 Review
Review

Affiliation:
1Fondazione Italiana FegatoONLUS, Area Science Park Basovizza SS14 km 163.5, 34149 Trieste, Italy
2Philippine Council for Health Research and Development, DOST Compound, Bicutan Taguig City 1631, Philippines
Affiliation:
1Fondazione Italiana FegatoONLUS, Area Science Park Basovizza SS14 km 163.5, 34149 Trieste, Italy
Affiliation:
1Fondazione Italiana FegatoONLUS, Area Science Park Basovizza SS14 km 163.5, 34149 Trieste, Italy
ORCID: http://orcid.org/0000-0001-6596-7595
Affiliation:
1Fondazione Italiana FegatoONLUS, Area Science Park Basovizza SS14 km 163.5, 34149 Trieste, Italy
Email: natalia.rosso@fegato.it
ORCID: http://orcid.org/0000-0002-4251-3547
Explor Med. 2020;1:51–74 DOI: https://doi.org/10.37349/emed.2020.00005
Received: March 11, 2020 Accepted: April 16, 2020 Published: April 30, 2020
Academic Editor: Amedeo Lonardo, Ospedale Civile di Baggiovara, Azienda Ospedaliero-Universitaria di Modena, Italy
The article belongs to the special issue Exploring NAFLD/NASH
Non-alcoholic fatty liver disease (NAFLD) remains a major cause of chronic liver disease worldwide. Despite extensive studies, the heterogeneity of the risk factors as well as different disease mechanisms complicate the goals toward effective diagnosis and management. Recently, it has been shown that sex differences play a role in the prevalence and progression of NAFLD. In vitro, in vivo, and clinical studies revealed that the lower prevalence of NAFLD in premenopausal as compared to postmenopausal women and men is mainly due to the protective effects of estrogen and body fat distribution. It has been also described that males and females present differential pathogenic features in terms of biochemical profiles and histological characteristics. However, the exact molecular mechanisms for the gender differences that exist in the pathogenesis of NAFLD are still elusive. Lipogenesis, oxidative stress, and inflammation play a key role in the progression of NAFLD. For NAFLD, only a few studies characterized these mechanisms at the molecular level. Therefore, we aim to review the reported differential molecular mechanisms that trigger such different pathogenesis in both sexes. Differences in lipid metabolism, glucose homeostasis, oxidative stress, inflammation, and fibrosis were discussed based on the evidence reported in recent publications. In conclusion, with this review, we hope to provide a new perspective for the development of future practice guidelines as well as a new avenue for the management of the disease.
Non-alcoholic fatty liver disease (NAFLD) is a wide spectrum disease that presents two phenotypes, namely non-alcoholic fatty liver (NAFL) and non-alcoholic steatohepatitis (NASH). While NAFL is considered a relatively benign and reversible accumulation of lipids within the hepatocytes (simple steatosis), NASH is considered a more severe form of the disease. NASH is characterized by cellular injury induced by sustained inflammation, oxidative stress, extracellular matrix remodeling, and hepatic stellate cells activation aside from simple steatosis. NASH tends to progress to cirrhosis and, eventually, to hepatocellular carcinoma (HCC) [1]. Nowadays NAFLD has emerged as a major cause of chronic liver disease. In particular, NASH is increasing as the etiology for end-stage liver disease as well as for HCC-related liver transplantation and is expected to surpass hepatitis C for this indication in the next years [2]. NAFLD is strongly related to insulin resistance (IR) and it is associated with clinical conditions such as overweight or obesity, type 2 diabetes mellitus (T2DM), hypertension, hypertriglyceridemia and low high-density lipoprotein (HDL)-cholesterol [all of which constitute essential elements in the spectrum of the metabolic syndrome (MetS)]. NAFLD prevalence shows a discrepancy among continents but the estimated worldwide prevalence is approximately 24–25% [3, 4]. In the general adult population, overall NAFLD prevalence is higher in men than women (22–42% in men vs. 13–24% in women) [5, 6]. However, if reproductive groups are considered, there is a higher prevalence of NAFLD in men compared only to women in premenopause (whose prevalence is 6% to 9%). Interestingly, in the postmenopausal age, the prevalence of NAFLD is fully comparable between men (22–42%) and women (19–31%), suggesting the protective role of estrogens [6–8]. The liver is a sexually dimorphic organ, exhibiting major sex differences in the profile of steroids and drug metabolism as well as the expression of several genes and proteins [9]. Thus, it is not surprising that the physiological differences between males and females are still present under pathological conditions. In 2014, the National Institutes of Health (NIH) made a policy to account for sex as a biological variable to their funded studies. This led to a steady increase in the number of publications on sex differences in different disease entities, particularly the features of MetS [5]. However, studies of sex differences in NAFLD are still minimal and most of them are focused on clinical and/or epidemiological aspects. Although it has been established that the prevalence, risk factors, and clinical outcomes of NAFLD differ between sexes, the molecular mechanisms by which sex modulates the pathogenesis and clinical outcome of NAFLD are poorly defined. Therefore, we aimed to review the current knowledge on sex differences in NAFLD particularly in those factors involved in lipid metabolism, glucose metabolism, oxidative stress, inflammation, fibrosis, and metabolomics. We hope to provide a new perspective for the development of future practice guidelines as well as providing a new avenue for the management of the disease.
As mentioned hereafter, the liver shows sexual dimorphisms in a complex set of genes related to lipid metabolism, drug metabolism, and glucose homeostasis among others (Table 1). Indeed, several studies reported that sexual dimorphism has a role in the risk, onset, progression, and treatment response of NAFLD. Therefore, a better understanding of the molecular mechanisms and the role of sexual dimorphism is of utmost importance to improve both prevention and therapeutic strategies of NAFLD.
Sexually dimorphic hepatic genes associated with NAFLD
| Gene | Function (s) | Model (s) Used | Female | Male | Effect (s) |
|---|---|---|---|---|---|
| JNK | Transcription factor; regulate fatty acid oxidation; improve insulin sensitivity | SHROB Rats | ↑↑ | ↑ | Less severe steatosis in females |
| AMPK | Transcription factor; regulate fatty acid oxidation; improve insulin sensitivity | SHROB Rats | ↑↑ | ↑ | Less severe steatosis in females |
| CD36 | Receptor; promotes fatty acid uptake into muscle and adipose tissues; fatty acid transport, β-oxidation | SHROB Rats; FXR-deficient mice; C57BL6/J HFD Mice | ↑↑ | ↑ | Less severe steatosis in females |
| PPARγ | Transcription factor; activates SREBP-1c, a master transcription factor that promotes de novo lipid synthesis (DNL) | SHROB Rats | ↑ | ↑↑ | More severe steatosis and inflammation in males |
| PNPLA3 | Enzyme; involved in the accumulation of lipid droplets; esterification | SHROB Rats; C57BL6/J HFD Mice | ↑ | ↑↑ | More severe steatosis and inflammation in males |
| AQP7 | The main channel in facilitating the release of lipolytic glycerol from adipocytes | C57BL/6 HFD mice, streptozotocin-induced DM rats | ↑↑ | ↑ | Increase plasma glycerol levels in both sexes |
| AQP9 | The principal facilitative pathway in liver uptake of glycerol | C57BL/6 HFD mice, streptozotocin-induced DM rats | ↓↓ (Expression and glycerol permeability) | ↓ | Less severe steatosis and hyperglycemia in females |
SHROB: obese spontaneously hypertensive; ↑ increased; ↑↑ highly increased; ↓ decreased; ↓↓ highly decreased
Several animal models mimicking MetS and NAFLD showed that clusters of hepatic genes involved in lipid and glucose metabolism are differentially regulated between sexes, the so-called sexually dimorphic genes. A study utilizing obese spontaneously hypertensive (SHROB) rats revealed sex-specific differences in the development of hepatic steatosis. The SHROB rats, a model of MetS without overt T2DM, are known to develop congenital obesity, glucoseintolerance, hepatomegaly, and fatty liver similar to many patients with MetS [10]. The genome of SHROB rat harbors a nonsense mutation in codon 763 of the gene encoding the leptin receptor, rendering it dysfunctional [11]. Female SHROB rats have higher expression of phosphorylated c-Jun NH<sub>2</sub>-terminal kinases (JNK) and AMP-activated protein kinase (AMPK) and enhanced expression of cluster of differentiation 36 (CD36) than male counterparts [12]. JNK and AMPK are key transcription factors involved in regulating de novo lipid synthesis (DNL) [13]. On the other hand, the CD36 gene (also known as fatty acid translocase or FAT) is known to be involved in fatty acid uptake into muscle and adipose tissues, and its deficiency is associated with insulin resistance [14] (Figure 1). The elevated CD36 mRNA expression is also congruent with farnesoid X receptor-deficient (FXR-deficient) female mice fed with western diet (WD) and female mice born to mothers with a high-fat diet (HFD), wherein they exhibited less severe steatosis and less inflammation as compared to males, respectively [15, 16]. FXR, a member of the nuclear receptor (NR) superfamily, functions as the primary sensor for endogenous bile acids (BA) and it is known to play a crucial role in hepatic triglyceride homeostasis [17]. In summary, the high expression of CD36 in female rodents points to a more efficient fatty acid transport, lipid utilization, and greater insulin sensitivity, all of which contribute to reducing steatosis in female livers.
Peroxisome proliferator-activated receptor-γ (PPARγ) participates in the regulation of fatty acid storage and glucose metabolism. The genes activated by PPARγ stimulate lipid uptake and adipogenesis by fat cells. It has been reported that mRNA levels are higher in SHROB males than females, indicating that in SHROB males, PPARγ contributes, in part, to the lipid accumulation [12]. The aberrant expression of PPARγ is associated with the pathogenesis of NAFLD by the activation of the sterol regulatory element-binding protein-1c (SREBP-1c), a key transcription factor that promotes DNL [18, 19]. In the same model, it has been also reported that male rats have a remarkable up-regulation of patatin-like phospholipase domain containing 3 (PNPLA3) gene (500-fold than females). These high levels of PNPLA3 expression in male rats and mice are linked to more severe steatosis [12, 20]. PNPLA3 codes for a nonspecific lipid acyl hydrolase that is abundant in the liver and adipose tissue [21]. The exact role of PNPLA3 in lipid metabolism remains controversial but several studies suggest that probably the accumulation of lipid droplets is a result of the elevated expression of ChREBP and SREBP-1c that transcriptionally activates the PNPLA3 gene [22]. This is further supported by studies showing that a genetic variant of PNPLA3 known as PNPLA3I148M is a major genetic risk factor for NAFLD. Expression of PNPLA3I148M, but not wild type (WT) PNPLA3WT, in female and male mice reproduces the fatty liver phenotype and its depletion resolved the hepatic fat accumulation [20, 23]. On the contrary, a clinical study revealed that there is a paradoxical dissociation between hepatic DNL and hepatic fat content due to the PNPLA3148M allele, indicating that increased DNL is not a key feature in all individuals with hepatic steatosis [24]. However, the sex differences with regards to the role of PNPLA3 variant on NAFLD pathogenesis remain elusive.
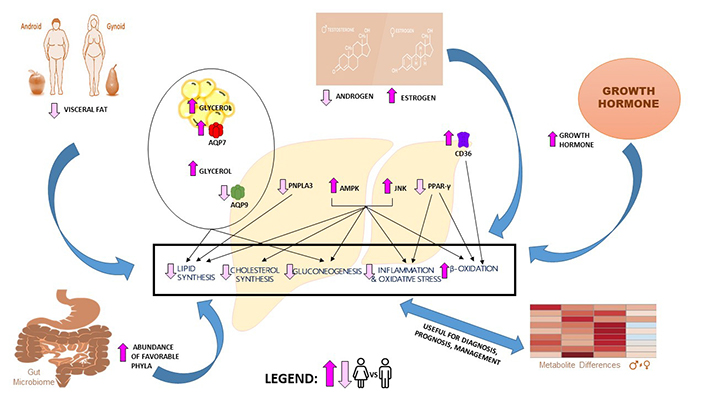
Sex differences in NAFLD pathogenesis. Compared to men, women exhibit higher plasma glycerol due to their higher lipolytic rate and abundance of AQP7 in visceral adipose tissue. Women also exhibit lower hepatic AQP9 expression and lower glycerol permeability than men. These differences in glycerol levels and AQP expression lead to a lower prevalence of NAFLD and insulin resistance in women. Lower CD36 expression in women is associated with increased β-oxidation. Increased JNK and AMPK in women are associated with decreased DNL, decreased cholesterol synthesis, decreased gluconeogenesis, decreased inflammation, and increased β-oxidation. Decreased PPARγ expression in women is associated with decreased inflammation and increased β-oxidation. Decreased PNPLA3 expression in women is associated with decreased accumulation of lipid droplets. Increased GH and estrogen levels in women are known to have a protective role in NAFLD development. The presence of favorable bacterial phyla in the female gut is associated with less steatosis development and less hepatic inflammation. In silico analysis showed several metabolites showing sex-associated differences in NAFLD patients that could be potential biomarkers and therapeutic targets. The conditions showing upregulation (bright pink) or downregulation (light pink) in women compared to men are indicated
Growing evidence suggests that other important players of sexual dimorphism in NAFLD are glycerol and aquaglyceroporins (AQP). Glycerol is the main precursor of triacylglycerol synthesis and glucose through hepatic gluconeogenesis during the states of negative energy balance, such as fasting or exercise [25]. Females are known to have higher plasma glycerol levels as well as higher sensitivity to lipolytic hormones and growth hormone (GH) as compared to men [26]. Apart from glycerol, much attention has also been directed to the role of AQP channels as important metabolic gateways in adiposity and insulin resistance control [27]. AQP are channel proteins known to transport glycerol and other neutral solutes across biological membranes. Among the several types of AQP channels, AQP9 has shown to play a pivotal role in the uptake of glycerol in the liver and adipose tissue [27, 28]. Accumulating evidence showed that sexual dimorphism exists in AQP9 expression in rats, where females present significantly lower hepatic AQP9 protein as compared to their male counterparts, in both fed and fasting conditions [29, 30]. This is further supported by the observation that AQP9-deficient mice are associated with defective hepatic glycerol metabolism, elevated plasma glucose, and steatosis [31, 32]. The possible role of estrogen in preventing the increased expression of AQP9 during starvation was explored. Ovariectomy in rats and the administration of estradiol (E2) in cultured hepatocytes led to increased hepatic AQP9 expression, indicating sex-specific different modulation [29]. For NAFLD, a recent study showed that there was a lower expression of AQP9 protein in a female rat model of metabolic syndrome and steatosis [33]. Similarly, in AQP9-deficient male rats, there was a less severe HFD-induced steatosis [34]. In humans, a lower expression of AQP9 has been found in a mixed-gender cohort of obese patients [35]. Aside from the lower expression of AQP9 protein, obese women are also associated with lower hepatic permeability to glycerol as compared to obese men [35] (Figure 1). The exact reason why such differences occur in glycerol permeability is still under investigation.
The prevalence and incidence of NAFLD are higher in men and postmenopausal women than premenopausal women, suggesting multiphasic effects of sex and hormones on NAFLD pathogenesis [36–38]. Over 1, 000 liver genes display a sex-bias in their expression and among them, lipid metabolism is one of the top biological pathways [39]. Furthermore, regulation of lipid metabolism is generally linked to the action of several hormones and growth factors [40–42]. Among the hormones implicated in the pathogenesis of NAFLD, GH, estrogen, and androgens are well studied. Estrogens regulate liver lipogenesis and insulin sensitivity [43, 44]. E2, the most potent estrogen for premenopausal women is predominantly synthesized by the ovaries and to some extent by non-reproductive tissues such as liver, adipose tissue, brain, heart, and bone [45]. In men, E2 is found in significant quantities in many tissues and aromatase, the enzyme that converts testosterone to estrogen is abundant in brain, penis, and testes [46]. Estrogen acts on both nuclear and membrane receptors. Estrogen receptor α (ERα) and estrogen receptor β (ERβ) are nuclear receptors that both activate genomic (regulate transcription of target genes) and non-genomic (works via second messengers) pathways. On the other hand, G protein-coupled estrogen receptor (GPER) has been reported to be present in the plasma membrane and mediate the non-genomic actions of estrogen [47, 48].
Several reports support a robust role of estrogen signaling through ERα in the metabolism of lipids and glucose. ERα is more abundant in hepatocytes and quiescent hepatic stellate cells (HSC) than ERβ (Table 2) [49, 50]. Gao et al. [51], revealed that 43 of the lipid genes are transcriptionally regulated by ERα, while Villa et al. [52], demonstrated that several liver genes involved in lipid metabolism vary in expression with the four-day estrous cycle of the mouse in an ERα-dependent manner. The association was further strengthened by data obtained in ERα-knockout mice model. In female mice lacking ERα was observed a higher visceral fat accumulation, and reduced energy expenditure while the treatment of E2 in ovariectomized mice improved insulin sensitivity and reduced lipid synthesis [44, 53]. E2 treatment also promoted fatty acid oxidation in the liver by increasing the expression of the fatty acid transport protein, carnitine palmitoyltransferase 1 (CPT-1) [44]. Male mice also express estrogen receptors in many tissues and similar to their female counterparts, loss of function or mutations in ERα are associated with impaired glucose tolerance, hyperglycemia, and hyperinsulinemia [54]. Moreover, both female and E2-treated male mice are protected against HFD-induced insulin resistance and adipose tissue inflammation [43]. Overall, in vitro and animal studies strongly suggest that E2 has a protective effect in females by enhancing fatty acid oxidation and reducing hepatic lipogenesis. In humans, several studies demonstrated that E2 therapy improves insulin sensitivity and glucose homeostasis in postmenopausal women [55–57]. However, limited studies have shown a beneficial effect of hormone replacement therapy on NAFLD [58].
Estrogen receptors and its role in sexual dimorphism of NAFLD
| Receptor | Female | Effect(s) | Male | Effect(s) | Notes |
|---|---|---|---|---|---|
| ERα | ERα-knockout mice | Higher visceral fat accumulation and reduced energy expenditure | ERα-knockout mice | Impaired glucose tolerance, hyperglycemia, hyperinsulinemia | Abundant in hepatocytes and inactivated HSCs |
| E2 treatment in ovariectomized mice | Promotes fatty acid oxidation; improves insulin sensitivity; reduce lipid synthesis; improves glucose homeostasis | E2 treatment | Protected against HFD-induced insulin resistance and adipose tissue inflammation | ||
| ERβ | ERβ-knockout mice | Increased body weight gain and fat deposition under HFD-treatment | ---- | ---- | Less expression in hepatocytes, high expression in fibrotic livers and activated HSCs |
| ---- | ---- | ERβ-selective agonists (β-LGND2 and WAY200070) | Inhibits body weight and white adipose tissue, and increases metabolism in male HFD mice; improves plasma insulin levels and glucose tolerance | ||
| ERβ-knockout mice | Protected against diet-induced IR and glucose intolerance | ERβ-knockout mice | No hepatic steatosis and decreased in serum cholesterol levels | ||
| GPER or GPR 30 | GPER-knockout mice | ↓ Levels of HDL; ↑ in fat liver accumulation | GPER-knockout mice | ↑ Levels of HDL; ↓ in fat liver accumulation; | Expressed in hepatocytes and quiescent HSCs |
| GPER-knockout mice | Moderate obesity and larger adipocyte size beginning later age | GPER-knockout mice | Moderate obesity and larger adipocyte size beginning earlier age | ||
| GPER-knockout mice | Protected from HFD-induced obesity, blood glucose intolerance, and insulin resistance | ---- | ---- |
ER: estrogen receptor; GPR30: G protein-coupled receptor 30; ↑ increased; ↓ decreased; ---- sex-balanced studies not available
For ERβ, its expression was observed both in normal and fibrotic livers, and in quiescent and activated HSC in both genders [50]. Several studies also mentioned ERβ role in lipid and glucose metabolism, but the results are contrasting. A recent report showed that ERβ-selective agonist, β-LGND2, inhibits body weight and white adipose tissue, and increases metabolism in male C57BL/6 mice fed with anHFD. This is further supported by a study whereinloss of ERβ, studied in ERβ -/- female mice showed increased body weight gain and fat deposition under HFD-treatment [59]. Contrary to these results, hepatic steatosis and an increase in serum cholesterol levels have been reported in ERα-knockout, ERα/β-knockout mice but not ERβ-knockout male mice [60].
In recent years, it has become clear that GPER can mediate the non-genomic action of estrogens in several biological pathways [47]. GPER has been functionally implicated with the regulation of body weight, feeding behavior, inflammation, as well as glucose and lipid homeostasis [61]. GPER has also been associated with sexual dimorphism in NAFLD but the exact mechanism is not yet fully understood (Table 2). A study on GPER-deficient mice fed with a HFD revealed that females rather than male mice had significantly lower levels of HDL-cholesterol along with an increase in liver fat accumulation [62]. Moreover, GPER-deficient male mice developed moderate obesity and larger adipocyte size beginning at 8 weeks of age while their female counterparts developed increased body weight 6 weeks later [63]. In contrast, GPER-knockout female mice were protected from HFD-induced obesity, blood glucose intolerance, and insulin resistance [64]. The authors suggest that an increase in GPER expression could counteract the role of ERα in regulating adipose tissue energy homeostasis, an aspect that needs to be further studied.
Testosterone is the predominant male hormone that circulates in the body as either free (2%), bound to albumin (38%), or linked to sex-hormone-binding globulin (SHBG) (60%) [65]. In women, testosterone is produced from its precursor, androstenedione, and total and free serum testosterone levels are far lower as compared with men [66]. In men, a low level of testosterone is associated with the features of metabolic syndrome and its supplementation posed beneficial effects [67, 68]. On the other hand, insulin resistance and obesity are associated with increased levels of testosterone in women [69, 70]. Several studies also attempt to determine the role of testosterone on the sexual dimorphism of NAFLD, but the relationship remains vague. A meta-analysis regarding the association of testosterone and NAFLD revealed that lower serum testosterone levels are associated with men with NAFLD and higher levels are associated with women with NAFLD [71, 72]. Moreover, a study found a strong correlation between low levels of testosterone with both the presence of NASH and increasing fibrosis stage [73]. Interestingly, when women were divided into reproductive groups, low testosterone levels were associated with NAFLD in the postmenopausal group only [74]. The difference in testosterone levels between men and women could explain its contrasting role in NAFLD pathogenesis. Moreover, after menopause, the principal source of estrogen is through its extra-gonadal conversion from androgens [65, 66]. Since it is established that estrogen has a protective role in NAFLD development, postmenopausal women with low levels of testosterone are at higher risk.
The GH, secreted by the anterior pituitary gland, is well-known for its action as a regulator of post-natal growth [75, 76]. Moreover, accumulating evidence suggests that it is also involved in the regulation of lipid and carbohydrate metabolism, particularly by the liver and adipocytes. Microarray analysis on GH-knockout mice revealed that several transcript profiles were altered including genes involved in lipid uptake, fatty acid synthesis, fatty acid oxidation as well as an alteration in the expression of the nuclear receptor, PPARγ. Furthermore, several animal and clinical studies revealed a strong association of GH deficiency with obesity and fatty liver disease [77, 78]. GH exerts its action by binding to its receptor or through inducing insulin growth factor-1 mediator (IGF-1), which is primarily produced in the liver. The pattern of GH secretion differs between sexes, in males being pulsatile and in females being continuous. These patterns resulted in a higher plasma average concentration of GH in females than males [76]. In rats, supplementation of testosterone produces higher GH pulses while estrogen suppresses the GH pulses, implying the pivotal role of sex hormones on the secretory pattern of GH [79]. Aside from pattern difference and the role of sex hormones, a recently identified family of transcription factors known as signal transducers and activators of transcription (STAT) plays a major role in the sexual dimorphism of hepatic genes regulated by GH. Much attention has been directed to the role of STAT5, particularly the types a and b in sexual dimorphism because they are known to regulate several key lipids metabolizing enzymes and genes (Table 3). STAT5 is phosphorylated by JAK2 and Src family kinase (SFK) after GH binds to its receptor in the cell membrane. Phosphorylation of STAT5 is followed by dimerization, nuclear translocation, and binding to DNA response elements in genes regulated by STAT5 [76, 80]. In the liver of male mice and rats, STAT5b is directly activated by pulsatile plasma GH, whereas in female rat liver, there is desensitization because of the continuous GH secretion leading to a lower STAT5b expression compared to males [76, 81].
Androgen and growth hormone-STAT5b axis role in sexual dimorphism of NAFLD
| Male | Effect(s) | Female | Effect(s) |
|---|---|---|---|
| ↓ Levels of androgen | Associated with insulin resistance, T2DM, obesity, and NAFLD | ↑ Levels of androgen | Associated with the features of metabolic syndrome, NAFLD, and severe fibrosis |
| STAT5b-knockout male mice | Postnatal growth-retardation;obesity;↑ hepatic steatosis;↑ PPARγ;enhance insulin sensitivity to further increase lipogenesis | STAT5b-knockout female mice | Near-normal postnatal growth |
| STAT5b inactivity in human | Obesity | ---- | ---- |
| Testosterone deficiency | ↓ STAT5b | ---- | ---- |
↑ increased; ↓ decreased; ---- sex-balanced studies not available
The importance of these transcription factors was supported by the findings that STAT5b-knockout male mice became obese later in life and STAT5b inactivity leads to obesity in the adult human [82, 83]. Moreover, hepatic steatosis was also observed in liver-specific STATa/b-knockout mice, implying its role in lipid metabolism and NAFLD pathogenesis. One study group developed a STAT5b biomarker gene set to identify genes, chemicals, and hormones that activate (masculinize) or suppress (feminize) STAT5b function in a large, annotated mouse liver and primary hepatocyte gene expression compendium. Their results revealed that liver STAT5b activation/masculinization occurred at puberty and suppression/feminization occurred during aging and in mutant mice with defects in GH signaling [81]. Masculinization of STAT5b in male mice liver causes the activation of male-specific genes and suppresses the female-specific genes, a pattern that is reversed by STAT5b deficiency and with the absence of testosterone [84]. Moreover, suppression/ feminization was associated with the upregulation of PPAR-γ but no other lipogenic transcription factors linked to steatosis [81, 85]. However, apart from the substantial role of STAT transcription factors in sexual dimorphism, it is also important to consider the regulatory actions of other transcription factors such as Myc, Fos, and NF1 [80]. This is in line with the observation that, only 20% of GH-stimulated genes show alteration in expression in the liver of STAT5b-deficient mice [86].
The metabolic variables associated with insulin resistance [insulinemia, homoestatic model assessment of insulin resistance (HOMA-IR), triglyceride concentration, and visceral adiposity index (VAI) scores] differ significantly in a sex-specific manner in patients with NAFLD. A recent study showed that Mexican women with NAFLD had a higher body mass index (BMI), waist circumference, and alanine aminotransferase (ALT), as well as higher insulinemia, HOMA-IR, and VAI [87] suggesting a sex-related difference in the pathophysiological mechanisms involved in IR. Similar to NAFLD, the incidence of T2DM in postmenopausal women is significantly increased as compared with premenopausal women. This is attributed to the loss of estrogen that is known to have a role in the occurrence and development of IR and dysregulation in glucose homeostasis. A study on male mice lacking ER-α (LERKO mice) showed elevated gluconeogenic activity and fasting hyperglycemia, suggesting the role of E2/ERα signaling in glucose metabolism [49]. Similarly, the aromatase knockout (ArKO) mouse model showed sexual dimorphism wherein only male mice exhibited impaired hepatic insulin sensitivity, glucose, and pyruvate intolerance, with consistent improvement upon estrogen treatment [88]. More recently Allard et al. [89], showed that the global deletion of nuclear ERα, and to a lesser extent membrane ERα, alters the central control of hepatic glucose production in female mice. On the other hand, only the absence of nuclear ERα impairs the central regulation of insulin secretion in male mice. Convincing evidence shows that GPER affects insulin secretion. For example, E2, as well as GPER agonist, improves insulin secretion in human and mice B cells [90, 91]. Similarly, E2 treatment increases insulin and decreases glucagon secretion in T2DM rat models and human pancreatic islets via GPER [92]. However, the exact role of GPER in glucose metabolism in each sex is poorly understood. Contrary to the results of other ERs, a pro-diabetogenic function of the ERβ was suggested by the observation that ERβ-deficient female mice fed with HF-diet were protected against diet-induced IR and glucose intolerance. The authors also suggested that the improvement in whole-body insulin sensitivity and glucose tolerance is due to increased adipose tissue mass which causes chronic stimulation PPARγ [59].
Both androgen excess in females and androgen deficiency in males are associated with an increased risk of obesity, IR, and NAFLD pointing to the role of sexual dimorphism in the modulation of glucose metabolism by androgens. A meta-analysis found that high testosterone levels are associated with increased risk of T2DM in women but with a decreased risk in men. Further, the inverse association of SHBG with T2DM risk was stronger in women than in men [93]. Similarly, a recent review mentioned that androgens stimulate insulin sensitivity and glucose utilization in skeletal muscle cells, in both men and more in women [94]. Moreover, it was reported that androgen receptors (AR) found in pancreatic B cells work differently in modulating glucose homeostasis between sexes. In males, testosterone-AR complex enhances glucose-stimulated insulin secretion by potentiating the insulinotropic action of glucagon-like peptide-1 while in females, excess testosterone promotes insulin hypersecretion leading to oxidative injury [95]. Unfortunately, studies on the specific role of androgens in glucose metabolism and its association with sexual dimorphism in NAFLD patients are limited.
It is well established that the GH acts as an insulin antagonist by rapidly decreasing the glucose uptake in skeletal muscles and adipocytes. As mentioned previously, GH and sex steroids can regulate liver gene transcription indirectly by interacting with the GH receptor (GHR)-STAT5b signaling pathway. In male post-pubertal rats, the activation of the GHR-STAT5b pathway is episodic, with low levels of circulating GH are required to achieve maximal activation of STAT5b-mediated transcription. On the other hand, the GH secretion in female rats is continuous with a reduced STAT5b activation compared with males [96]. Aside from its role in lipid metabolism, emerging evidence also found that STAT5b has a crucial role in maintaining glucose homeostasis. One study revealed that insulin inhibits GH signaling via the GHR/JAK2/STAT5b pathway [97]. Another study using C57BL/6 male mice without functional STAT5 showed enhanced insulin sensitivity and glucose disappearance rate as mechanisms to further increase lipogenesis suggesting that GH-mediated STAT5b activation acts at multiple levels in the major insulin-responsive tissues to promote insulin sensitivity [98]. This is further supported by feminization/suppression of STAT5b in models of diabetes and obesity, wherein plasma glucose was shown to be elevated and was at least partially reversed by leptin or resveratrol treatment (Table 3) [85].
It is well accepted that females are protected against obesity-induced IR even if the exact mechanism(s) accounting for this difference is still not fully understood. As discussed above, sex hormones are involved in the regulation of glucose and lipid metabolism, and specifically in the control of insulin sensitivity. Therefore, it is interesting to consider sex differences in the inflammation occurring during the progression of NAFLD. Several findings indicate that inflammation of the white adipose tissue (WAT) is one of the driving forces that promotes an increased supply of fatty acids to other tissues due to the inability of insulin to suppress increased lipolysis. In line with this, Camporez et al. [43], explored plasma and WAT cytokines concentration both in males and females in a murine model fed with HFD. The authors showed that the plasma and WAT levels of IL-6, IFNγ, and TNF-α were significantly lower in females than in males, and these differences were, most probably, due to the presence of E2. Indeed, HFD-fed males treated with E2 showed a reduced inflammation both in the release of inflammatory cytokines and in the absolute number of macrophages in WAT compared with HFD male mice. Specifically, in this study, the authors demonstrated that the presence of E2 in a cell line of macrophages could counteract the increased release of TNF-α and IL-6 induced by palmitate and that the inhibition of ERα prevented the protective effect of E2 against palmitate-induced inflammatory cytokine release. Macrophages present several receptors which differ in males and females (reviewed here [6]); in males, the receptors activate a pro-inflammatory response, while females macrophages induce the production of anti-inflammatory prostanoids [99] suggesting that innate immunity promotes the activation of the inflammatory response in males while macrophages display a more protective and anti-fibrotic phenotype in females. Undoubtedly sex hormones play a crucial role in the differences observed in NAFLD incidence between males and females. In line with this, there is a growing evidence on the strong association between polycystic ovary syndrome (PCOS) and NAFLD/NASH. PCOS is the most common endocrinopathy among women of early reproductive age; it is characterized by chronic anovulation, clinical/biochemical hyperandrogenism, and the presence of polycystic ovaries [100]. Most of the endocrine and metabolic characteristics of women with PCOS, including elevated androgen levels, IR, dyslipidemia, and elevated low-grade inflammation levels, are thought to contribute to the presence and progression of NAFLD. There is a strong association between hyperandrogenism and inflammation in PCOS [101, 102]. It has been reported that various pro-inflammatory markers (TNF-α; MCP-1; MIP-1; and IL-18) are specifically elevated in women with PCOS and may contribute to the low-grade inflammation in this population. The chronic inflammatory status might contribute to hepatic cell injury. Moreover, proteins secreted mainly by hepatocytes in response to inflammation [C-reactive protein (CPR), follistatin, and ferritin] have been reported to be higher in women with PCOS than women without PCOS [103, 104].
It has also been reported that estrogens also affect the activity of adipokines. For instance, leptin is produced in the adipose tissue and its amount is directly related to the extent of body fat. Leptin regulation is tightly related to insulin and glucocorticoids. Leptin has a pro-inflammatory activity, enhances energy expenditure and inhibits food intake [105]. The leptin receptor (LepRb) co-localizes with ERα, suggesting cooperation between estrogens and leptin. Data have been provided indicating that estrogens induce and testosterone inhibits leptin synthesis, respectively [106]. The serum level of leptin differs between males and females and it is tempting to conclude that the proinflammatory properties of leptin may lead to the development of more severe NAFLD in men. Conversely adiponectin, which is an adipokine with anti-diabetic, anti-atherogenic and anti-inflammatory, also presents a sex disparity. Adiponectin serum levels are lower in males than those observed in females [105]. Interestingly, higher levels of androgens are associated with lower levels of adiponectin, and serum levels of this adipokine are decreased in NAFLD and even more reduced in NASH [107].
These are just some of the main aspects involved in sex-related differences in inflammation. Since the inflammatory process is the result of a complex balance between cytokines, adipokines, receptors and downstream responses, clearly defined sex differences in this process would be extremely relevant for the development of new therapeutic strategies.
Environmental factors such as the diet can increase hepatic inflammation and oxidative stress. Compared to males, female mice demonstrate significantly lower inflammatory protein expression (ICAM-1, MCP-1 and COX-2), lower expression of the DNA damage marker (Gadd153) and the oxidative stress marker HO-1. Moreover, female mice showed higher expression of antioxidant enzymes (SOD-1; SOD-2 and Ref-1) and lipogenic enzymes (FASN, ACLY) compared to male mice [108]. These differences might be attributed to the stimulatory effects of estrogens on the expression of these proteins [109–111].
Mitochondrial dysfunction is a common feature of NAFLD leading to oxidative stress, but the connection linking FFA to altered mitochondrial function is still undefined. Normally, damaged or low-functioning mitochondria are removed or recycled though the activation of mitophagy, a mitochondrial specific form of macroautophagy. Disruption of the process of macroautophagy and mitophagy may contribute to hepatic fat deposition by increasing the accumulation of dysfunctional organelles, leading to a subsequent increase in the generation of reactive oxygen species (ROS), inflammation, and apoptosis. Females possess inherent differences in hepatic mitochondrial metabolism that are paired with protection from diet-induced steatosis. A recent study performed in mice exposed to HFD [112] shows that females present increased hepatic mitochondrial respiratory coupling control, lower H2O2 emission, and protection against steatosis vs. males. These data suggest that female mitochondria can more efficiently utilize increased dietary lipids for energy production as compared to males. The proliferative-activated-receptor-γ coactivator-1 (PGC-1) family of transcriptional coactivators plays a central role in regulating mitochondrial biogenesis and function. This regulation is achieved by both PGC-1α and PGC-1β. PGC-1α levels in the liver are minimal under fed conditions but are dramatically increased during fasting and in diabetes. Thereby PGC-1α plays a determinant role in the regulation of gluconeogenesis [113] and mitochondrial biogenesis [114]. PGC-1β regulates hepatic fatty acid metabolism by controlling the expression of gene regulating both β-oxidation [115] and de novo synthesis of fatty acids [116]. Both PGC-1α and PGC-1β are critical for estrogen signaling to anti-oxidant genes that protect against liver injury [117, 118]. Impairment of mitochondrial biogenesis and function, with increased lipogenesis and reduced β-oxidation, has been observed in ovariectomized rats and those effects have been ameliorated by the E2 replacement. These findings explain why females exhibit greater oxidative capacity and mitochondrial differentiation compared to males.
Interestingly, it has been shown that female mice present a lower susceptibility to acetaminophen (APAP) hepatotoxicity than males. Even if APAP induced similar hepatic glutathione (GSH) depletion in both sexes, females presented a higher and faster GSH recovery both in the mitochondria and in the total liver which was correlated with greater induction of glutamate-cysteine ligase and attenuated mitochondrial oxidative stress [119]. A possible explanation for the observed differences in the GSH recovery and the susceptibility to APAP overdose might be in part attributed to the estrogen effect. In this study, authors observed that E2 exerted a moderated protection by reducing oxidative stress in males, even if the effect was not accompanied by improved recovery of GSH levels, protein adduct formation or JNK activation. Thus, the estrogen treatment of male mice does not mimic the exact mechanism observed in female mice, and most probably other mediators are playing a protective role.
Chronic endoplasmic reticulum (ER) stress is strongly related to the pathogenesis of some metabolic diseases [120]. The ER stress leads to the accumulation of unfolded or misfolded proteins in the ER lumen. To deal with this stress, cells activate a signal transduction system linking the ER lumen with the cytoplasm and nucleus, called the unfolded protein response (UPR) [121]. It has been suggested that there is sexual dimorphism in the hepatic ER stress that might be related to sex hormones. Testosterone treatment in prepuberal male rats stimulates the UPR in hepatic tissue by regulating ER markers [122], suggesting that this process is sexually dimorphic before puberty due to the lack of testosterone in males.
The human gut microbiota is a highly diverse ecosystem that is extremely variable among individuals. The liver receives 70% of its blood from the intestine and is constantly exposed to intestinal-derived metabolites. Therefore, it is not surprising that gut dysbiosis plays a crucial role in the development of metabolic diseases. Several studies have reported physiological differences in the diversity and composition of the gut microbiota between females and males both in mice [123] and humans [124]. These sex-differences in the microbial communities may be mediated by sex hormones. Hepatic inflammation is associated with intestinal bacterial overgrowth, increased gut permeability, and reduced immunological defenses [125]. BA are jointly produced by hepatic and bacterial enzymes and play a crucial role in regulating the absorption of nutrients, steroids, and drugs as well as the immune response. BA signaling also plays a crucial role in the regulation of metabolic homeostasis via the gut-to-liver axis. BA activates FXR to regulate lipid, glucose, and energy metabolism. Moreover, BA control gut bacteria overgrowth to protect the intestinal barrier function. At the same time, the microbiota can modulate metabolism through the modification of BAs as well as FXR signaling. In a recent study performed in FXR knock-out (KO) and wild type mice fed with WD [126], it has been shown that sex differences exist in hepatic steatosis, IR, BA, and microbiota profilesand that these changes were FXR-dependent. In this model, authors showed that both WD and FXR inactivation induced more severe steatosis in males than in females and that FXR-KO male mice presented a massive lymphocyte infiltration, serum lipopolysaccharides (LPS) and ALT levels than females, males were more susceptible to the development of steatohepatitis and fibrosis due to a substantial increase in inflammatory cytokines and Col1a1. Due to the relative abundance in females, Lachnospiraceae, Clostridiaceae, Streptococcaceae, and Christensenellaceae are more likely to have protective effects against hepatic inflammation (Figure 1). This aspect is very promising, and recently the study of gut microbiota gained much attention. However, the studies are usually complicated and the comparison among the different results is sometimes inconclusive.
Considering the previously mentioned differences between sexes in the severity of NAFLD, it is plausible that liver fibrosis would follow the same pattern. Unfortunately, the current available data do not provide consistent information about it. In a cross-sectional study that investigated the influence of sex and menopause on the severity of liver fibrosis using a large single-center prospective database, it was reported that of 541 patients with biopsy-proven NASH [38] the risk of more severe fibrosis was higher in men compared to premenopausal women. Moreover, postmenopausal women and men had similar severity of liver fibrosis. Conversely, in a recent study [127] conducted in NAFLD patients, the authors showed similar frequencies of advanced fibrosis for both sexes (present in 36% of 284 younger men and 31% of 132 younger women, while in 64% of 120 older men and 63% of 226 older women). In line with these observations, in an in vivo juvenile murine model of NASH characterized by our group [128], we observed that male and female mice fed with HFD for 16 weeks developed the same histological extent of liver fibrosis; Col1a1 overexpression (both gene and protein) and α-SMA. Interestingly and despite the similar fibrosis score, we observed that during the early stages of the disease the prevalent driving mechanisms were different between sexes. In males, the HFD induced aprogressive portal and lobular inflammation in the absence of lipid-peroxidation products, and the opposite in females. Moreover, differently from males, females presented hyperglycemia with no insulin-resistance. Altogether these data suggest that sex differences exist in the regulation of different mechanisms that converge into liver fibrosis, but not directly in fibrogenesis. However, further studies are still necessary to understand if these assumptions are real or just speculations.
In a CCl4-induced liver injury model of adult male and female mice, it has been shown that more severe liver fibrosis was found in male’s liver sections, with higher activation of HSC by enhanced immunostaining of α-SMA [129]. Conversely, Col1a1 synthesis and TGF-β1 were similar between sexes. While male mice castration improved liver damage, testosterone supplementation in this group aggravated it, mainly by regulating the activation of NLRP3 inflammasome, which influence the subsequent progression toward fibrosis. Similarly, in another study performed in CCl4-treated male mice [130], the authors showed that supplementation with 2-Methoxyestradiol (2-ME) inhibited CCl4-induced liver toxicity, with an apparent reduction in liver fibrosis with reduced collagen deposition and α-SMA expression in HSC. The anti-fibrotic effect of 2-ME is mainly due to an inhibitory effect on macrophage infiltration to the injured liver and with an alternative activation (M2 phenotype). Even if CCL4-induced liver damage is a harsh model of hepatotoxicity which might differ from NAFLD, the current evidence reported here indicates the key role of sex hormones in the pathological processes involved in the progression of liver injury. Further studies in the context of NAFLD will provide further information on this specific process.
In the last two decades, the use of high-throughput technologies enables the generation of thousands of biological data which nowadays are being utilized in multi-omics integrative approaches.
An example of in silico omics integration is available through Mergeomics, a useful platform to model the molecular mechanism underlying the sexual dimorphism in NAFLD [131]. Another example is LiverSex [132], a large-scale liver metabolic computational model, which allows to delineate sex-dependent complex liver pathologies and to perform prediction about how genetic and environmental factors interact with sex in disease appearance and progression.
Beyond the above-mentioned integrative pipelines, when we refer exclusively to metabolomics datasets, the studies regarding NAFLD were performed without examining the sexual dimorphism factor [133], and consequently, further studies considering sex will be necessary to optimize the computational models.
In this section, we will revise studies investigating sex-differences in healthy and NAFLD subjects obtained via serum metabolomics. One of the first large population study included 3, 300 serum samples and the quantitative determination of 132 metabolites through LC/MS/MS system [134]. Using a linear regression approach, it was reported that 102 of the total metabolites presented significant differences between males and females. Among those analytes, most of the amino acids were significantly higher in males except for the concentrations of glycine and serine, which displayed the highest levels in females. The same trend was observed for acylcarnitines, whereas phosphatidylcholines and sphingomyelins presented the opposite.
In a serum metabolomics study, Saito et al. [135], investigated the gender and age-associated differences in a Japanese population. A total of 516 endogenous metabolites were identified in sera; with the 31 and 25% of metabolites showing sex-associated differences in the young and old populations, respectively. The highest differences were observed for metabolites involved in redox homeostasis or been part of the steroid and purine-nucleotide metabolism pathways.
Similar findings were observed by Krumsiek et al. [136], in a large-scale serum metabolomics analysis, where 1, 756 samples from independent participants were investigated. One-third of the total metabolites (507 analytes) showed significant differences between males and females. The greatest difference was observed in components of steroid metabolism, fatty acids, lipids, a large fraction of amino acids, oxidative phosphorylation, purine metabolism, and gamma-glutamyl dipeptides.
Li et al. [137], performed a plasma metabolomics analysis on 236 participants using a TQ MS mass spectrometry system. The study quantified a total of 168 metabolites for each subject, and after performing differential correlation network analysis, 10 phosphatidylcholines and 4 acylcarnitines were identified as the key metabolites depicting differences between sexes.
When considering metabolomics studies that take into account sex-differences in NAFLD, a limited amount of information has been reported. Recently, Meda et al. [138], investigated the contribution of liver ERα in the hepatic sexual dimorphism using, transcriptomics and metabolomics, in a mice model of diet-induced obesity. They demonstrated that in females, ERα keeps lipid uptake low preventing hepatic lipid deposition, inhibiting lipid synthesis, and promoting mtFAO. Authors argue that a proper liver ERα activity should be preserved in females to maintain the metabolic flexibility and counteract the metabolic impairments in the postmenopause period.
It is interesting to point out that several components of purine metabolism were associated with sexual dimorphism in the studies mentioned above. Therefore, it is essential to investigate further the relationship between altered serum uric acid (UA) levels (the final oxidation product of purine metabolism) and the development of metabolic disorders such as gout’s pathogenesis, obesity, cardiovascular disease, T2DM, and NAFLD [138–141].
It is well known that UA levels are higher in men than women; however, the molecular mechanisms regulating UA homeostasis are still not fully understood, involving genetic, environmental and lifestyle factors. In this context, Albrecht et al. [142], performed a metabolomics study by which 335 metabolites were analyzed in 1, 764 individuals (males and females) and throughout metabolite network analysis established that 25 analytes showed considerable differences between sexes. Additionally, their data suggested that dipeptides, amino acids, and steroid hormones are relevant regulators of purine catabolism and consequently of serum urate levels.
As briefly mentioned above, data have been reported on the relationship between serum UA levels and NAFLD development. In a large study of 100, 275 subjects from America and Asia, Darmawan et al. [143], found that the risk of NAFLD was increased almost 2-fold in the highest serum UA group compared to the lowest; and that women presented a higher risk in a dose-dependent manner with UA concentration, as previously reported [144–146]. These findings make UA level (or other metabolites from purine catabolism) an attractive parameter to be included in the various existing non-invasive algorithms to improve specificity in NAFLD diagnosis or for a better disease stratification. In this context, we recently published a study in which by the surface-enhanced Raman spectroscopy (SERS) of plasma [147], and demonstrated that the UA/ hypoxanthine ratio was significantly different between NAFL and NASH in women.
Culafic et al. [148], suggested HUFA index, a simple non-invasive algorithm to predict NASH from simple steatosis in a NAFLD cohort. The predictive model which includes four parameters (HOMA-IR, UA, ferritin, and ALT) demonstrated to differentiate NASH from simple steatosis. Ballestri et al. [149], also identified UA together with other morpho-biochemical parameters as good predictors for the diagnosis of NASH and with HOMA-IR and serum total cholesterol for the differential diagnosis of moderate-to-severe steatosis.
When considering fibrosis in NAFLD, the reported evidence is less extensive and controversial. In a prospective study including 113 NAFLD patients stratified into moderate-severe steatosis vs. significant liver fibrosis [using controlled attenuation parameter (CAP) – transient elastography (TE)], Sandra et al. [150], demonstrated that hyperuricemia was an independent risk factor for significant fibrosis. Moreover, Huang et al. [151], reported a study in which 76 biopsy-proven NASH patients were categorized according to their fibrosis stage. Results indicated that CK18 combined with UA levels could be used for the prediction of advanced fibrosis in NASH patients. On the contrary, Jaruvongvanich et al. [152], performed a meta-analysis to investigate the association between hyperuricemia and fibrosis stage in NAFLD. A total of 749 NAFLD patients were included from five observational studies. The study strongly suggested the absence of a causal relationship between hyperuricemia and advanced liver fibrosis in NAFLD.
Whilst the listed literature reviewed herein indicate that the inclusion of UA or purine metabolites could have a potential in the improvement of diagnostic NAFLD algorithms, however, additional observational and retrospective work considering sex differences is needed to reduce bias, improve reproducibility and maximize the ability of diagnostic indexes in distinguishing minimal to severe variants of NAFLD.
NAFLD is a progressive wide spectrum disease that involves several factors and requires interaction with different organs. Despite the intrinsic complexity of NAFLD pathogenesis, some promising therapeutic alternatives have emerged recently to halt NASH progression to cirrhosis, and therefore preventing serious liver-related outcomes. Recently, arandomized placebo-controlled global phase 3 study (REGENERATE) [153] has shown that obeticholic acid (OCA) improved histological features and improved laboratory parameters in NASH patients. It has been also shown in vitro that OCA reduces the extracellular collagen deposition and regulates the activity of metalloproteinases and their inhibitors [154]. Other trials in late phase 2 (such as FGF19 [155], FGF21 [156], elafibranor [157] among others) have suggested being beneficial against NASH and liver fibrosis. However, to date, there are still no approved therapies to NASH. Moreover, non-invasive diagnostic tools are still missing to provide an early diagnosis of NASH. To make this complex scenario even more complicated, sex differences exist between males and females, and among females depending on their reproductive age. Considering the existing differential biological functions between males and females under physiological conditions, especially related to the metabolism, it is somehow obvious and expected that these differences persist during pathological metabolic alterations. However, these relevant aspects are usually not considered by the current guidelines for the diagnosis and management of the disease. Also, the available information from experimental models is still insufficient to draw solid conclusions about this issue.
In the present review, we tried to summarize the sex differences reported in the most relevant aspects related to the progression of NAFLD such as glucose and lipid metabolism, inflammation, oxidative stress, gut microbiota, and abundance of different metabolites. The role of hormones is undeniable in the regulation of several of the pathological aspects involved in NAFLD between males and females. However, other parameters might also be playing a significant role in the pathogenesis of the disease. To enlighten the underlying mechanisms, males and females should be included as separate groups in future experimental models. Based on the evidence of the role of sex hormones, age must not be disregarded, especially considering the increasing prevalence of pediatric NAFLD. Collectively the sex related differences in the pathogenesis and clinical outcome of NAFLD and NASH is a puzzle that is still far to be assembled.
2-ME: 2-Methoxyestradiol
ALT: alanine aminotransferase
AMPK: AMP-activated protein kinase
APAP: acetaminophen
AQP: aquaglyceroporins
AR: androgen receptors
ArKO: aromatase-knockout
BA: bile acids
BMI: body mass index
CAP: controlled attenuation parameter
CD36: cluster of differentiation 36
CPR: C-reactive protein
CPT-1: carnitine palmitoyltransferase 1
DNL: de novo lipid synthesis
E2: estradiol
ER: endoplasmic reticulum
ERα: estrogen receptor α
ERβ: estrogen receptor β
FXR: Farnesoid X receptor
GH: growth hormone
GHR: growth hormone receptor
GPER: G protein-coupled estrogen receptor
GPR30: G protein-coupled receptor 30
GSH: glutathione
HCC: hepatocellular carcinoma
HFD: high-fat diet
HOMA-IR: homeostatic model assessment of insulin resistance
HSCs: hepatic stellate cells
IGF-1: insulin growth factor-1 mediator
IR: insulin resistance
JNK: phosphorylated c-Jun NH2-terminal kinases
KO: knock-out
LepRb: leptin receptor
LPS: lipopolysaccharides
MetS: metabolic syndrome
NAFL: non-alcoholic fatty liver
NAFLD: non-alcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
NIH: National Institutes of Health
NR: nuclear receptor
OCA: obeticholic acid
PCOS: polycystic ovary syndrome
PGC-1: proliferative-activated-receptor-γ coactivator-1
PNPLA3: patatin-like phospholipase domain containing 3
PPARγ: peroxisome proliferator-activated receptor-γ
ROS: reactive oxygen species
SERS: surface-enhanced Raman spectroscopy
SFK: Src family kinase
SHBG: sex-hormone-binding globulin
SHROB: obese spontaneously hypertensive
SREBP-1c: sterol regulatory element-binding protein-1c
STAT: signal transducers and activators of transcription
T2DM: type 2 diabetes mellitus
TE: transient elastography
UA: uric acid
VAI: visceral adiposity index
WAT: white adipose tissue
WD: western diet
NCS; PJG; CT and NR contributed conception and design of the review and in the writing of the different sections of the review. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
NCS is funded by the Department of Science and Technology-Philippine Council for Health Research and Development (DOST-PCHRD), Philippines; PJG is supported by Post-doctoral Fellowships 2020 from Fondazione Umberto Veronesi, Italy; NR is funded by Fondazione Cassa di Risparmio di Trieste (CRTrieste) and by Fondazione Italiana Fegato ONLUS.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Amedeo Lonardo, Stefano Ballestri
Marvin Ryou ... Gyorgy Baffy
Michael Doulberis ... Stergios A. Polyzos
Marica Meroni ... Paola Dongiovanni
Ivana Mikolasevic ... Sandra Milic
Valerio Rosato ... Marcello Persico
Carlo Acierno ... Ferdinando Carlo Sasso
Rosa Lombardi ... Anna Ludovica Fracanzani
Angelo Di Vincenzo ... Marco Rossato
Angelo Colucci ... Claudia Mandato
