 Review
Review

Affiliation:
1Medical Laboratory Center, Shenzhen Luohu District People’s Hospital, Shenzhen 518005, Guangdong, China
2Department of HIV/AIDS Prevention and Control, Shenzhen Center for Disease Control and Prevention, Shenzhen 518020, Guangdong, China
ORCID: http://orcid.org/0009-0001-2072-8248
Affiliation:
2Department of HIV/AIDS Prevention and Control, Shenzhen Center for Disease Control and Prevention, Shenzhen 518020, Guangdong, China
Email: wxhszcn@aliyun.com
ORCID: http://orcid.org/0000-0002-8683-7180
Explor Med. 2025;6:1001348 DOI: https://doi.org/10.37349/emed.2025.1001348
Received: November 27, 2024 Accepted: June 06, 2025 Published: July 24, 2025
Academic Editor: Lee M. Wetzler, Boston University School of Medicine, USA
The article belongs to the special issue Global Perspectives on the Clinical Diagnosis, Treatment, and Functional Cure of HIV Infection in the Post-ART Era
CD4+ T lymphocytes are an important part of human immune cells and the main target cells of human immunodeficiency virus (HIV) infection. Acquired immune deficiency syndrome (AIDS) is a chronic infectious disease that usually takes 2–8 years from infection to onset. Research has found that only a small number of CD4+ T cells in HIV infected individuals are in an HIV infected state, and the dynamic changes of HIV-infected CD4+ T lymphocytes (HICTLs) at different stages of HIV infection are still unclear. Meanwhile, HICTLs are the source of the HIV reservoir. Therefore, analyzing the percentage and dynamic changes of infected cells in infected individuals is of great significance for AIDS research. However, due to the high variability of the HIV gene and the lack of highly sensitive detection methods, the standard method for distinguishing HIV infected and uninfected CD4+ T cells has not been established so far. This article summarizes the markers and detection methods of HICTLs at present.
Acquired immune deficiency syndrome (AIDS) is an infectious disease caused by human immunodeficiency virus (HIV) infection, with serious harm to the body’s immune function [1]. According to the data of the United Nations AIDS Programme (UNAIDS) [2], by the end of 2023, there were over 39.9 million reported surviving HIV infected individuals worldwide, and 1.29 million people living with HIV in China. HIV belongs to the Lentivirus genus of Retroviridae [3], which mainly infects CD4+ T lymphocytes [4]. HIV enters CD4+ T cells, where its RNA is reverse transcribed into HIV DNA and integrated into the genome of CD4+ T cells, forming HIV-infected CD4+ T lymphocytes (HICTLs) [5]. As shown in Figure 1, some HICTLs undergo death through activation [6], exhaustion [7], apoptosis, or pyroptosis [8, 9], while a small fraction forms the HIV latent reservoir [10–13]. A portion of the HIV latent reservoir was established early in infection when HIV enters resting memory CD4+ T cells, where it remains dormant [14]. A small number of HIV latent reservoir cells have complete HIV gene information [15], which poses great challenges to the cure of HIV. Current antiretroviral therapy (ART) effectively suppresses HIV replication in the body to undetectable levels but fails to eliminate the HIV reservoir. If treatment is discontinued, the HIV reservoir can be reactivated, leading to the production of new viral particles [16]. Therefore, accurately detecting HICTLs not only aids in exploring the mechanisms underlying HIV reservoir formation but also offers potential strategies for clearing the HIV viral reservoir.
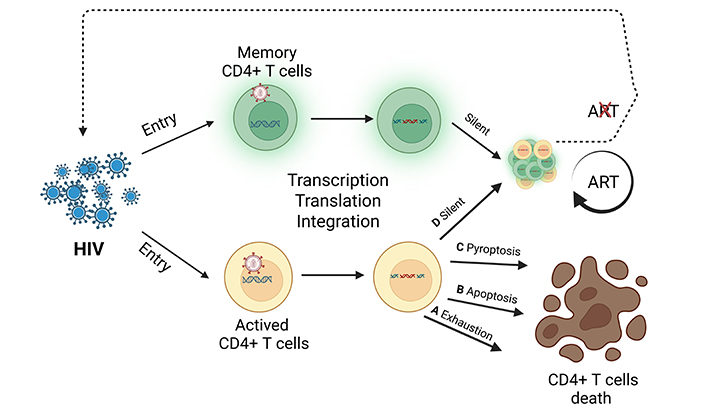
HIV-infected CD4+ T lymphocytes death pathway and the HIV reservoir formation process in HIV infection. ART: antiretroviral therapy; HIV: human immunodeficiency virus. Created in BioRender. Miya, H. (2025) https://BioRender.com/4uq0czh
In this study, the keywords “detect”, “HIV infected CD4+ T lymphocyte”, and diagnostic biomarkers such as p24, HIV DNA, and HIV RNA were employed to search the PubMed database. During the search, the sample types were strictly limited to human CD4+ T cells or peripheral blood mononuclear cells (PBMCs), and studies involving animal samples or cultured cell samples were excluded. After filtering, the resulting papers were further searched in Connected Papers for highly related or similar studies to obtain research outcomes from different research teams. In this paper, the detection methods will be categorized and described according to the different diagnostic biomarkers used.
HIV completes processes such as translation and transcription within CD4+ T cells, integrating its DNA into the host genome and subsequently undergoing replication [17]. Therefore, when detecting HICTLs, specific HIV-associated markers should be targeted.
HIV is a spherical enveloped virus, with an outer lipoprotein envelope containing two glycoproteins: the surface protein gp120 and the transmembrane protein gp41. Beneath the envelope lies the matrix protein p17, while the viral core consists of the capsid protein p24, genomic RNA, and essential enzymes including reverse transcriptase, integrase, and protease [18, 19]. Due to the high variability of gp120 and gp41, these proteins are currently not considered reliable diagnostic markers [20]. In contrast, p24, viral RNA, and HIV DNA produced during transcription and reverse transcription are more stable and thus serve as common targets for detection.
The following sections will discuss the proportions and characteristics of HICTLs detected using different markers and methodologies, including flow cytometry (FCM), microfluidic technology, in situ hybridization, and single-cell sequencing.
The p24 protein, encoded by the Gag gene of the HIV genome, is a structural protein and a major component of the viral nucleocapsid. It plays a critical role in viral packaging and maturation and is abundantly produced during the early stages of infection when viral replication is highly active. Due to the relatively low genetic variability of p24, its detection in CD4+ T cells via FCM has been one of the earliest and most widely used methods for determining the frequency of HICTLs.
Detection of p24 antigen represents the earliest methodology for HICTLs identification. As demonstrated by Costigliola et al. [21], this approach employs fluorescein-conjugated p24 monoclonal antibodies in flow cytometric analysis (FCA) to specifically identify HIV-infected CD4+ T cells, with p24-FCA positive cells operationally defined as HICTLs. Their quantitative analysis revealed significant differences in HICTLs frequencies across study groups: Healthy group (n = 24): 0.160 ± 0.152%; Untreated group (n = 22): 1.685 ± 1.902% of CD4+ T cells; ART group (n = 25): 0.825 ± 0.910%. Gao et al. [22] performed standardized FCM using commercially available anti-p24 antibodies, revealing markedly different HICTLs frequencies between groups (Mann-Whitney U test, p < 0.001): Untreated group (n = 63): 6.33% [IQR (interquartile range): 3.90–9.05%] of CD4+ T cells; Healthy group (n = 42): 0.93% (IQR: 0.55–1.30%).
However, during the early development of this methodology, researchers had already demonstrated that relying solely on a single p24 antibody as a marker could not effectively exclude false-positive results caused by non-specific antibody binding [23]. Both aforementioned research teams indeed detected low but measurable positivity rates in their healthy control groups (Costigliola et al. [21]: 0.16%; Gao et al. [22]: 0.93%). To address this limitation, subsequent investigators implemented a dual p24 antibody approach, wherein only cells demonstrating simultaneous positivity for two distinct p24 antibodies were classified as genuine HICTLs. This refinement significantly enhanced detection specificity.
Pardons et al. [24] utilized a combination of two antibodies targeting distinct epitopes of the HIV p24 protein (KC57 and 28B7) as markers to detect the frequency of p24 dual-positive cells (KC57+, 28B7+). In the ART group (n = 33), no p24+ cells were detected, while in the untreated group (n = 11), the frequency of dual-positive CD4+ T cells was approximately 0.001% (10/106 cells). Similarly, Tessema et al. [25] employed the same p24 antibody combination (KC57 and 28B7) for detection. Their results showed that in the untreated group (n = 6), the double-positive CD4+ T cells frequency was approximately 0.01% (100/106); in the ART group (n = 6), the dual-positive CD4+ T cells frequency was approximately 0.0001% (1/106). Subsequently, after stimulating these two groups of cells with phorbol-12-myristate-13-acetate (PMA)/ionomycin and repeating the detection, they observed a significant increase in the proportion of double-positive CD4+ T cells in both groups. The researchers then sorted the dual-positive cell populations and performed HIV DNA testing, revealing that each p24 dual-positive cell contained viral DNA at a frequency of approximately 1.25 copies/cell. This confirmed that the detected cells were replication-competent HICTLs.
The results demonstrate that p24 antigen can serve as a biomarker for detecting HICTLs via FCM. However, single-antibody detection introduces false-positive events, as evidenced by detectable signals in healthy controls due to nonspecific antibody binding, leading to overestimation of the true HICTLs frequency. To improve specificity, a dual-antibody approach (using two p24 antibodies labeled with different fluorophores) was adopted, wherein only double-positive cells (KC57+, 28B7+) were considered true HICTLs. While this method enhances specificity, it concurrently reduces sensitivity, potentially underestimating the actual infected cell frequency, particularly in ART-treated individuals, where HICTLs levels are either undetectable or extremely low.
Currently, the detection of HIV-infected CD4+ T cells through genetic material analysis employs fluorescence in situ hybridization (FISH) and single-cell sequencing technologies to identify HIV-RNA within infected cells.
Grau-Expósito et al. [26] used RNA flow FISH to detect the percentage of HIV RNA+ CD4+ T cells in untreated infected and ART-treated patients (< 20 copies/mL). After the cells were stained and fixed, they were hybridized with 50 highly sensitive target-specific probes spanning the entire Gag-Pol HIV mRNA sequence (1,165–4,402 bases of the common genome of HXB2), then amplified, and finally detected by RNA flow FISH. After analysis, it was found that the average number of HIV RNA+ CD4+ T cells in the untreated infected patients was 165/106 cells, accounting for 0.001–1% of the total CD4+ T cells. While in the ART group, the number of RNA+ CD4+ T cells was 10/106 cells, accounting for 0.001–0.01% of the total CD4+ T cells. In addition, researchers simultaneously detected HIV RNA and p24 antigen in some samples of HIV infected individuals. Double marker positive cells (RNA+, p24+) were not or rarely detected in all analyzed populations.
Baxter et al. [27] used RNA flow FISH technology to combine HIV protein detection with FCM. The researchers chose a combination probe set targeting Gag and Pol genes, and combined it with HIV p24 (KC57) antibody to detect the transcription and translation products of HIV. After analysis, it was found that approximately 123/106 HIV RNA+/p24+ CD4+ T cells were detected in the untreated group, and 311/106 cells were detected under PMA/ionomycin stimulation. Sannier et al. [28] designed a set of 5' exon RNA probes, complementary to Nef RNA or Gag RNA probes, and used RNA flow FISH technology to detect cells co-expressing Gag RNA+ or Nef RNA+ or p24+ with 5' exon RNA. The results showed that the average number of HIV RNA+ CD4+ T cells in the healthy control group was 2/106 cells, the untreated group had an average of 82/106 cells, while the ART group had an average of 5/106 cells.
Collora et al. [29] used expanded Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-compatible cellular indexing of transcriptomes and epitopes by sequencing (ECCITE-seq) technology to capture surface protein expression, cell transcriptome, HIV RNA, and T cell receptor (TCR) sequences within individual cells. They detected 267 HIV RNA+ cells (1,239/106) and 68 (315/106) in vivo cloned HIV RNA+ T cells from a total of 215,458 CD4+ T cells in 6 HIV-1-infected individuals (during viremia and after suppressive ART).
The identification of HIV-infected CD4+ T cells through genomic material relies primarily on two advanced techniques: FISH and single-cell RNA sequencing (scRNA-seq), both targeting intracellular HIV RNA. FISH employs fluorescently labeled probes to visualize HIV-specific RNA or DNA within intact cells at single-cell resolution. This method permits simultaneous detection of cellular markers (e.g., CD4, CD3), making it invaluable for spatial analysis in tissue samples such as lymph nodes. However, FISH exhibits limitations, including moderate throughput (typically hundreds of cells per assay) and stringent requirements for probe design to minimize cross-reactivity. In contrast, scRNA-seq offers unparalleled sensitivity (approximately 0.001%), enabling genome-wide profiling of both viral and host transcripts in rare infected cells. This high-throughput approach can process thousands of cells, uncovering clonal expansion patterns and potential therapeutic targets. Nevertheless, scRNA-seq demands substantial computational resources and may underrepresent latent infections with minimal transcriptional activity due to its reliance on RNA capture.
Recent advances in HIV reservoir monitoring have witnessed the emergence of HIV DNA as a novel biomarker, with single-cell sequencing technologies representing the predominant analytical approach. The remarkable progress in this field has been further enhanced through integration with established methodologies, including FCM and microfluidic platforms. For the purpose of this review, we will focus specifically on detection techniques applicable to human CD4+ T cells or PBMCs, while acknowledging that a comprehensive discussion of all available technologies exceeds our current scope.
Clark et al. [30] developed a microfluidic method to detect HIV-DNA+ cells in memory CD4+ T cells of 5 HIV infected individuals who received ART for a long time, known as Focused Interrogation of Cells by Nuclear Acid Detection and Sequencing (FIND-seq) technology. Researchers first isolated millions of single cells from oil in water droplets and immediately lysed them. Then, polyadenylation RNA sequences were recovered and sorted based on HIV-DNA detection results. At the same time, the complete transcriptome profile of HIV-DNA+ cells in their natural state was analyzed. The number of HIV-1+ cells in memory CD4+ T cells of ART patients is 534–2,153/106 cells; HICTLs exhibit inhibition of six transcriptome pathways under antiviral therapy, including inhibition of death receptor signaling, necrotic apoptosis signaling, and anti-proliferative Gα12/13 signaling.
Wu et al. [31] applied Assay for Transposase Accessible Chromatin with sequencing (ATAC-seq), a high-throughput sequencing technique that uses transposase to study chromatin accessibility technology, and utilized the property of transposase Tn5 that can bind to open chromatin. Tn5 enzyme was used to capture HIV pre-viral DNA sequences, and cell surface protein expression was identified by ASAP-seq (ATAC with select antigen profile by sequencing). 54 (5,950/106 cells) HIV+ cells were detected in 9,075 lymph node CD4+ memory T cells of two untreated infected individuals, and 213 (1,278/106 cells) HIV+ cells were detected in 166,357 peripheral blood CD4+ T memory cells of four ART-treated individuals.
Sun et al. [32] developed Phenotype and Proviral sequencing (PheP-seq), an advanced single-cell technique derived from CITE-seq, to characterize HIV-1-infected cells through simultaneous phenotypic and proviral analysis. By examining 530,143 CD4+ T cells from PBMCs of five individuals (4 ART-treated, 1 elite controller), they established a novel classification system: Category 1 (cells with any HIV-1 provirus, n = 2,859), Category 2 (cells containing intact HIV-1 genomes, n = 193), Category 3 (cells from large viral clones, n = 125), and Category 4 (total provirus-containing cells, n = 398). Their findings revealed that peripheral blood cells harboring intact proviruses and large clones displayed distinct surface markers conferring resistance to cytotoxic T and natural killer cell-mediated killing, along with elevated immune checkpoint expression (e.g., PD-1, LAG-3) that may suppress viral transcription. This unique phenotype likely promotes viral persistence by minimizing immunogenic exposure and reducing elimination by host immune responses, providing new insights into HIV-1 reservoir maintenance mechanisms and potential therapeutic targets for eradication strategies.
Artesi et al. [33] pioneered Pooled CRISPR Inverse PCR sequencing (PCIP-seq), an innovative methodology combining selective cleavage of circular DNA fragments harboring proviral DNA with a library of CRISPR guide RNAs. Applying this technique to DNA extracted from CD4+ T cells of two long-term ART-treated HIV-1 patients, they identified 73 and 158 unique integration sites in the respective patient samples, while simultaneously generating approximately 500 kb of HIV proviral sequences linked to specific integration loci. This technological advancement significantly enhances our understanding of HIV DNA integration patterns in the viral reservoir. However, when CRISPR-based approaches are coupled with single-cell sequencing methodologies, current limitations prevent comprehensive characterization of individual HICTLs features, highlighting an important area for future methodological refinement (Table 1).
Detection of HICTLs using different detection methods and biomarkers
| Biomarkers | Detection methods | Results (cells) | Sensitivity | Specificity | Reference | ||
|---|---|---|---|---|---|---|---|
| Healthy | ART | Untreated | |||||
| p24 | Flow cytometry | 1,600/106 | 8,250/106 | 16,850/106 | Low | Low | [21] |
| p24 | Flow cytometry | 9,300/106 | - | 63,300/106 | Low | Low | [22] |
| p24 + p24 | Flow cytometry | - | 0 | 10/106 | Low | High | [24] |
| p24 + p24 | Flow cytometry | - | 1/106 | 100/106 | Low | High | [25] |
| RNA | RNA flow FISH | - | 10/106 | 165/106 | Medium | Medium | [26] |
| RNA + p24 | RNA flow FISH | - | - | 123/106 | Medium | High | [27] |
| RNA + p24/Gag RNA/Nef RNA | RNA flow FISH | 2/106 | 5/106 | 82/106 | Medium | High | [28] |
| RNA | ECCITE-seq | - | a1,239/106 (Mix) | High | High | [29] | |
| DNA | Microfluidic + FIND-seq | - | 534–2,153/106 | - | High | Medium | [30] |
| DNA | ATAC-seq + ASAP-seq | - | 1,278/106 | 5,950/106 | High | Low | [31] |
| DNA | PheP-seq | - | - | - | - | - | [32] |
| DNA | PCIP-seq | - | - | - | - | - | [33] |
Note: “a” indicates that the number is obtained from a joint sample of two populations (ART and untreated individuals). “-” indicating that the researcher did not conduct testing or did not write down. “Healthy” indicates that they are not infected with HIV; “ART” indicates that HIV patients are receiving antiretroviral therapy; “Untreated” refers to HIV infected individuals who have not received ART treatment. Some references list the duration of ART treatment or HIV infection, but this article does not provide a summary. ART: antiretroviral therapy; ASAP-seq: ATAC with select antigen profile by sequencing; ATAC-seq: Transposase Accessible Chromatin with sequencing; ECCITE-seq: expanded CRISPR-compatible cellular indexing of transcriptomes and epitopes by sequencing; FIND-seq: Nuclear Acid Detection and Sequencing; FISH: fluorescence in situ hybridization; HICTLs: HIV-infected CD4+ T lymphocytes; HIV: human immunodeficiency virus; PCIP-seq: Pooled CRISPR Inverse PCR sequencing; PheP-seq: Phenotype and Proviral sequencing
The advent of single-cell detection technologies has enabled researchers to move beyond merely quantifying the frequency of HICTLs and instead investigate the mechanisms and characteristics of viral reservoir formation. These approaches provide unprecedented resolution for analyzing proviral integration sites, clonal expansion, and host-virus interactions at the single-cell level. However, single-cell sequencing-based detection of HIV-infected CD4+ T cells presents significant challenges, including technical complexity in sample processing, stringent experimental requirements, and high costs, which currently limit its application to large-scale cohort studies.
The primary obstacle to achieving an HIV cure lies in the persistence of the viral reservoir, which remains a major focus of recent scientific advancements [34–36]. HICTLs serve as both the origin of the viral reservoir and the outcome of reservoir reactivation, making their monitoring crucial for understanding viral persistence and eradication strategies. Consequently, the development of accurate and sensitive HICTLs detection methods holds substantial scientific and clinical importance in HIV cure research.
Each biomarker detection modality offers unique advantages: the p24 antibody assay combines operational simplicity with multi-immunophenotype detection capability, RNA-based methods provide superior sensitivity for monitoring active viral replication, while DNA testing has strong advantages in virus reservoir quantification. Nevertheless, these techniques collectively face shared limitations, including standardization challenges, sensitivity thresholds in clinical samples, and technical variability across laboratory implementations.
Current methodologies for detecting HICTLs employ diverse biomarkers (p24, DNA, RNA) and detection platforms (FCM, flow FISH, microfluidics, single-cell sequencing, CRISPR-based assays), yet reported HICTLs frequencies vary substantially across studies due to technical and biological factors. Single p24 antibody-based detection shows particularly wide variability in HICTLs estimates [21, 22]. In Table 1, we summarize the proportions of HICTLs detected by different biomarkers and corresponding detection methods. while multi-parameter approaches (combining p24 with other markers) yield more consistent results: approximately 0.1% of CD4+ T cells in untreated individuals versus 0.001% in ART-treated patients [24–28]. Notably, single-cell sequencing of HIV DNA/RNA demonstrates even greater (100-fold) discrepancies [29–31], primarily because conventional sequencing cannot distinguish replication-competent proviruses from defective ones, thereby inflating apparent reservoir sizes. This limitation underscores the critical need for intact proviral discrimination in reservoir quantification.
Beyond the influence of biomarkers and detection methods, the selection of study populations significantly affects HICTLs measurement outcomes. Most notably, in ART-treated individuals, HICTLs detection primarily identifies the latent HIV reservoir—yet these reservoir cells themselves constitute only a minor fraction of total HICTLs populations. In ART-treated individuals, HICTLs frequencies undergo progressive decline to exceptionally low levels (< 1 cell per 1,000 CD4+ T cells in peripheral blood), with most residual cells demonstrating minimal viral RNA transcription [37]. Consequently, conventional detection methods with limited sensitivity (e.g., standard FCM or dual-marker assays) frequently fail to identify HICTLs in this population. Furthermore, the dynamic changes in HICTLs percentages across different infection stages (acute/chronic infection, elite controllers) remain poorly characterized, necessitating careful consideration of cohort characteristics—including ART duration, virological control status, and time since infection—when designing and interpreting reservoir studies. These observations highlight the critical importance of both methodological optimization for low-abundance detection and standardized reporting of participant clinical profiles in HIV persistence research.
A critical limitation of existing HICTLs detection approaches—whether targeting intracellular antigens (e.g., p24) or HIV nucleic acids (DNA/RNA)—is their inherent incompatibility with cell viability, as these methods require cell fixation/permeabilization, thereby precluding the isolation and functional characterization of live HIV-infected CD4+ T cells. Investigating the lifecycle dynamics of these cells remains a highly compelling but technically challenging research direction. Presently, the quantitative viral outgrowth assay (QVOA) stands as the sole “gold standard” method capable of detecting replication-competent virus from live HICTLs. However, QVOA’s reliance on large quantities of healthy donor PBMCs, extended culture durations (typically 2–3 weeks), and substantial costs severely constrain its widespread application and scalability for in-depth studies of viral reservoir biology.
Current detection methods and biomarkers exhibit certain limitations, and screening for higher-specificity p24 antibodies and optimized nucleic acid probes may help address these challenges. More importantly, developing approaches for the direct detection of live HICTLs would open new avenues in AIDS research. We propose that screening for high-specificity HIV envelope protein antibodies, conjugated with bright fluorophores, to detect HIV envelope proteins on the surface of CD4+ T cells via FCM represents a promising and practical strategy. This method would enable: (1) separation of HICTLs from uninfected CD4+ T cells, (2) discrimination between direct HIV-mediated cytopathic effects and immune-mediated bystander damage to uninfected cells, and (3) observation of differential cellular fates between these populations. Such an approach would provide novel insights into HIV infection processes and pathogenesis mechanisms.
AIDS: acquired immune deficiency syndrome
ART: antiretroviral therapy
ATAC-seq: Transposase Accessible Chromatin with sequencing
CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats
FCA: flow cytometric analysis
FCM: flow cytometry
FISH: fluorescence in situ hybridization
HICTLs: HIV-infected CD4+ T lymphocytes
HIV: human immunodeficiency virus
IQR: interquartile range
PBMCs: peripheral blood mononuclear cells
PMA: phorbol-12-myristate-13-acetate
QVOA: quantitative viral outgrowth assay
scRNA-seq: single-cell RNA sequencing
ZH: Writing—original draft, Writing—review & editing. XW: Conceptualization.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by Shenzhen Committee of Scientific and Technical Innovation grant [JCYJ20180508152244835]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5076
Download: 47
Times Cited: 0
Yadessa Tegene Woldie ... Mark Spigt
Violetta Vlasova ... Konstantin Shmagel
Werner Krause
Yun-Meng Yan ... Qi-Wen Yang
Yang Zhou ... Hongzhou Lu
Faisal Gunu Abdul-Samed ... Gifty Apiung Aninanya
