result from a combination of cytotoxic effects of the conditioning regimens, donor allogeneic immunity during graft-versus-host reactions, and the gradual dilution of the pool of infected cells during immune cell replacement [79, 80].
However, although stem cell transplantation did work, it is not feasible for a broader range of HIV patients because the procedure is high-risk with a high mortality rate. It is extremely expensive and can only be performed in dedicated centers, and lastly, there are not enough potential CCR5Δ32/Δ32 donors available (< 1% of the Caucasian population).
In gene therapy, a healthy gene is inserted into the cell using a vector, for example, a polymer, a plasmid, or a viral vector, with the objective of initiating the expression of the new gene. Alternatively, using recombinant DNA technology, the mutant gene can be knocked out and replaced by a new gene. Another method is RNA interference (RNAi), where a double-stranded RNA neutralizes targeted mRNA molecules using small/short interfering RNAs (siRNAs) and small/short hairpin RNAs (shRNAs). A review has been published by [81]. Gene editing (ex vivo and in vivo) of CCR5 or of the virus itself has been actively explored as a strategy to cure HIV. The approaches are using zinc finger nucleases (ZFN) to target CCR5, transcription activator-like effector nucleases (TALEN), and Clustered Regularly Interspaced Short Palindromic Repeats/Cas-9 (CRISPR/Cas-9). mRNA encapsulation in lipid nanoparticles (LNPs), as already widely used in other indications, such as COVID-19, may facilitate delivery of gene therapy, including mRNA encoding CRISPR–Cas9 and other approaches [68]. So far, success, in terms of HIV cure, has not yet been reported. In Table 4, an overview of gene therapy studies is provided.
Gene therapy studies.
| Method | Drug | Reference(s) |
|---|---|---|
| Modified T cells | AGT103-T | NCT05529342, NCT05540964, NCT04561258 |
| ZFN-modified T cells | SB-728-T or SB-728mR-T | NCT04201782, NCT01044654 |
| Modified T cells | Vector VRX496 | NCT00295477 |
| Modified lymphocytes | Lymphocytes, Gemini study | NCT04799483 |
| Modified CD4+ and CD8+ cells | CD4+ and CD8+ cells | NCT00001409 |
| Modified CD34+ cells | CD34+ cells ± busulfan | NCT03517631 |
| CD4 CAR + CCR5 ZFN T-cells | T cells | NCT03617198 |
| Modified lymphocytes | Lymphocytes + NeoR gene | NCT00001353 |
| Naked DNA with human IL-12 mutant as an immune adjuvant | GX-12 | NCT00517569 |
| Modified T cells | pHIV7-shI-TAR-CCR5RZ treated CD4 cells | NCT01153646 |
| Modified stem cells HSPC | SB-728mR-HSPC after conditioning with busulfan | NCT02500849 |
| Modified T cells | CD4-zeta gene-changed T cells and/or IL-2 (recombinant interleukin-2) | NCT01013415 |
| CRISPR/Cas9 gene editing | EBT-101 in AAV9 vector | NCT05144386 |
| Anti-HIV-1 ribozyme transduced cells | OZ1 | NCT00074997, NCT01177059 |
ZFN: zinc finger nuclease; AAV9: adenovirus-associated virus vector serotype 9; HSPC: hematopoietic stem cell transplantation.
ART is able to effectively protect uninfected cells from viral infection, but has no activity against cells that are already infected and harbor chromosomally-integrated proviral DNA [50]. Antibody-dependent cell-mediated cytotoxicity (ADCC) might be a path towards killing infected cells. An example of effective ADCC and complement-dependent cytotoxicity is the CD52-targeting monoclonal antibody, alemtuzumab. CD52 is a peptide with 12 amino acids and a glycosylphosphatidylinositol (GPI) anchor. It is highly negatively charged and can be found on sperm cells and lymphocytes. Its function is presumably anti-adhesion, allowing cells to freely move around [82–87]. In humans, CD52 is expressed at high levels on B and T lymphocytes and at lower levels on NK cells [84]. CD52 expression on immune cells is retained in HIV-1 infection regardless of CD4 cell count, viral load, and treatment status, and is amenable to alemtuzumab-induced depletion. In an in vitro experiment, Ruxrungtham et al. [88] studied the ADCC efficacy in blood samples of HIV patients and found extensive cell killing. Alemtuzumab also kills non-infected immune cells circulating in the blood, thereby preventing further infection. Rasmussen describes the effects of alemtuzumab treatment in an HIV infected individual on ART with Sezary syndrome [89]. The antibody effectively depleted circulating T cells and decreased the frequency of latently infected CD4+ T cells, but HIV DNA remained detectable even after extensive CD4+ T cell depletion. In a monkey model using SIV-infected rhesus macaques, alemtuzumab was administered either at the time of ART initiation (at 42–46 weeks after infection) or during chronic ART treatment (at 42 weeks after infection). Alemtuzumab treatment resulted in substantial depletion (> 95%) of CD4+ T cells in blood. In lymph nodes, the depletion was only modest [90]. Similarly, in cynomolgus monkeys who received alemtuzumab following cardiac transplant, there was a much more profound reduction of CD4+ T-cells in peripheral blood (99% reduction) than in lymph nodes (70% reduction) [91]. The lessons learned from these experiments are, first, that alemtuzumab efficiently kills normal and infected immune cells and, second, as expected, latent reservoirs, such as lymph nodes, are only incompletely accessible for the antibody. The conclusion is that alemtuzumab alone is not suitable for treating HIV infection, as soon as latent reservoirs have been formed. At the moment, we cannot exclude that treatment could be possible before latency occurs. To get a closer opinion on this statement, a monkey study in SIV-infected animals should be performed with antibody treatment on the day of infection. Alternatively, alemtuzumab might be a suitable partner for either “shock and kill” or “block and lock” approaches. These combination strategies will be addressed later in this review.
Elite controllers are patients who are able to avoid rebound of HIV after discontinuation of ART due to the initiation of potent CD8+ T cell responses, which result in the production of bNabs targeting up to 99% of tested HIV variants [92]. This observation heavily triggered research into bNAbs. One of the bNabs detected in elite controllers is VRC01, which is targeting the CD4-binding site on the Env glycoprotein gp120 [93]. The Env glycoprotein complex described above is the primary target of the HIV virus available for antibody attachment. It is built from three gp41 and three gp120 proteins and is heavily protected by glycosylation. Furthermore, it shows a great sequence heterogeneity in its Env proteins. First-generation antibodies were directed against this epitope [94] with the intention to prevent virions from entering host cells, resulting in bNabs such as VRC01, 3BNC117, and BH10 gpl20 [12]. However, their clinical effect on HIV was disappointing [95]. On the other hand, HIV and other viruses, such as influenza and hepatitis, do harbor highly conserved exposed sites, usually associated with function, which can be targeted by bNabs [96]. In order to circumvent the limited accessibility of the Env epitope, bNabs have been developed that target epitopes that are preserved in spite of heavy mutation of the rest of the virus. The epitopes of interest (with examples of bNabs in parentheses) were the following: MPER of gp41 (2F5) [97], outer domain glycan, the V1V2-glycan site (PGDM1400 [98], CAP256-VRC26.25), V3-glycan (PCDN76-33A [99], 1B2530 [100], BF520.1 [101]), and the CD4 binding site [102], CH103 [103], 12A12 [104]. Reviews are available by Haynes et al. [100] and by Abana et al. [105]. In SIV monkey studies, bNabs have shown different grades of efficacy [106]. Further studies of bNabs in monkeys showed that the emergence of virus in plasma and lymph nodes was delayed by bNAb treatment [107] and that bNAbs given early in infection and before natural antibodies occur may lead to the formation of immune complexes, which, following cross presentation to CD8+ T cells, may boost antiviral immune responses and lead to viral control. bNAbs with increased affinity to FcγRs could be shown to shape innate and adaptive cellular immunity [108]. bNabs alone will probably not be able to lead to an HIV cure, although a combination of bNabs [3BNC117 + 10–1074] showed good results in a clinical trial [107]. Long-term suppression was observed in the absence of ART. According to Galvez et al. [109], individual bNAbs have limited activity due to antibody half-life or escape, whereas vectored delivery of a single bNAb, well-matched to the infecting strain, is capable of continuously suppressing a subset of viremic humanized mice. Several well-characterized bNAbs can facilitate the killing of HIV-infected cells through NK cell-mediated ADCC [110, 111]. The bNAbs that arise during natural HIV-1 infections tend to have unusual features likely to pose challenges for vaccination, such as high levels of somatic mutation, autoreactivity with self-proteins, and long complementarity-determining regions [112]. In summary, bNAbs can create a vaccine-like effect and are a potential alternative to vaccines, but they alone will most likely not be able to cure HIV.
For vaccination, two potential applications are conceivable: prophylactic or therapeutic use, which means either preventing infection with HIV or treating HIV infection. In the latter case, treatment could either be stimulatory by using the host’s own immune system or indirectly by administering a vaccine that results in compounds that can either fight the virus by themselves or in a joint effort with the host’s immune system. An example of the stimulatory approach is T cells, and for the second approach, bNabs.
Some of the hurdles that need to be overcome in HIV vaccination have already been touched upon in the section on bNabs, since vaccination is one of the tools to produce bNabs. Challenges in vaccine development include the lack of known immune correlates, suitable animal models, and the high mutation rate of the virus [17], resulting in an extremely high genetic viral diversity [112]. Overcoming immune evasion [113] and the integration into host immune cells are making the virus resistant to host immunity and treatment [114]. Potential epitopes that can be addressed by vaccination are Env, V2apex, MPER, and the CD4 receptor. The first step in the vaccination procedure is to find an appropriate immunogen addressing these epitopes. For example, for the Env target, a major progress was the development of a stable trimer (SOSIP Env trimer) [115]. Meanwhile, the number of potential vaccine candidates has increased considerably, and some of them have been tested in clinical trials. Table 5 provides an overview of the latest studies.
Summary of recently initiated clinical trials on HIV vaccination according to ClinTrials.gov.
| Vaccine | Study | Procedure | Status |
|---|---|---|---|
| 426c.Mod.Core-C4b | NCT06006546 | Germline (GL)-targeting HIV-1 envelope (Env) protein -derived immunogens to activate naïve B cells that express the unmutated GL forms of VRC01-class bnAbsAdjuvant: 3M-052-AF + alum | Recruiting |
| VRC07-523LS + PGDM1400LS + ChAdOx1.tHIVconsv1, ChAdOx1.HIVconsv62 prime, MVA.tHIVconsv4 + A244d11gp120 | NCT06484335 | Determine safety and impact on viral load setpointAdjuvant: ALFQ | RV630ACHIEVPhase INot yet recruiting |
| VRC07-523LS | NCT06484335 | VRC07-523LS (bNab) + PGDM1400LS (bNab) + ChAdOx1.tHIVconsv1 + ChAdOx1.HIVconsv62 prime + MVA.tHIVconsv4 + A244d11gp120Adjuvant: ALFQ | Phase INot yet recruiting |
| CH505 TF chTrimer + 3M-052-AF | NCT06680479 | Induce new HIV-1 Env B-cell neutralizing immune responsesAdjuvant: alum | A5422Phase 1Recruiting |
| GRAdHIVNE1 | NCT06617091 | Gorilla-isolated (GRAd32) adenovirus vectored networked epitopes vaccine | IAVI C114Phase 1Not yet recruiting |
| A244 + B.63521 | NCT05423418 | A244 consists of the gp120 Env glycoprotein HIV-1 subtype CRF_01AE A244 derived from the CM244 CRF_01AEAdjuvant: ALFQ | RV575Phase 1Recruiting |
| IHV01 + A244 + AHFG ± ALFQ | NCT04658667 | IHV01: full-length single chain (FLSC) gp120-CD4 chimera subunit HIV-1 vaccine, encoded by a synthetic gene, which contains a human codon-optimized HIV (BaL) gp120 sequence followed by human CD4D1D2, with a flexible 20-amino acid linkerAdjuvant: aluminum phosphatePatients had previously received a late boost of AIDSVAX®B/E ± ALVAC in RV306 | RV546Recruiting |
| Maraviroc + Dolutegravir + Dendritic Cell Vaccine + Auranofin + Sirtuin Histone deacetylase inhibitor | NCT06805656 | Multi-interventional approaches | Not yet recruiting |
| 426c.Mod.Core-C4b | NCT06613789 | Adjuvant: 3M-052-AF + alum | HVTN316Phase 1 |
| Hiltonol: Poly-ICLC-adjuvanted CD40. HIVRI.Env (VRIPRO) | NCT06665646 | Who previously participated in the HVTN706/MOSAICO study and remain without HIV | Phase 1Not yet recruiting |
| 426c.Mod.Core-C4b | NCT06006546 | Germline (GL)-targeting HIV-1 Env protein-derived immunogens to activate naïve B cells that express the unmutated GL forms of VRC01-class broadly neutralizing antibodies (bnAbs)ATI for elicitation of VRC01-lineage antibodies adjuvant 3M-052-AF + alum | Recruiting |
| Ad4-Env150KN + VRC-HIVRGP096-00-VP (Trimer 4571) with alumAd4-Env145NFL + VRC-HIVRGP096-00-VP (Trimer 4571) with alum | NCT03878121 | Adenovirus serotype 4 (Ad4)-based HIV vaccinesNasal administration | Phase 1Recruiting |
| Stabilized CH505 TF chTrimer | NCT06680479 | Inducing new HIV-1 Env B-cell neutralizing immune responsesadmixed with 3M-052-AF + alum | A5422Phase 1 |
| UVAX-1107 ± UVAX-1197 | NCT06905275 | Glycan-trimmed HIV-1 nanoparticle vaccine (UVAX-1107), followed by homologous or wild-type HIV-1 nanoparticle vaccine (UVAX-1197) boost, each adjuvanted with 3M-052-AF + alum | Phase 1Not yet recruiting |
| Trimer 4571 | NCT04985760 | To generate bNabs against HIV trimer 4571 adjuvant: alum | Active, not recruiting |
AHFG: aluminum hydroxide fluid gel; ATI: (antiretroviral) analytical treatment interruption; Alum: aluminum hydroxide; ALF: army liposome formulation; ALFQ: army liposome formulation mixed with the saponin QS-21(Quillaja saponaria-21) adjuvant.
Another direction of research includes viral vaccine vectors, such as adenoviruses and poxviruses, which allow for prime/boost sequences, where an initial vaccination is followed by boosters using either homologous or heterologous vectors to deliver the same antigen [17]. Homologous boosters induce humoral responses, and heterologous boosters improve Th1 and memory T cell responses [116]. A cytomegalovirus (CMV) vaccine containing SIV Gag, Rev/Tat/Nef, Env, and Pol inserts has been tested in an SIV model, resulting in undetectable SIV DNA after 30 months and a decline in latent reservoirs [117]. SAV001‑H is a modified, dead HIV-1 virus and has been tested in a phase I study (NCT01546818). Antibodies against the p24 capsid antigen and against gp120 were found [118]. Another target is the regulatory protein TAT, which is part of the HIV transcription and replication system [17]. A vaccine would disrupt the capacity of the virus to replicate and propagate.
DNA vaccines mimic live attenuated vaccines. They induce innate, humoral, and cellular immune responses [119]. HIVIS is a multigene, multi-subtype HIV-DNA vaccine. It is a combination of plasmid DNA constructs and encodes several components, including structural and non-structural genes from several subtypes of HIV-1 [120]. This HIV-DNA has been studied extensively in terms of preclinical safety and immunogenicity. Homologous DNA immunizations augmented by electroporation stimulated strong cellular immune responses. When considering both cellular and humoral immune responses, a combination of DNA, modified vaccinia Ankara viral vector expressing HIV-1 genes env/gag/pol (MVA-CMDR), and rgp140C immunizations induced the overall most potent immune responses and the highest avidity of HIV Env-specific antibodies [121]. After the experience with the COVID-19 pandemic, RNA vaccines moved into the scene. They have many advantages over other types of vaccines. They are easy to design, stable in vitro and in vivo, scalable in production, and can lead to durable T and B cell responses while also stimulating the innate immune system [122, 123]. Preclinically, several Env-based RNA vaccines have been shown to generate robust antigen-specific responses [124, 125]. Clinical trials have not yet been initiated.
Since CD8 T cells target HIV infected cells, vaccines based on T cells are being developed both for prophylaxis and for therapy [15]. The following vaccines have already reached efficacy trials: VAX003 and VAX004 are aiming at nAbs, MRKAd5 is trivalent with Gag, Pol, and Nef genes, HVTN505 with 6 DNA plasmids, RV144, a heterologous prime-boost combination, and HVTN702.
DC vaccines are produced by collecting DCs of the patient, cultivating them in vitro together with an appropriate antigen, and reinjecting them together with an adjuvant into the patient. Clinical trials have shown safety and immunogenic efficacy [126].
Currently, HIV vaccines for therapeutic purposes can boost the immune response in HIV-1 infection clearances but have not yet induced sustained HIV remission. One of the major reasons is that they are not able to thoroughly eliminate the latent HIV reservoir [127]. Prophylactic vaccination might be the lower-hanging fruit, but it is much harder to verify. Further challenges are the lack of known immune correlates, suitable animal models, and the high mutation rate of the virus. Instead of using single-agent strategies, the combined use of synergistic anti-HIV-1 agents (LRAs and vaccination) is more likely to be successful.
The CAR cell family comprises the following members: CAR T cells, CAR NK cells, and CAR-encoding hematopoietic stem/progenitor cells [128]. CARs are synthetic proteins with an extracellular antigen recognition domain, for example, a single-chain fragment of an antibody, and a domain for intracellular signaling, for example, CD3 or other CDs. In autologous CAR T or CAR NK therapy, T or NK cells are isolated from the blood of the patient, genetically modified to express CARs, expanded, and re-injected into the patient to recognize and kill HIV-infected cells and eliminate HIV reservoirs [129].
CAR T cells are able to recognize and eliminate infected cells [130], but have failed in clinical trials. In CAR T-cell therapy, CAR T cell expansion, persistence, off-target effects, and severe cytokine release syndrome have been reported [127]. 2nd-generation CARs have additional domains, such as CD28 or 4-1BB. The 3rd-generation of CARs have been produced by adding multiple costimulatory molecules to the 2nd-generation molecules, for example, OX40. In the 4th generation, a signal for the production of cytokines has been inserted, such as IL-7, IL-12, or others, increasing the efficacy further [131]. CAR NK cells have advantages over CAR T cells. There is no need for human leukocyte antigen (HLA) compliance, and there is a reduced risk of cytokine release syndrome [132]. Hematopoietic stem cells have also been used in the CAR approach, leading to CAR HSPCs [133]. The downsides of the CAR approach are potential off-target effects, such as cytokine release syndrome, high fever, hypotension, and neurotoxicity, high costs, and the development of CAR cells at a large scale.
The viral reservoirs are one of the major hurdles that need to be overcome for an HIV cure. There are three different approaches possible to address this issue:
Latency reversal, which means initiating transcription in dormant cells so that subsequently either conventional ART, the host’s own immune system, or other agents can kill them.
Reservoir shut-down or sealing of the reservoirs forever.
Killing of infected cells, dormant or not, by, for example, ADCC.
The objective of LRAs is to initiate transcription in dormant cells. As described above, latency depends on multiple systems that need to be affected by the virus to induce dormancy of the host cells, which is mirrored in the mechanisms of LRAs. Table 6 provides a brief overview of LRAs using different mechanisms of action. No LRA has yet been identified that can induce expression of all latent HIV in a safe and effective manner [40] due to an inadequate reactivation and/or lack of an effective kill [105].
Latency reversing agents.
| Mechanism | Compounds | Remarks | Reference(s) |
|---|---|---|---|
| HDAC inhibitors | Valproic acid | NCT00289952, NCT00614458, NCT00000629, NCT00312546, NCT03525730 | [134] |
| Mocetinostat | Various cancers | [135] | |
| Givinostat | Increased p24 by 15-fold in ACH2 cells | [136] | |
| Oxamflatin | Activates HIV-1 gene expression in latently infected cells | [137] | |
| Belinostat | Approved for PTCL | [138] | |
| Panobinostat | NCT01680094, NCT02471430, NCT06240520 | [139] | |
| Vorinostat | Class I HDACs [HDAC-1, -2, -3 and -8]NCT01365065, NCT01365065, NCT02475915, NCT03198559, NCT03382834, NCT03212989, NCT05700630, NCT02336074, NCT02707900, NCT03803605 | [140] | |
| Romidepsin | NCT02850016, NCT01933594, NCT02616874, NCT03041012, NCT03619278, NCT02092116 | [141] | |
| Nonhistone chromatin modulators | Pyrimethamine | BAF inhibitor, SMAC inhibitorNCT03525730, NCT06240520, NCT00132535 | [105] |
| Decitabine | DNA methyltransferase inhibitorNCT05230368 | [142] | |
| Zebularine | DNA methyltransferase inhibitor | [143] | |
| JQ1 | BET bromodomain inhibitor | [144] | |
| NF-κB stimulators | Bryostatin-1 | PKC agonistNCT02269605, NCT00022555 | [145, 146] |
| Pyrimethamine | NCT03525730, NCT06240520, NCT00132535 | [144] | |
| Ingenol-B | PKC agonist | [146] | |
| Prostratin | PKC agonist | [147] | |
| Extracellular stimulators | TNFα | Activation of caspase protease leading to apoptosis | [148, 105] |
| TLR agonists | Vesatolimod (GS‑9620) | TLR5 stimulator | [149] |
| Others | Disulfiram | Aldehyde dehydrogenase inhibitor NCT01944371, NCT01286259, NCT00878306, NCT01286259, NCT00002065 | [150] |
HDAC: histone deacetylase; SMAC: second mitochondrial-derived activators of caspases (non-canonical NF-κB); BAF: GBRG1/BRM-associated factor; ACH2: human T lymphocyte cell line chronically infected with HIV-1; PKC: protein kinase C; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; TLR: toll-like receptors; PTCL: peripheral T-cell lymphoma; BET: bromodomain extra-terminal motif.
An alternative to reactivating latent reservoirs is the possibility of blocking the reactivation of the virus by pushing the virus into a deep latent state. Possible mechanisms include inhibition of Tat, tyrosine kinase, heat shock protein 90 (HSP90), or the Janus activating kinase-signal transducer and activator of transcription (JAK-STAT) pathway. Examples are summarized in Table 7.
Latency-promoting agents.
| Mechanism | Compound | Remarks | Reference(s) |
|---|---|---|---|
| Tat inhibitors | Levosimendan | Marketed as a Ca sensitizer (Simdax) for heart failure | [151] |
| Didehydro-cortistatin A (dCA) | Locks a transient conformer of Tat | [152–154] | |
| Spironolactone | Approved as a diureticShuts off the Tat-dependent transcription | [155] | |
| Triptolide | Accelerates Tat protein degradationNCT01817283, NCT03403569, NCT02002286, NCT02219672 | [156] | |
| Tyrosine kinase inhibitors | Ponatinib | Akt-mTOR inhibitorChronic myeloid leukemia and Philadelphia chromosome–positive acute lymphoblastic leukemia | [157] |
| HSP90 inhibitors | AUY922 | Cancer research | [158] |
| 17-AAG | Cancer research | [159] | |
| JAK-STAT inhibitors | Ruxolitinib | JAK1/2 inhibitorApproved for rheumatoid arthritis, GVHD NCT02475655 | [160] |
| Tofacitinib | JAK1/2 inhibitorApproved for rheumatoid arthritis | [161] | |
| siRNA | PromA | Induces transcriptional gene silencing by disrupting the regulation of chromatin structure | [162–164] |
| LTR-362 | Targets the tandem NF-κB sites in the HIV-1 promoter | [165] | |
| S4-siRNA | Targets the unique NF-κB binding sequences in HIV-1 subtype C | [130] | |
| FACT | Curaxin 100 (CBL0100) | Blocks HIV-1 replication and reactivation | [166] |
| LEDGINs | CX05045 | Reduces HIV-1 integration, shifts integration towards the inner nucleus, and retargets the provirus away from H3K36me2/3 | [167] |
Tat: trans-activator of transcription; Akt-mTOR: protein kinase B-mammalian target of rapamycin; HSP90: heat shock protein 90; JAK-STAT: Janus activating kinase-signal transducer and activator of transcription; siRNA: small interfering RNA; GVHD: graft versus host disease; FACT: facilitates chromatin transcription complex; LEDGINs: lens epithelium-derived growth factors.
As observed for LRAs, latency-promoting agents alone are not sufficiently effective for HIV cure.
Currently available ART uses combinations of different drugs that, in the majority, aim to inhibit the reproduction of the virus by either attacking early in the process, such as attachment to the host cell, or late, for example, by inhibiting capsid formation. Combinations of other mechanisms are the next step in the search for an HIV cure. Various approaches have been investigated to diminish viral rebound by either reactivating viral reservoirs and subsequently killing the virus (named “shock and kill” or “kick and kill” or “shock and clear”), by suppressing the reservoirs “forever” so that reactivation of the dormant virus is blocked (“block and lock”) or by locking the virus and killing it in the infected cell (“block and clear”). Figure 3 provides an illustration of the three approaches.
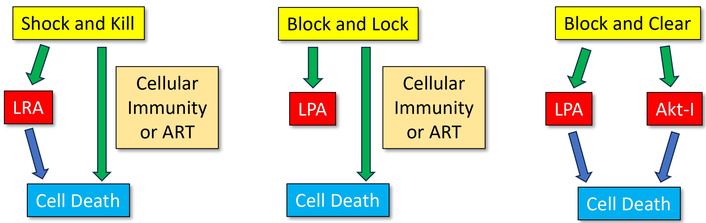
Illustration of the “shock and kill”, “block and lock”, and “block and clear” approaches. LRA: latency reversing agent; LPA: latency promoting agent; Akt: protein kinase B; Akt-I: Akt inhibitor.
In Table 8, some examples of the different approaches are listed.
Some examples of the different combination approaches.
| Method | Components | Remarks | Reference(s) |
|---|---|---|---|
| Shock and kill | OKT3 + IL-2 | Clinically not effective | [168] |
| ART + vorinostat (LRA) + AGS-004 (DC vaccine) | NCT02707900Safe but not effective | [169, 170] | |
| ART + vorinostat (LRA) + VRC07-523LS (bNab) | NCT03803605 | [171] | |
| ART + vorinostat (LRA) + disulfiram | NCT03198559Evidence of latency reversal, but not safe | [172] | |
| Venetoclax (pro-apoptotic) + anti-CD3 plus anti-CD28 T cell stimulation | Bcl-2 antagonist | [173] | |
| CRISPRa + gRNA-V | In vitro, an effective all-in-one approach | [174] | |
| Vorinostat + acitretin | RIG-I inducerReactivates latent HIV and induces apoptosis | [175] | |
| LEDGINs + JQ1 | Inhibits the HIV-1 integrase and LEDGF/p75 interaction | [167] | |
| YSE028 + JQ1 | PKC activator + BET bromodomain inhibitor | [176] | |
| Block and lock | dCA + ART | Tat inhibitor | [177] |
| LEDGINs + ZL0580 | BRD4 modulator | [167, 178] | |
| Triplitode + ART (Tenofovir + lamivudine + Lopinavir / ritonavir + raltegravir) | Immune modulator | A | |
| ZL0580 + ART | BRD4 modulator | [179] | |
| Block and clear | LPA + Capivasertib | Akt inhibitor | [180] |
CRISPRa: CRISPR activated; RIG-I: retinoic acid-inducible gene I; dCA: didehydro-cortistatin A; LRA: latency reversing agent; Akt: protein kinase B; A: NCT02219672 (not published yet).
AIDS was first identified in 1981, and its connection to HIV was established in 1983. And, still in 2025, there is no cure for the disease available except for the few cases with CCR5Δ32 donor stem cell transplantations. The reasons for the lack of a general cure are the latent reservoirs where the virus can hide in the form of a provirus until ART is stopped, the high mutation rate, and the avoidance of any attacks by the patient’s immune system, which is either no longer functioning or cannot detect and fight the virus. These characteristics are very similar to cancer, in particular regarding oncogenes or cancer stem cells.
However, in contrast to many types of cancer, HIV infection is no longer a destination of death, at least in those parts of the world that can afford treatment. ART has made HIV a life-long treatment with relatively high safety. Measured at this standard, any HIV cure needs to provide significantly greater benefits and/or diminished side effects in comparison to lifelong ART, because with improved treatment, HIV has become a chronic but manageable condition.
Huge efforts are still being undertaken in the search for a cure. The steps necessary for achieving a cure have been identified and described by a Working Group in 2021 [67]. Most probably, the process would comprise three generations of drugs until the final goal of “one drug one shot”. The first generation includes combinations of compounds with different mechanisms, excluding ART, such as, for example, the “shock and kill” approach, which is currently considered by most researchers as the preferred procedure to move on. But also, bNabs and vaccines fall into that category. The second generation includes modifications of cells, such as CAR T cells, NK cells, or B cells, without the need for prior myeloablation. And the third generation comprises in vivo modifications of the patient’s cells using gene therapies or other methods available at that time.
In the current first generation, combinations of drugs have not been able to completely deplete the latent reservoirs since the LRAs are either not strong enough or not able to reach all the reservoirs. Likewise, the kill components of the “shock and kill” approach have not been able to clear all newly produced viruses. In the kill phase after HIV-1 reactivation, it is necessary to inhibit replication of the reactivated virus and prevent new infection. This could be done, for example, by specific cytolytic T cells, but so far this procedure has not been effective [174]. Sealing the reservoirs, as described in the “block and lock” approach, did not lead to a permanent lockdown of the reservoirs. Combinations of LRAs with drugs exhibiting kill power, nevertheless, should be further evaluated.
Vaccines have not fulfilled the expectations that had been put into this procedure, although much progress has been made in B-cell lineage immunogen design, germline-targeting, immunofocusing, and structure-based immunogen design [100]. However, the learnings from the COVID-19 pandemic and the huge success of mRNA vaccine technology should be applied to HIV in a broad effort. The advantages of the mRNA approach are multifold, ranging from simultaneous use of many different components, scalability, the use of the LNP technology, and the large safety margin. The COVID-19 vaccines were injected intramuscularly and nevertheless were able to reach the lungs in sufficient concentrations to be effective. This should provide hope that, in the case of HIV, also hard-to-reach reservoirs could be a target. Another aspect of the vaccine technology is the use of adjuvants, which have made great progress, but further efforts are still needed.
Gene editing might be the ultimate goal because it could offer a permanent solution through either completely excising the HIV provirus or by modifying susceptible cells to resist infection. However, it must reach all reservoirs and must avoid off-target effects.
In summary, the road to a cure for HIV is heavily populated, but until the final goal is reached, there are still quite a number of stations on the way. My personal favorite to achieve a cure, defined as the eradication of HIV, is mRNA technology. On the way to a cure, I would also like to see a monkey study with alemtuzumab, aiming at two objectives. First, is treatment right after infection effective in avoiding the formation of latent reservoirs? And second, is the treatment with alemtuzumab at the point of rebound able to kill all newly infected cells and simultaneously deprive the virus of its site of survival, namely any cells in which it could hide?
ADCC: antibody-dependent cell-mediated cytotoxicity
AIDS: acquired immunodeficiency syndrome
AML: acute myeloid leukemia
ART: antiretroviral therapy
BET: bromodomain extra-terminal motif
bNabs: broadly neutralizing antibodies
CAR: chimeric antigen receptor
CRISPR: clustered regularly interspaced short palindromic repeats/Cas-9
CXCR4: chemokine receptor type 4
DCs: dendritic cells
HLA: human leukocyte antigen
HSP90: heat shock protein 90
LRAs: latency-reversing agents
SIV: simian immunodeficiency virus
ZNF: zinc finger
WK: Conceptualization, Investigation, Methodology, Supervision, Visualization, Writing—original draft, Writing—review & editing. The author read and approved the submitted version.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 14610
Download: 60
Times Cited: 0
Yadessa Tegene Woldie ... Mark Spigt
Violetta Vlasova ... Konstantin Shmagel
Zhimin Huang, Xiaohui Wang
Yun-Meng Yan ... Qi-Wen Yang
Yang Zhou ... Hongzhou Lu
Faisal Gunu Abdul-Samed ... Gifty Apiung Aninanya
