 Review
Review

Affiliation:
Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
†These authors contributed equally to the manuscript.
ORCID: https://orcid.org/0000-0002-1239-7012
Affiliation:
Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
†These authors contributed equally to the manuscript.
ORCID: https://orcid.org/0000-0001-7761-7533
Affiliation:
Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
ORCID: https://orcid.org/0000-0002-0310-3529
Affiliation:
Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
ORCID: https://orcid.org/0000-0003-3320-3878
Affiliation:
Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
Email: luigielio.adinolfi@unicampania.it
ORCID: https://orcid.org/0000-0001-8453-4912
Affiliation:
Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
ORCID: https://orcid.org/0000-0002-9142-7848
Explor Med. 2020;1:287–306 DOI: https://doi.org/10.37349/emed.2020.00019
Received: July 01, 2020 Accepted: July 19, 2020 Published: October 30, 2020
Academic Editor: Lindsay A. Farrer, Boston University School of Medicine, USA
The article belongs to the special issue Exploring NAFLD/NASH
The pathophysiological mechanisms underlying the close relationship between nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus (T2DM) are multiple, complex and only partially known. The purpose of this paper was to review the current knowledge of these mechanisms in a unified manner. Subjects with NAFLD and T2DM have established insulin resistance (IR), which exacerbates the two comorbidities. IR worsens NAFLD by increasing the accumulation of free fatty acids (FFAs) in the liver. This occurs due to an increase in the influx of FFAs from peripheral adipose tissue by the activation of hormone-sensitive lipase. In addition, there is de novo increased lipogenesis, a transcription factor, the sterols regulatory element-binding transcription factor 1c (SREBP-1c), which activates the expression of several genes strongly promotes lipogenesis by the liver and facilitate storage of triglycerides. Lipids accumulation in the liver induces a chronic stress in the endoplasmic reticulum of the hepatocytes. Genome-wide association studies have identified genetic variants associated with NAFLD severity, but unrelated to IR. In particular, the alteration of patatin-like phospholipase domain-containing protein 3 contributes to the susceptibility to NAFLD. Furthermore, the lipotoxicity of ceramides and diacylglycerol, well known in T2DM, triggers a chronic inflammatory process favoring the progression from hepatic steatosis to steatohepatitis. Reactive oxygen species produced by mitochondrial dysfunction trigger both liver inflammation and beta-cells damage, promoting the progression of both NAFLD and T2DM. The close association between NAFLD and T2DM is bidirectional, as T2DM may trigger both NAFLD onset and its progression, but NAFLD itself may contribute to the development of IR and T2DM. Future studies on the mechanisms will have to deepen the knowledge of the interaction between the two pathologies and should allow the identification of new therapeutic targets for the treatment of NAFLD, currently substantially absent.
Non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes mellitus (T2DM) could be considered two sides of the same coin. NAFLD is currently the most widespread liver disease in the world and is the most common among patients with T2DM [1]. A recent meta-analysis showed that patients with NAFLD have a risk of developing T2DM at least twice as high as those without NAFLD and that patients with “severe” NAFLD had an even greater risk of developing T2DM [2].
NAFLD encompasses a broad spectrum of diseases, from pure hepatic steatosis in the absence of inflammation, to non-alcoholic steatohepatitis (NASH), liver cirrhosis and potentially hepatocellular carcinoma (HCC) [3, 4].
We currently know that metabolic hepatic steatosis is a phenotype that recognizes diverse and complex causes and a wide spectrum of clinical severity, as well as considerable inter-patient variability and, therefore, the NAFLD terminology used to identify the disease it is generic and includes numerous subtypes. The acronym NAFLD therefore seems obsolete and does not reflect the current knowledge on the pathology, on the diagnostic criteria, on the lack of consideration of the different risk profiles and on the difficulty of interpreting the response to the treatments. A consensus has recently been reached among a group of experts who proposed replacing the acronym “NAFLD” with “MALFD”, “metabolic-dysfunction-associated fatty liver disease” as a more appropriate general term [5]. The acronym MAFLD shifts the diagnosis criteria from an exclusion disease to an inclusion disease. Diagnosis is based on the recognition of metabolic abnormalities with the acceptance that MAFLD can coexist with other conditions. The metabolic abnormalities proposed are liver steatosis diagnosed either by histology, imaging or blood markers, and at least one among the following: overweight/obesity, T2DM or evidence of metabolic syndrome (MS) [5]. Insulin resistance (IR) appears to be the main physiopathological link between NAFLD and T2DM [6]. Both NAFLD and T2DM patients share the same qualitative and quantitative multi-organ IR patterns, affecting not only glucose metabolism but also fatty acids and amino acid metabolism that is typical of the patient with overt T2DM [7]. In fact, it has been known for years how MS may underpin both NAFLD and T2DM. IR appears to explain many of the MS-related conditions, including NAFLD and T2DM [8]. This hypothesis is tempting, but too simplistic and reductive, since NAFLD also occurs in lean and non-diabetic individuals [9]. Therefore, NAFLD cannot be explained by IR alone, as other factors such as lipid function, mitochondrion, innate immunity, gut microbiota, genetic determinants, nutritional factors and lifestyle are involved in the development of the disease [10–12].
In any case, the close epidemiological association between the two conditions appears unequivocally clear. Over 70% of T2DM patients also have NAFLD [13, 14] and up to 20% of T2DM patients also have overt hepatic fibrosis [15–17]. Furthermore, the data show that the association between NAFLD and T2DM is a prognostic poor condition, as it carriers a high risk of mortality from liver cirrhosis and HCC [18, 19].
As already mentioned, the pathophysiological thread of this process is mutual. Patients with liver cirrhosis are in fact characterized by profound alterations in insulin-mediated glucose metabolism [20], which leads to impair glucose tolerance in 60–80% of cases, and overt diabetes in 10–20% of patients [21–23].
At the same time, T2DM patients may also exhibit a continuum of liver damage, ranging from a simple increase in circulating levels of liver enzymes, up to liver cirrhosis, HCC and liver failure. T2DM not only increases the risk of NAFLD, but the reverse is also true, with all the mutual complications and a reduced prognosis. Based on epidemiological and pathophysiological observations, a close association emerges between T2DM and NAFLD, with diabetes negatively affecting both severity and progression of NAFLD, and NAFLD increasing the risk of developing T2DM and its cardiovascular and renal complications [24, 25]. Therefore, a combined approach is needed to explain the pathophysiological basis of these two conditions.
Therefore, NAFLD and T2DM cannot be dissociated, due to the double thread that binds them. On the one hand, this relationship is supported by IR, whereby not only does one influence the other, but also its mechanisms exasperate and enhance the other, causing these two entities to make the same coin.
Currently, as schematized in Figure 1 the pathophysiological mechanisms underlying the close relationship between NAFLD and T2DM are multiple, complex and only partially clarified. Therefore, the purpose of this review was to make an in-depth and unitary analysis of the current knowledge of these mechanisms.
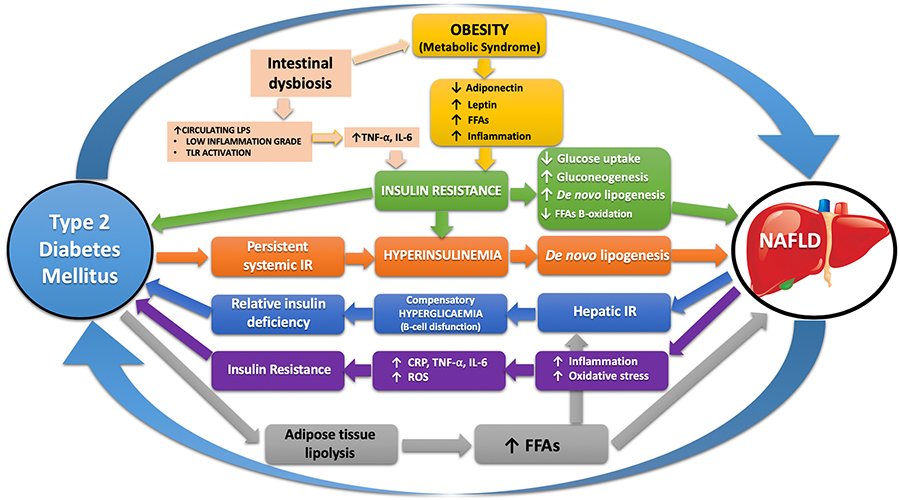
Schematic representation of the pathophysiological mechanisms shared between NAFLD and T2DM that connect the two sides of the coin. LPS: lipopolysaccharides; CRP: C reactive protein; TNF-α: tumor necrosis factor; IL-6: inteleukine-6; ROS: reactive oxygen species; TLR: toll like receptor
Hepatic steatosis, characterized by fat accumulation in more than 5% of hepatocytes, occurs due to an imbalance between fatty acids storage and their digestion in the liver. Triglycerides, also called triacylglycerols, are glycerol with the chains of three fatty acids in place of hydrogens from the hydroxyl groups [26]. Triglycerides are produced by the liver from the acyl-coenzyme A which, in turn, derives from free fatty acids (FFAs), thanks to the acyl-CoA synthetase and L-glycerol 3-phosphate from glycolysis [27].
FFAs reach the liver via three pathways [28]: (i) lipolysis of peripheral adipose tissue, (ii) fatty acids synthesis in the liver (de novo lipogenesis), and (iii) diet, grouped into chylomicrons and released by the tissue lipoprotein-lipase. Once in the hepatocytes, FFAs have two destinations. They can undergo a beta-oxidation in mitochondria, which brings either to ATP production, or to the synthesis of ketone bodies. Alternatively, they can be esterified to produce triglycerides, then either stored as droplets into the hepatocytes or packaged in apolipoproteins and secreted as very low-density lipoproteins (VLDL) in the serum.
As during T2DM the deposit of triglycerides into the liver increases, causing steatosis, it requires further investigation, both from a biochemical and pathophysiological point of view. It should be noted that IR, in the course of the disease, is able to increase the content of fatty acids in the liver, thanks to which there is a greater synthesis and deposition of triglycerides.
The strong association between IR, hyperinsulinemia, glucose intolerance, reduced high density lipoprotein cholesterol levels and hypertension has been well-known since the end of 80s.
It was immediately clear that both glycemic compensation and maintaining plasma FFAs levels within normal ranges could be achieved with a state of hyperinsulinemia by increased its production by the pancreatic beta-cells, a condition known as IR [29]. Overall, abdominal obesity, IR, dyslipidemia, and a high blood pressure are enclosed in the definition of MS. This condition appears to be associated with other comorbidities, including a pro-thrombotic status, NAFLD and reproductive disorders [30, 31]. In this regard, it seems evident that overeating, usually associated with obesity and genetic predisposition, leads to IR which, in turn, is invariably coupled with NAFLD. MS attempts to worsen the condition of the liver to steatohepatitis (NASH), mainly due to lipotoxicity [32].
Hormone-like fibroblast growth factor (FGF)-21 appears to be deeply involved in the development of IR and NAFLD. FGF-21 is highly expressed in the liver and regulates liver glucose production and lipid metabolism, as well as the energy metabolism of cells. Hepatic expression of FGF-21 is controlled by peroxisome proliferator-activated receptor (PPAR)-α pathway [33]. Lack of FGF-21 or resistance to its action can trigger glucose intolerance. High circulating levels of FGF-21 have been reported in patients with obesity, T2DM, IR and NAFLD and appear to be related to both hepatic and peripheral IR [34]. Many clinical and experimental studies have shown a correlation between circulating levels of FGF-21 and the fat content in the liver [35]. Furthermore, experimental studies have shown that an altered FGF-21 signal plays an important role in the development and progression of NAFLD. It has recently been reported that the administration of an FGF-21 analogue improves NAFLD [36]. Therefore, FGF-21 could be both a NAFLD biomarker and a possible new therapeutic approach [35].
T2DM is a metabolic disease characterized by hyperglycemia as part of IR and relative insulin deficiency [37]. The failure of pancreatic beta-cells to attempt the hyperinsulinemia necessary to compensate IR represents a critical step in the pathogenesis of T2DM. Thus, the key role of IR in the onset of this condition is evident and the lack of compensation of the beta-cells of IR occurs in the pathogenesis of T2DM [38]. In the case of IR, beta-cells keeps a normal glucose tolerance by increasing insulin secretion. Only when they are not able to release an adequate amount of insulin in the presence of IR, glucose concentration increases [39]. Current evidence may help in establishing IR as leading cause of T2DM when pancreatic beta-cells exhaust their ability to keep a state of hyperinsulinemia [38].
However, an alternative gaining ground view is the potential role of IR in the protection of critical body tissues from a metabolic damage in case of nutrients oversupply. To say, the dogma that IR is harmful for the body should be countered by every means has been argued. Thus, the thought that IR may represent a defense mechanism of critical cardiovascular tissues against injury induced by chronic nutrients excess would be overturned [40–43]. In other words, IR during MS, although it represents a biomarker of metabolic poor health, could only represent an epiphenomenon rather than the root cause. Therefore, if IR plays an adaptive protective role, any attempt to circumvent it in treatment could potentially result in harm. Therefore, there should be a change in the interpretation of IR, T2DM etiology, the therapeutic management of patients with either MS, T2DM and related conditions.
Insulin is a peptide hormone produced by pancreas beta-cells in the islets of Langerhans [44]. Insulin binds its receptor, a homodimer with an alpha and a beta subunit placed on the cell membrane. The consecutive activation of the receptor triggers a signaling chain through which the phosphorylation of tyrosine residues occurs which, in turn, results in the activation of AKT, called protein kinase B (PKB), a protein of the cytosol. PKB finally induces cell growth and resistance to apoptosis, by activating mTOR. AKT, once activated, is also able to inhibit FOXO1, a so-called “fork-head box” O1, which acts as a DNA transcription factor, suppressing the hepatic production of glucose and allowing metabolism in the post-prandial state [45]. FOXO1 once activated by phosphorylation, moves from the nucleus to the cytoplasm via the insulin dependent PI3K/AKT pathway. Consequently, the transcription of glucose-6-phosphatase (G6P) is reduced, and the amount of both gluconeogenesis and glycogenolysis decreased [46]. FOXO1 is also able to negatively regulating de novo lipogenesis by binding PPAR-γ promoter and thus hindering its transcription. Indeed, lipogenesis initially requires elevated levels of PPAR-γ. During the active signaling of insulin, FOXO1 moves outside the nucleus, becoming unable to both prevent PPAR-γ transcription and inhibit lipogenesis [47].
Briefly, the consequences of insulin on glucose and lipid metabolism are: (1) increased glycogen synthesis, reduced gluconeogenesis and glycogenolysis, resulting in an increase in the amount of glycogen, and (2) increased lipogenesis and decreased lipolysis with an increase in the level of triglycerides [48]. In light of this, it seems paradoxical that during T2DM, usually characterized in its early stages by IR and hyperinsulinemia, glycogen levels are low and triglycerides levels high. Such a behavior can be explained by a passage of FOXO1-mediated metabolism towards lipogenesis. Indeed, FOXO1 increases transcription of the sterol regulatory element-binding transcription factor 1c (SREBP-1c), which is involved in lipogenesis. At the same time, once FOXO1 is translocated out of the nucleus, it hinders the transcription of G6P, triggering an increase in lipogenesis against glycogen production [49]. Insulin-mediated suppression of hepatic glucose production plays a critical role in the pathogenesis of T2DM. The production of hepatic glucose occurs mainly due to the reduction of hepatic Acetyl-CoA, suppressing lipolysis in the white adipose tissue, resulting in a reduction in the flow of pyruvate carboxylases.
Overall, these findings establish that hepatic Acetyl-CoA from white adipose tissue lipolysis is the main regulator of insulin-mediated hepatic glucose production. Thus, hepatic acetyl-CoA is associated with inflammation-induced hepatic IR which, in turn, is related to both obesity and T2DM.
Finally, IL-6 activates the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB)-c-Jun N-terminal kinase (JNK)-ceramide pathways, which inhibit insulin signaling and, in turn, they increase the transcription of gluconeogenic protein [50]. Consequently, the hepatic concentration of Acetyl-CoA increases, due to the increased beta-oxidation of fatty acid, which triggers hepatic gluconeogenesis by activating the mitochondrial pyruvate carboxylases pathway [50]. The rapid reduction in insulin-mediated hepatic glucose production (i.e. few minutes), as well as the lack of any relationship between the hepatic expression of gluconeogenic proteins and fasting hyperglycemia in obese subjects, with or without T2DM, supports the clinical relevance of such a mechanism [51].
Whatever the cause, either the leading one or a simple epiphenomenon, NAFLD and T2DM subjects have an established IR, which exacerbates the two comorbidities. First, IR worsens NAFLD by increasing deposition of FFAs inside the liver, through an increase in the influx of FFAs from peripheral adipose tissue trough activation of hormone-sensitive lipase (HSL) [52]. Second, there is an increased de novo lipogenesis, mediated by SREBP-1c, whose maturation inside endoplasmic reticulum (ER) depends on insulin. SREBP-1c activates the expression of several genes involved in the glycolysis pathway (e.g., glucokinase, L-pyruvate kinase, acetyl-CoA carboxylases). In this way, glucose metabolism and lipogenesis genes (e.g., FFA synthase, sterol-CoA-desaturase) are activated, together with the carbohydrate-response element-binding protein (ChREBP), which strongly promotes lipogenesis [53]. Finally, IR can also suppress beta-oxidation. There is an increase in the production of Malonyl-CoA, capable of inhibiting carnitine palmitoyl transferase-1, reducing the influx of fatty acids inflow into the mitochondria and inhibiting the subsequent oxidation of FFAs in hepatocytes [53].
The accumulation of lipids in the liver induces a chronic endoplasmic reticulum stress of the hepatocytes. These events trigger a harmful vicious circle by furtherly promoting the accumulation of self-destructive lipids in the hepatocytes [54]. Other elements, such as bile acids are involved in the relationship between steatosis and IR. Bile acids, in fact, by binding to the transcription factor farnesoid X receptor (FXR), act on different targets of lipid metabolism (i.e. SREBP-1c or PPAR-γ) and counteract hepatic steatosis. Treatment of patients with a FXR agonist like obeticholic acid is an effective therapeutic option for patients with advanced NAFLD.
Besides an increased in systemic IR, NAFLD and T2DM patients also have an elevated hepatic IR, most likely mediated by intrahepatic fats accumulation, as well as an increase in proinflammatory cytokines [27, 55, 56].
Inflammatory pathways are critically involved in the development of IR. IR is associated with a low degree of chronic inflammation and several mediators released by various types of cells, including immune system cells and adipocytes, are involved. The most frequent mediators are TNF-α, IL-1, IL-6 and numerous adipocytokines.
Currently, it is not yet clear which sites are affected by inflammatory processes. Beyond the adipose tissue, the gastrointestinal tract, due to its highly altered microbiota, could also be one of the first events in the development of both IR and NAFLD. Insulin binds to its specific receptor activating its intracellular signaling pathway. Once bound, the insulin receptor phosphorylates itself and several members of the insulin receptor substrate (IRS) family. In the liver, IRS1 and IRS2 are the main mediators of insulin signal by regulating insulin sensitivity. Some studies have reported that TNF-α is involved in the obesity-related systemic IR by inhibiting insulin receptor tyrosine-kinase in the two tissues primarily responsible of the insulin-induced glucose uptake: muscle and adipose tissue [57]. Therefore, IR is a complex metabolic disorder, most likely involving numerous pathways. There is substantial evidence that the activation of the transcription factor NF-kB, a protein complex that acts as a transcription factor, is involved in hepatic IR. In fact, I-kappa-B kinase β (IKK-β, either named inhibitor of nuclear factor kappa-B kinase subunit beta), once activated, phosphorylates NF-kB inhibitor, thus hindering the normal transcription role of NF-kB. TNF-α is able to activate IKK-β, thus leading to a reduction in insulin signaling, which manifest itself in IR. These data corroborate the hypothesis of a role of inflammation in the pathogenesis of IR, T2DM and obesity. In this case, IKK-β could be a potential therapeutic target for insulin sensitization [58].
The accumulation of lipids in the liver causes activation of NF-kB and the production of downstream cytokines resulting in subacute hepatic inflammation, determining both hepatic and systemic IR [59].
The NF-kB ligand receptor activator (receptor activator of nuclear factor kappa-Β ligand, RANKL) contributes to this pathway. Its high serum concentration represent an independent risk factor for T2DM. RANKL binds its receptor in the liver and activates the NF-kB pathway, increasing local inflammation and causing IR. RANKL can be considered a potential valid therapeutic target, similar to what happen for osteoporosis [60]. This would suggest that bone have an endocrine regulatory function on glucose homeostasis and, therefore, IR [61].
Adipose tissue has emerged as the main site of inflammation in obesity-related disorders. Proinflammatory cytokines (TNF-α, IL-6) are produced by the human adipose tissue, in relation to the degree of obesity. In particular, TNF-α and IL-6 mRNA expression is greater in adipose tissue than in the liver in patients with a severe obesity [62, 63].
The mechanisms of the progression of adipose tissue inflammation has not yet been understood. Local mechanic factors, as well as various adipokines, could be critically involved [64]. In obese individuals, adiponectin levels are reduced, contributing to the development of both IR and T2DM. Therefore, oral AdipoR agonists may represent a valid therapeutic approach [65]. The data seem to confirm the hypothesis of a crosstalk between adipose tissue and liver, as well as between IR and chronic inflammation [66].
In recent years, adipose tissue has emerged as a real endocrine organ, capable to producing various biochemical compounds, called adipokines, which exert autocrine, paracrine and endocrine functions by acting on the body’s energetic metabolism [67]. Therefore, adipokines play a crucial role in the development of MS, as well as in the progression of nonalcoholic fatty liver (NAFL) to NASH and cirrhosis [68]. In addition to adipokines, adipose tissue also contains many immune cells, such as macrophages, B and T cells, and neutrophils which, in case of obesity, may trigger a chronic low-grade inflammation, capable to triggering changes in the production of adipokines and, therefore, in the metabolic regulation of adipose tissue [69].
Adipokines produced by the adipose tissue can play a positive or a negative role towards NAFLD. For example, adiponectin reduces IR and has both anti-steatosic and anti-inflammatory effects, while conversely, TNF-α and IL-6 have pro-inflammatory effects [70]. However, the continuous crosstalk between positive and negative adipokines makes the situation extremely dynamic, as these are influenced by various factors (e.g., genetic susceptibility and lifestyle) [71]. In particular, the relationship between Adiponectin and TNF-α seems to be reciprocal, as one inhibits the synthesis, transcription and biochemical activity of the other, thus being able to act on IR and systemic energy metabolism [72]. Under normal conditions Adiponectin and TNF-α have a balanced ratio, while in obesity, an imbalance towards TNF-α develops leading to IR and NAFLD [70].
The reality is much more complex. In fact, the modification of adipokines during the expansion of the adipose tissue represent a homeostatic compensation mechanism to preserve insulin sensitivity. However, vice versa, when produced in largequantities they exert the counterproductive adverse effect of increasing IR and liver steatosis, this is the case of leptin, as well as adiponectin and resistin, thus becoming the negative trigger of NAFLD [73].
Leptin, the first adipokine described [74], is produced by the adipose tissue in proportion to its mass and, beyond the neuroendocrine functions, is responsible of energetic homeostasis and regulation of metabolism [75]. Leptin and insulin communicate within the liver with significant metabolic overlap, resulting in activation of JAK-2, STAT-3, MAPK/ERK and PI3K/AKT/mTOR pathways [76]. At low levels it exerts a protective role on hepatic steatosis, due to its insulin-sensitizing effects, suppression of gluconeogenesis, de novo lipogenesis, as well as by stimulating FFAs beta-oxidation [77]. However, when produced in larger quantities, leptin acts as a pro-inflammatory and fibrogenetic factor acting on metalloproteinases [TIMP metallopeptidase inhibitor 1 (TIMP-1) and matrix metalloproteinase-1 (MMP-1)], stimulating TGF-β, and upregulating CD14 on liver Kupffer cells [78–81]. Furthermore, leptin increases hepatic fibrosis by reducing PPAR-γ expression in hepatic stellate cells [82]. Higher levels of circulating leptin were observed both in NAFL patients and even more so with NASH [83].
Adiponectin is the most widespread adipokine produced by the adipose tissue [84]. Once secreted, it is able to bind its two trans-membrane receptors, AdipoR1 and AdipoR2, mainly expressed at the level of skeletal muscle and liver [85]. Adiponectin also exerts anti-steatosic effects, increasing oxidation of FFAs and decreasing both gluconeogenesis and de novo lipogenesis. It also protects hepatocytes from apoptosis [86].
In addition to these metabolic effects, adiponectin also has anti-inflammatory and anti-fibrotic effects trough suppressing of pro-inflammatory cytokines (TNF-α and IL-6) and inducing anti-inflammatory cytokines (IL-10) [72], as well as reduction of both activation and proliferation of liver stellate cells by downregulating TGF-β, reducing both connective tissue expansion and promoting matrix degradation [87]. Unlike other adipokines, it decreases as the adipose tissue increases [71]. NAFLD patients have low circulating adiponectin levels. However, these levels paradoxically increasing when NASH progresses to cirrhosis [88, 89], possible due to decreased hepatic clearance or as compensatory effect against a massive production of pro-inflammatory cytokines occurring during liver cirrhosis.
Finally, resistin is a peptide produced during differentiation of adipocytes, most likely by macrophages that infiltrate the adipose tissue rather than by adipocytes themselves [90, 91]. Resistin appears to play a role in liver inflammation and fibrosis. It can trigger IR [90], mediated on the one hand by the activation of adenosine monophosphate activated protein kinase (AMPK), which acts on the expression of liver gluconeogenesis enzymes, and partially by the modulation of VLDL secretion and hepatic lipogenesis [92]. Furthermore, resistin acts as a true pro-inflammatory cytokine, stimulating both production and action of TNF-α, IL-1 and IL-6 [93, 94]. It is also capable to influencing hepatic fibrosis, through the activation of hepatic stellate cells, as well as the increase in the production of TGF-β and type I collagen through Kuppfer cells [87]. In vivo resistin, IR, steatosis and liver fibrosis seem to be closely associated [95], although the data are controversial [96].
So far, it seems clear that IR plays a key role in worsening both T2DM and NAFLD. However, other factors independent of IR have emerged that can influence these conditions. Indeed, genome-wide association studies have established genetic variants associated with NAFLD severity, but not related to IR.
Alteration of patatin-like phospholipase domain-containing protein 3 (PNPLA3) contributes to the difference in both liver fats content and susceptibility to NAFLD. The missense mutation Ile148Met into PNPLA3 (either named adiponutrin) modifies the progression of NAFLD [97]. In particular, the substitution of methionine with isoleucine at the residual 148 would limit the access of the substrate to the catalytic serine in position 47, thus trigger an altered hydrolysis of hepatic triglycerides and a consequent increase in the content of cellular triglycerides. However, it does not associate with IR [98].
It thus evident how PNPLA3 (I148M) polymorphism is an important determinant for the accumulation of hepatic fat predisposing to NAFLD [99]. In addition to PNPLA3, the TM62F protein, and especially the Glu167Lys (E167K) variant, also appears to influence the hepatic fats content, as their activity is mandatory for a normal secretion of VLDL, while its reduced functioning can contribute to the pathogenesis of NAFLD.
The TM6SF2 variant encoding p.Glu167Lys causally contributes to NAFLD [100]. In particular, carriers of the variant Glu167Lys (E167K) of TM62F protein seem to be associated with a distinct NAFLD subtype, characterized by a preserved insulin sensitivity regarding lipolysis, hepatic glucose production and lack of hypertriglyceridemia, although there is an increased content of hepatic fatty acids [101].
A study on mice assessed the association between NAFLD and IR and increased plasmatic apolipoprotein C3 (APOC3) concentration, reporting that an increased concentration predisposes mice to both NAFLD and hepatic IR [102]. These results underline the still unclear role of IR as a risk factor or marker of NAFLD.
A study on subjects with familial hypobetalipoproteinemia (FHBL) also reported that hepatic steatosis in this subpopulation is decoupled from IR, suggesting that the increase in intrahepatic lipids content could be a marker rather than a cause of metabolic dysfunction [103].
NASH is defined as presence of steatosis along with hepatocellular injury, which is characterized by ballooning degeneration and lobular inflammation. Liver steatosis, either with or without lobular inflammation is not sufficient for the diagnosis of NASH, so these patients should be considered to have NAFL.
In the past, “two-hits hypothesis” was the most accepted theory about the progression from NAFLD to NASH [104]. The “first hit” is excessive accumulation of lipids in the hepatocytes, known as liver steatosis (NAFL), which promotes lipotoxicity, thus making the liver more sensitive to potential damage. The “second hit”, comes from immune responses caused by the excessive accumulation of toxic intermediate fatty acids in the liver that triggers the progression of NASH.
In reality, the pathophysiological pattern is much more complex, a “multiple parallel hits hypothesis” [12] has been proposed, which underlines the close relationship between liver, adipose tissue, intestinal tract and skeletal muscle [105] in the development of NASH [66].
A role in the onset and progression of NAFL towards NASH is also played by the end products of advanced glycation (AGE) which are formed by the non-enzymatic reaction between reducing sugars and proteins or lipids. The overproduction of intracellular and extracellular AGEs can be induced by various conditions including hyperglycemia and T2DM [106]. AGE induces the production of its RAGE receptor. The AGE-RAGE axis contributes to the accumulation of fat in the liver leading to inflammation, fibrosis, IR, and oxidative stress [107]. Inhibition of AGE formation and mitigation of AGE-mediated effects can be a good therapeutic target for improve both NAFLD and diabetic complications.
During early stages of NAFLD, triglycerides accumulate in the hepatocytes. Diacylglycerol acyltransferase 2 (DGAT2) catalyzes the final stage of hepatic triglycerides synthesis. In fact, triglycerides stored in the liver are inherently inert and non-toxic. However, other bioactive intermediates, such as ceramides and diacylglycerols (DAGs), can induce lipotoxicity, thus causing potentially inflammation and liver fibrosis [108].
Therefore, in the liver, there will observe an excessive accumulation of toxic lipids (e.g., DAGs and ceramids) during NASH. Similar, this also occurs within liver macrophages, i.e. Kupfer cells. These macrophages will attract T-CD4+ and B lymphocytes, in response to lipopolysaccharide (LPS) stimulation, thus producing proinflammatory cytokines and chemokines. In other words, there is a shift of the Kupfer cells phenotype towards a proinflammatory M1 state [109]. Activated Kupffer cells not only secrets excessive amounts of proinflammatory cytokines (e.g., TNF-α and IL-6), but also promote the activation of hepatic stellate cells which, in turn, produce specialized collagen with tissue formation scarring.
NAFL progresses into NASH when the mechanisms that protect hepatocytes from fatty acids-mediated lipotoxicity are overwhelmed. In this case, dead hepatocytes trigger repair responses within the liver, establish the activation of hepatic stellate cells and myofibroblasts. The latter begin to create an excessive matrix and produce growth factors favoring collagen deposition and, therefore, fibrosis. Furthermore, the cytokines produced in this context attract numerous other inflammatory cells to the liver, thus exacerbating the fibrosis processes [110]. It is thus evident that lipotoxicity triggers a chronic inflammatory process promoting, over time, the evolution from NAFL to NASH.
In addition to intrahepatic lipotoxicity, during T2DM, NASH can be exacerbated by a series of vicious circles triggered by obesity-induced IR in adipose tissue, muscle and liver, and is a crucial risk factor for the development of T2DM.
At a molecular level, in NASH patients IR is promoted by the transition of macrophages from the M2 state (kept by STAT6 and PPARs) to an activated M1 state, driven by NF-kB, AP1 and other transcription factors that act in the innate immune system. In fact, during IR, the lipolysis induced by the white adipose tissue favors the release of FFAs, thus resulting in the enrolment and activation of macrophages in the adipose tissue. M1 activates macrophages which, in turn, secrete huge amounts of cytokines, including the chemoattractanting protein monocyte-1, TNF-α and IL-1-beta. These cytokines not only induce the vicious circle of local IR, but also enhance systemic inflammation, thus leading to a higher infiltration of proinflammatory cytokines in the liver [111].
Mitochondrial dysfunction is an additional element that favor the progression of NAFL to NASH. In fact, as aforementioned, intrahepatic FFAs can undergo a beta-oxidation process in the mitochondria. The entry of FFAs in mitochondria, a crucial step in initiating beta-oxidation, is controlled by carnitine O-palmitoyl transferase 1 (CPT1). CPT1, which is inhibited by insulin, malonyl-CoA and acyl-CoA, activates PPAR-α, thus stimulating beta-oxidation of FFAs [112]. Reduced mitochondrial function could exacerbate IR. An association between NAFLD and a decreased muscle mitochondrial activity has been reported [113]. Therefore, the muscle mitochondrial adaptation could represent one of the mechanisms capable of modulating lipids deposition in the liver during T2DM development [114]. Impaired hepatic energy metabolism and IR could prevent NASH development [115]. Therefore, NAFLD is not only associated with IR, although it could rather be preemptive of T2DM and of its cardiovascular complications. In T2DM patients, IR is related to impaired hepatic energy metabolism, partially justified by lipids deposits [116].
NASH patients are characterized by a higher mitochondrial mass. During NASH, increased hepatic oxidative stress and oxidative DNA damage are accompanied by a reduced antioxidant defense ability and increased inflammatory response, suggesting “hepatic mitochondrial flexibility” during the early phases of IR, related to obesity and subsequently causing NASH [117].
Mitochondrial dysfunction could play a crucial role in NASH pathogenesis. Indeed, NASH patients present hepatic mitochondrial abnormalities, most likely related to liver fibrosis [118] These include a higher loss of protons by the electrons transport chain, due to an increased expression of mitochondrial uncoupling protein 2 (MUCP-2), a depletion of mitochondrial DNA and an abnormal oxide-reductive mitochondrial homeostasis, with the subsequent production of ROS [119, 120].
Finally, excessive lipid overload will undermine the antioxidant ability and accelerate the oxidative stress, resulting in mitochondrial protons loss, NASH development and IR exacerbation. Again, the link between liver disease and T2DM appears to be reciprocal, as mitochondrial dysfunction exacerbates both NASH and T2DM. Indeed, ROS produced by mitochondria provide liver inflammation and beta-cells damage, thus establishing the progression of both pathological conditions.
Lifestyle plays a important role in the qualitative and quantitative change of the intestinal microbiota [121], which contributes, both, directly (through chemical mediators produced by bacteria) and indirectly (through interference with the biochemical metabolic pathways), to the development of T2DM and NAFLD [122–125].
In fact, the increase in intestinal ethanol production, due to microbial dysbiosis, leads to both liver toxicity and an increase in intestinal permeability, secondary to the loss of the tight junction. Thus, gut-derived pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide, reach the liver through blood flow. As a result, proinflammatory pathways activate specific binding tool such as receptors and lead to liver inflammation and fibrosis. Furthermore, the gut microbiota increases the metabolism of choline, thereby leading to an increase triglycerides levels in the liver, due to the lack of VLDL excretion. Furthermore, dysbiosis has been associated with decreased secretion of fasting-induced adipocyte factors [FIAF and angiopoietin-like 4 (ANGPTL4)], which results in the inhibition of endothelial lipoprotein lipase and consequently the lack of hydrolyzation in the liver of triglycerides from VLDL particles. Gluconeogenesis and lipogenesis are upregulated due to an increased number of substrates, particularly short-chain fatty acids, thus promoting hepatic FFAs accumulation by inhibiting AMPK [126].
We have reported above that NASH is associated with the accumulation of lipotoxic mediators in the liver, such as DAGs and ceramides. These also appear to play a crucial role in the onset of hepatic IR. Both DAGs and ceramides stimulate the translation of protein kinase C (PKC) towards the cell membrane, hence its activation (PKC-ζ for ceramides; PKC-ε for DAG) [127, 128]. Activated PKCs will phosphorylate insulin receptor substrate-2 (IRS2) on serine and threonine residues, thereby preventing thyroxine phosphorylation. This will inhibit insulin signaling resulting in an IR status [129].
NF-κB is among the major transcriptional regulators of inflammation. Deletion of NF-κB activator, IKK-β, either in the myeloid cells or in the hepatocytes, prevented mice on a lipid-rich diet from developing IR [130]. Conversely, constitutive overexpression of IKK-β in hepatocytes produced hyperglycemia and a deeper hepatic IR [59].
An up-regulation of hepatic NF-kB p65 expression has been reported during NASH [110]. Once activated, NF-κB in turn transactivates proinflammatory cytokines, including TNF-α and IL-6, reported to induce IR [131].
Toxic peroxidation of lipid appears to produce necroinflammation. NASH is also strongly associated with an increased peroxidation of hepatic microsomal lipids. Indeed, an experimental model of steatohepatitis, induced by a methionine- and choline-deficient diet (MCD), was associated with an increase in the expression and activity of CYP2E1, a microsomal enzyme that triggers oxidative stress [132, 133]. Hepatic expression of CYP2E1 and its activity have also been found to be up-regulated in NASH individuals [134].
Once activated, CYP2E1 contributes to the pathogenesis of NASH by producing ROS which, in turn, undermine the insulin signaling pathway by inhibiting IRS-1 and IRS-2, then downstream enzymes, including PKB, glycogen synthase 3 and FOXO1A [135].
The close association between NAFLD and T2DM is evident and surprisingly it is their bidirectionality. Hence, NAFLD contributes onset to the development of IR in T2DM. Some studies [2, 136, 137] have reported an association between the presence of NAFLD and an increased risk of developing T2DM (2.4–3.5 times higher). Therefore, a diagnosis of hepatic steatosis could be a risk factor for the onset of T2DM, assuming that NAFLD may anticipate the diagnosis of T2DM [136, 137]. Patients with NAFLD exhibit impaired glucose metabolism compared to non-NAFLD subjects, which represents an independent risk factor for development of T2DM [138]. Patients with NASH, diagnosed by biopsy, were shown to have a 3-folds higher risk of developing T2DM or impaired glucose tolerance [132] and progression to liver fibrosis and cirrhosis was much more evident in patients with IR [139].
Furthermore, transplanted fatty liver have been shown to be a risk factor for the development of post-transplant T2DM, although it is not known how many of these donors had pre-transplant NASH [140].
A retrospective cohort study of approximately 400 NAFLD patients diagnosed with liver biopsy, without diabetes at baseline and followed for a mean time of 18.4 years, reported a higher incidence of T2DM in patients with severe fibrosis (stages 3–4) compared to those with mild fibrosis (stages 1–2) [141].
Mechanisms underlying NAFLD-induced T2DM include several mediators, such as lipotoxicity, inflammation and oxidative stress.
In addition to T2DM, prediabetes, an intermediate stage between normal glucose tolerance (NGT) and T2DM, is also associated with NAFLD. Prediabetes is associated with portal inflammation and liver fibrosis in NAFLD patients. Yilmaz et al. [142], showed that in patients with NAFLD, portal inflammation and fibrosis were significantly more severe in pre-diabetic subjects than those with NGT, while diabetic patients had much more severe portal inflammation and fibrosis. Those with prediabetes share the same pathophysiological mechanisms as T2DM, i.e. IR as the main dysfunction. It is also been reported that almost 10% of NAFLD and pre-diabetes patients will develop T2DM [143, 144].
It seems clear that IR, by itself, cannot explain the complex bidirectional relationship between NAFLD and T2DM. First, numerous rodent models have developed liver steatosis without IR, albeit rather due to the inhibition of VLDL efflux by MCD diet [145], an increased in fatty acids storage by DGAT overexpression [146] and inhibition of triglycerides hydrolysis by CGI-58 knockdown (a triglycerides lipase activator) [147].
Furthermore, studies on human genetic polymorphisms have reported that several genes associated with steatosis are not related to IR. Among these, we find the adipose triglycerides lipase, CGI-58, PNPLA3, etc. [148]. For example, a human variant of the PNPLA3 gene increases hepatic lipids content without inducing IR. Interestingly, significant differences were found in hepatic lipid composition compared to a group with similar but insulin resistant steatosis and NASH. In detail, in the IR group the lipids were abundant in ceramides, saturated and mono-unsaturated triglycerides, while the lipids in the PNPLA3 group were rich in poly-unsaturated fats. These results underline that the type of lipids accumulated in the liver is much more important for altering insulin signaling than for lipid accumulation itself [128]. This difference in lipid profile could explain why only some populations with NAFLD are associated with a higher risk of T2DM.
As evidence that not all NAFLDs are associated with IR, 263 NAFLD patients were recently studied by liver biopsy, of which 53 (20.2%) had no evidence of IR. The latter had a lower BMI and fewer metabolic risk factors (MS, diabetes mellitus, hypertension, hyperlipidemia and waist circumference) and in any case hepatic histological damage [149]. However, liver histology showed that in subjects without IR there was less severe liver damage with a lower prevalence of NASH and fibrosis than in subjects with IR. Obesity has been shown to be an independent predictor of fibrosis in NAFLD patients without IR [149].
In conclusion, the relationship between NAFLD and T2DM is so tight and complex that it is difficult to distinguish which is the consequence and which the cause. T2DM provides with an optimal metabolic condition for NAFLD progression, while NAFLD may in turn promote the risk of T2DM. Both these pathological conditions share numerous common factors, which are more likely to be involved in the progression of the disease, including abundance of fatty acids, pro-inflammatory cytokines, oxidative stress, etc. A comprehensive view of the underlying mechanism of NAFLD and T2DM could shed light on future therapeutic choices for these diseases.
In conclusion, the relationship between NAFLD and T2DM is not only epidemiological, as evident especially in the context of MS, but above all pathophysiological. The study of these mechanisms will probably help to deepen the knowledge of the association between the two pathologies and, above all, will allow the definition of pharmacological targets for the treatment of NAFLD, which are still fundamentally absent today.
AGE: advanced glycation end product
AMPK: adenosine monophosphate activated protein kinase
APOC3: apolipoprotein C3
DAGs: diacylglycerols
FFAs: free fatty acids
FGF: fibroblast growth factor
FXR: farnesoid X receptor
G6P: glucose-6-phosphatase
HCC: hepatocellular carcinoma
IKK-β: I-kappa-B kinase β
IL-6: inteleukine-6
IR: insulin resistance
IRS: insulin receptor substrate
MAFLD: metabolic-dysfunction-associated fatty liver disease
MCD: methionine- and choline-deficient diet
MS: metabolic syndrome
NAFL: nonalcoholic fatty liver
NAFLD: nonalcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells
PKB: protein kinase B
PKC: protein kinase C
PNPLA3: patatin-like phospholipase domain-containing protein 3
PPAR: peroxisome proliferator-activated receptor
RANKL: receptor activator of nuclear factor kappa-Β ligand
ROS: reactive oxygen species
SREBP-1c: sterol regulatory element-binding transcription factor 1c
T2DM: type 2 diabetes mellitus
TNF-α: tumor necrosis factor alpha
VLDL: very low-density lipoproteins
LEA, FCS, and RN contributed conception and design of the review, CA, AC, and PCP conducted selection of literature and wrote the first draft of the manuscript, and all the authors revised the manuscript and approved the final version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Noel C. Salvoza ... Natalia Rosso
Amedeo Lonardo, Stefano Ballestri
Marvin Ryou ... Gyorgy Baffy
Michael Doulberis ... Stergios A. Polyzos
Marica Meroni ... Paola Dongiovanni
Ivana Mikolasevic ... Sandra Milic
Valerio Rosato ... Marcello Persico
Rosa Lombardi ... Anna Ludovica Fracanzani
Angelo Di Vincenzo ... Marco Rossato
Angelo Colucci ... Claudia Mandato
