 Review
Review

Affiliation:
1First Laboratory of Pharmacology, School of Medicine, Aristotle University of Thessaloniki, 54124 Thessaloniki, Macedonia, Greece
2Division of Gastroenterology and Hepatology, Medical University Department, Kantonsspital Aarau, 5001 Aarau, Switzerland
†These authors contributed equally to the work, thus sharing first co-authorship
ORCID: http://orcid.org/0000-0002-0396-5081
Affiliation:
1First Laboratory of Pharmacology, School of Medicine, Aristotle University of Thessaloniki, 54124 Thessaloniki, Macedonia, Greece
†These authors contributed equally to the work, thus sharing first co-authorship
Affiliation:
1First Laboratory of Pharmacology, School of Medicine, Aristotle University of Thessaloniki, 54124 Thessaloniki, Macedonia, Greece
3Department of Gastroenterology, University Hospital of Larissa, 41334 Larissa, Thessaly, Greece
†These authors contributed equally to the work, thus sharing first co-authorship
ORCID: https://orcid.org/0000-0002-3563-4973
Affiliation:
4Second Medical Clinic, School of Medicine, Aristotle University of Thessaloniki, Ippokration Hospital, 54642 Thessaloniki, Macedonia, Greece
ORCID: http://orcid.org/0000-0001-6459-5136
Affiliation:
1First Laboratory of Pharmacology, School of Medicine, Aristotle University of Thessaloniki, 54124 Thessaloniki, Macedonia, Greece
Email: spolyzos@auth.gr
ORCID: http://orcid.org/0000-0001-9232-4042
Explor Med. 2020;1:170–183 DOI: https://doi.org/10.37349/emed.2020.00012
Received: May 16, 2020 Accepted: June 10, 2020 Published: August 31, 2020
Academic Editor: Lindsay A. Farrer, Boston University School of Medicine, USA
The article belongs to the special issue Exploring NAFLD/NASH
Pooled prevalence of nonalcoholic fatty liver disease (NAFLD) globally is about 25%. Nonalcoholic steatohepatitis (NASH) with advanced fibrosis has been linked with substantial morbidity and mortality, without having to-date any licensed treatment. C-C chemokine receptor (CCR) antagonists have been investigated as candidates for the treatment of NASH. Inhibition of CCR2 is expected to mitigate hepatic inflammation, through reducing the activation of Kupffer cells, as well as the infiltration of monocytes and macrophages into the liver. Inhibition of CCR5 is expected to mitigate hepatic fibrogenesis, through impairing the activation of hepatic stellate cells, as well as to mitigate hepatic inflammation, through impairing the activation of Kupffer cells and macrophages. Cenicriviroc (CVC) is the first in class, dual inhibitor of CCR2 and CCR5. After exhibiting favorable results in animal models, CVC was shown to be beneficial in NASH patients with more severe fibrosis at a phase 2b trial (CENTAUR) and is currently at a phase 3 clinical trial (AURORA). Apart from CVC, other CCR5 mono-antagonists, such as maraviroc, are under evaluation in clinical trials with human immunodeficiency virus patients with NAFLD. The aim of this review was to summarize existing evidence on CVC and other CCR antagonists in NASH patients, primarily focusing on their clinical efficacy and safety.
Pooled prevalence of nonalcoholic fatty liver disease (NAFLD) globally is approximately 25% [1]; it is currently the most common chronic liver disorder in the US, affecting around one out of four US people [2]. NAFLD refers to a wide spectrum of interconnected phenotypes, starting from simple steatosis (nonalcoholic fatty liver; NAFL) to nonalcoholic steatohepatitis (NASH) characterized by inflammation with hepatocellular injury, which may progress to liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC) [3]. NAFLD is pathophysiologically linked to insulin resistance (IR), which plays a critical role in its multifactorial pathogenesis, thus NAFLD dynamically interacting with components of the IR syndrome or metabolic syndrome (MetS), including obesity and type 2 diabetes mellitus (T2DM) [4]. Therefore, the prevalence of NAFLD rises in parallel with the epidemics of obesity and T2DM [5]. NASH, especially with advanced fibrosis, has been linked with substantial morbidity and mortality [6]. Nonetheless, there is to-date no licensed medication for NASH and/or fibrosis, which renders NASH an appealing target for the researchers, but also for the pharmaceutical industry.
C-C chemokine receptor (CCR) antagonists have been investigated as candidates for the treatment of NASH [7, 8]. Cenicriviroc (CVC), a dual CCR type 2 and type 5 antagonist, has been gaining increasing interest, owing to its anti-inflammatory and anti-fibrotic properties shown in animal models [9, 10]. In clinical terms, CVC showed favorable results in NASH patients with more severe fibrosis at a phase 2b trial [11, 12] and is currently at a phase 3 clinical trial (Table 1) [13]. Apart from CVC, other CCR antagonists are under evaluation for the treatment of NASH, including maraviroc (MVC).
Registered ongoing clinical trials on CVC in patients with NAFLD
| ID, registration date (DD/MM/YYYY) | Title | Design | Arms | Patients (n) | Duration | Primary outcome(s) |
|---|---|---|---|---|---|---|
| NCT03517540 09/04/2018 OR EUCTR2017-004208 24 21/06/2018 OR CTRI/2019/01/017014 09/01/2019 | A Randomized, Double-blind, Multicenter Study to Assess the Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and CVC in Adult Patients With NASH and Liver Fibrosis (TANDEM) | Multicenter, randomized, double-blind, phase 2 | Tropifexor 140 μg vs. CVC 150 mg vs. Tropifexor 140 μg + CVC 150 mg vs. Tropifexor 90 μg + CVC 150 mg | 200 | 12 months | Number of participants with adverse events |
| NCT03376841 2/8/2017 | An Open-Label Study to Evaluate the Effect of Severe Hepatic Impairment on the Pharmacokinetics of CVC and Its Metabolites Following Single Dose Administration | Single-center, non-randomized, open label, phase 1 | CVC (single dose; NA mg) | 16 | 6 days | 1) AUC from time 0 to time t (AUC0-t) 2) AUC from time 0 to infinity (AUC0-∞) 3) Maximum plasma drug concentration (Cmax) |
| NCT03059446 3/2/2017 | Open-label Rollover Study of CVC for the Treatment of Liver Fibrosis in Adult Subjects With NASH | Multicenter, single group, open label, phase 2 | CVC 150 mg | 560 | 24 months | Number of participants with adverse events |
| NCT03028740 13/1/2017 OR EUCTR2016-004566-26 22/02/2017 OR PER-041-17 02/04/2018 | A Phase 3 Study to Evaluate the Efficacy and Safety of CVC for the Treatment of Liver Fibrosis in Adult Subjects With NASH (AURORA) | Multicenter, randomized, double-blind, phase 3 | CVC 150 mg vs. placebo | 2000 | 12 months (for endpoint #1); 60 months (for endpoint #2) | 1) Improvement in fibrosis ≥1 stage without worsening of NASH 2) Composite outcome of all-cause mortality, cirrhosis, HCC, liver thransplantation and other liver-related events |
| NCT02342067 12/1/2015 | A Phase 1, Multiple-Dose, Open-Label, Randomized, Crossover Study in Healthy Subjects to Assess the Effect of PGZ on the PK of CVC and the Effect of CVC on the PK of PGZ | Single-center, randomized, open-label, phase 1 | CVC 150 mg, then PGZ 45 mg, then CVC + PGZ vs. PGZ 45 mg, then CVC 150 mg, then CVC + PGZ | 20 | 40 days | PK Assessment of CVC and PGZ, as measured by Cmax, Cmin and AUC |
| NCT02330549 22/12/2014 | Effect of CCR2 and CCR5 Antagonism by CVC on Peripheral and Adipose Tissue Insulin Sensitivity in Adult Obese Subjects With Prediabetes or Type 2 Diabetes Mellitus and Suspected NAFLD (ORION) | Multicenter, randomized, double-blind, phase 2 | CVC 150 mg vs. placebo | 45 | 6 months | Changes in insulin sensitivity (peripheral and adipose tissue) |
References are presented in date of registration order (newer to older); AUC: area under the curve; Cmax: maximum plasma drug concentration; Cmin: minimum plasma drug concentration; NA: not available; PGZ: pioglitazone; PK: pharmacokinetics
Aim of this review was to summarize existing evidence on CVC and other CCR antagonists in NASH patients, primarily focusing on their clinical efficacy and safety.
Chemokines, also known as chemotactic cytokines, are components of the innate immune system [14] and encompass a group of small (8–12 kDa) heparin-binding cytokines with pleiotropic roles [15–17]. Categorization of chemokines can be based on function according to the role they play principally in the context of inflammation and homeostasis, or on structure and the position of cysteine residues. Inflammatory chemokines are induced by a wide range of cell types in response to pro-inflammatory cytokines, tissue damage, or contact with pathogens. Such chemokines are released early on epithelial, stromal, and immune cells to recruit the first wave of innate immune effectors, including neutrophils, monocytes, and natural killer (NK) cells [17]. Likewise, chemokines promote the migration of activated dendritic cells (DCs) into secondary lymphoid tissues to activate adaptive immunity and the recruitment of T-effector cells to the location of damage. Regulatory T-cells are recruited in a comparable manner and the balance between T-effector and regulatory cell recruitment will determine the outcome of the immune reaction. Homeostatic chemokines are expressed in lymphoid and non-lymphoid tissues, where they mediate the physiological trafficking and positioning of cells involved in antigen sampling and immune surveillance. Functional distinctions are not absolute and chemokines initially thought to be homeostatic are usually induced and up-regulated at locations of chronic inflammation [17], including obesity and NAFLD [3].
Depending on the positions of the first two cysteine residues in their molecules, chemokines are classified into four motifs: CC-chemokines, CXC-chemokines, CX3C-chemokines, and C-chemokines [16]. In CC motif, the first two cysteine residues are adjacent and near the N-terminal of the peptide. In CXC and CX3C motives, the first two cysteine residues are separated by one (X) or three (X3), respectively, dissimilar amino acids, lying near the N-terminal of the peptide. In C-chemokines the two cysteine residues are remote, with only one cysteine near the N-terminal [16]. Chemokines orchestrate migration and trafficking of diverse cell populations, predominantly of the mentioned leucocytes, during immunological responses [15, 18]. Thus, migratory properties, including chemokinesis, haptotaxis, and haptokinesis, have been attributed to chemokines, as well as cell arrest or adhesion [18]. Through their actions, chemokines have been implicated in angiogenesis, hematopoiesis, lymphoid tissue maturation, tissue remodeling, and fibrosis [15, 16].
Chemokines act through cell surface G-protein-coupled (heptahelical) chemokine receptors, named CCR [18], consisting of an extracellular N-terminal, three extracellular and three intracellular loops and a C-terminal [16]. Two of the most substantial CCR with pharmacological interest and potential clinical implication are CCR2 and CCR5; they bind the CC-chemokine ligand (CCL)2 [also known as monocyte chemoattractant protein (MCP)-1] and CCL5 [also known as regulated on activation, normal T expressed and secreted (RANTES)], respectively [12, 19]. CCR2 and CCR5 have a structural affinity, sharing sequence homology to a significant extent (73%). CCR2 is expressed on monocytes/macrophages and hepatic stellate cells (HSCs) [20]. CCR2 may be expressed on the Kupffer cells of some animal models, whereas its expression on human Kupffer cells remains conflicting [21, 22]. The activation of CCR2 has been implicated in inflammatory processes [15], especially the initiation of hepatic inflammation [20], through the induction of monocyte/macrophage recruitment and a macrophage-dependent inflammatory response [9]. CCR5 is more widely expressed, i.e. on immune cells (macrophages, T-cells, NK cells, polynuclear granulocytes, DCs), HSCs, epithelial cells, endothelial cells, vascular smooth muscle cells and neural cells (neurons, astrocytes, microglia) [15, 23, 24].
Chemokines and their receptors have been implicated in the pathogenesis of IR and NAFLD [25]. Relative studies have reported that preadipocytes and adipocytes secret CCL2 and induce the infiltration of bone marrow-derived macrophages into adipose tissue of obese, thus leading to IR [26, 27]. Accumulation of immune cells in adipose tissue, specifically macrophages, plays an essential role in obesity-related inflammation and contributes to the development of IR and NAFLD [3, 28]. Likewise, there is evidence that chemokines and chemokine receptors contribute to other disorders related to IR and MetS, including T2DM, atherosclerosis, and cerebro-cardiovascular diseases [25, 29].
Mice overexpressing CCR2 are characterized by higher infiltration of macrophages into the adipose tissue, elevated IR, and hepatic steatosis [26, 30]. On the contrary, deletion of the CCR2 gene decreases the inflammation of adipose tissue, thus mitigating diet-induced IR [26, 27]. In mice with diet-induced obesity, propagermanium, a CCR2 inhibitor, decreases the accumulation of macrophages at the proinflammatory state (M1) into the adipose tissue, thus improving glucose tolerance, IR and hepatic steatosis [9]. Likewise, upregulation of the expression of CCL2 in the liver and monocytes has been reported in obesity [14] and NASH [23]. Regarding CCR5 pathway, its inhibition results in the attenuation of hepatic inflammation and fibrosis in animal models of obesity and NAFLD; importantly, mice that were knockout for CCL5 gene exhibited reduced HSC activation and immune cell infiltration together with decreased hepatic fibrosis [31]. In line, patients with chronic liver diseases have higher hepatic CCR5 expression [14, 23, 24].
In the liver, macrophages, conventionally signified Kupffer cells, together with liver sinusoidal endothelial cells, HSCs, and resident immune cells (particularly, unconventional T cells, NK cells, and liver DCs), comprise the non-parenchymal hepatic cells, which are producers of and responders to chemokines. Activated Kupffer cells secrete interleukin (IL)-1b and CXC chemokines, including CXCL1, CXCL2, and CXCL8 (IL-8). CXCL1, CXCL2, and CXCL8 attract neutrophils, predominantly through the chemokine receptors CXCR1 and CXCR2, which release reactive oxygen species and proteases, thereby evoking liver cells necrosis [32]. Chemokines have been proposed to control the migration and activity of liver resident cells (Kupffer cells, HSCs, endothelial cells) and immune cells infiltrating the liver [16]. Macrophages (Kupffer cells and those infiltrating the liver) and HSCs are regarded as key cells in the hepatic inflammation and fibrogenesis, respectively [3].
Infectious or noninfectious injury of liver cells leads to the induction of variable danger signals, called pathogen or danger connected molecular patterns, which bind to Toll-like receptor (TLR)4 and other TLRs on Kupffer cells, thus inducing the production of proinflammatory cytokines (e.g., tumor necrosis factor), chemokines and reactive oxygen and nitrogen species [33].
Mechanistically, liver injuries induce the activation of Kupffer cells and the chemo-attraction of circulating monocytes to the liver, where they are also activated to macrophages at M1 (or proinflammatory) state [3]. In NAFLD, Kupffer cells are exposed to a diversity of signals that change their phenotype toward an M1 state [34]. Likewise, cytokines released by M1-polarized Kupffer cells may stimulate triglyceride synthesis and inhibit fatty acid oxidation [35, 36]. Triglyceride accumulation is indicated by evidence showing that pharmacological or genetic nullification of CCL2 or CCR2 decreases macrophage infiltration in mice with experimental steatohepatitis and inhibits steatosis [37, 38].
Macrophages are crucial players in the pathogenesis of NASH, as indicated by the resistance to steatosis, inflammatory cell recruitment, hepatic injury, and fibrosis in mice, when Kupffer cells are depleted [39, 40]. Macrophages, which express CCR2 on their surface, infiltrate periportal areas of hepatic parenchyma upon injury and exert inflammatory properties, thus promoting NASH [37]. Upon their activation, Kupffer cells and infiltrating macrophages release cytokines and chemokines, which, in turn, attract additional monocytes from the circulation and activate more Kupffer cells, thus creating a vicious cycle of a low-grade, but chronic smoldering inflammation in NASH [5]. Furthermore, Kupffer cells interplay with HSCs, leading to their activation and differentiation to myofibroblasts [3]. The production of platelet derived growth factor (PDGF) and transforming growth factor (TGF)-β by Kupffer cells and infiltrating macrophages plays a key role in the activation of HSCs [15, 41]. Upon their activation, HSCs produce matrix proteins, including actin and collagen types I and III, contributing to the initiation and progression of hepatic fibrosis [10], which is the main histological prognostic factor of advanced disease in NAFLD [42]. The above considering, the inhibition of CCR2 and/or CCR5 are regarded as pharmaceutical targets against NASH (Figure 1) [8]. Inhibition of CCR2 is expected mainly to reduce the activation of Kupffer cells, as well as the infiltration of monocytes and macrophages into the liver, thus mitigating hepatic inflammation [43]. Inhibition of CCR5 is also expected to impair the activation and proliferation of HSCs, thus attenuating hepatic fibrogenesis, but also to impair the activation of Kupffer cells and macrophages, thus mitigating the hepatic inflammation (Figure 1) [20].
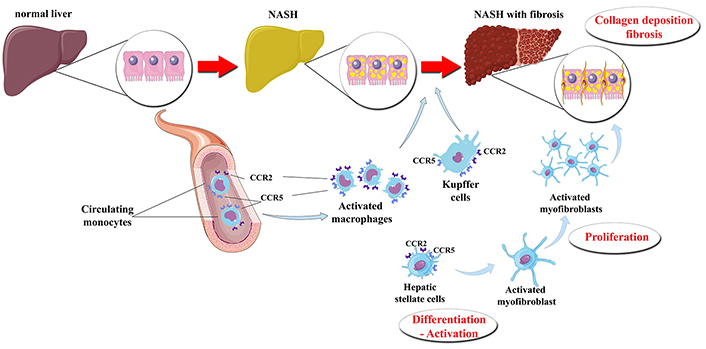
The presumptive effect of CVC on the liver of patients with NASH. Under certain pathogenetic factors (“hits”), a normal liver may subsequently progress to steatosis, inflammation and fibrosis. The activation of CCR2 and CCR5, which are possibly located on the Kupffer cells*, macrophages infiltrating the liver and HSCs, may result in their transformation and activation. Thus, hepatic inflammation and fibrosis are initiated and propagated. CVC is a dual CCR2 and CCR5 antagonist that seems to attenuate the activation of Kupffer cells, infiltrating macrophages, and HSCs, thus possibly mitigating hepatic inflammation and fibrosis. * CCR2 may be expressed in Kupffer cells of animal models, whereas CCR2 may not be expressed or may be not functional in human Kupffer cells
CVC mesylate (also known as TBR-652, TAK-652) is the first in class, oral, potent dual inhibitor of CCR2 and CCR5 [8]. Hepatic anti-inflammatory and anti-fibrotic properties have been attributed to CVC in preclinical studies [10, 21]. More specifically, CVC lessened the recruitment of monocytes and macrophages in animal models of NASH and fibrosis, thus reducing NAFLD activity score (NAS), collagen type I, and collagen deposition [10]. Other authors also reported that CVC treatment decreased the recruitment of monocyte-derived macrophages, IR, hepatic steatosis, NASH activity, and fibrosis in experimental models on NASH and fibrosis [21]. Importantly, the same authors reported that Kupffer cells may be implicated in hepatic lipid metabolism and the initiation of hepatic inflammation, whereas monocyte-derived macrophages that infiltrate the liver control growth factors and cytokines were associated with the progression of fibrosis [21]. Furthermore, other groups reported that CVC decreased the hepatic infiltration of macrophages after four weeks of treatment in a dietary mouse model of NASH [44]. A more prolonged treatment (14 weeks) did not further affect the intrahepatic macrophage infiltration, but was associated with less hepatic fibrosis [44]. This sequence seems to be rational, implying that targeting fibrosis may require a more prolonged treatment than targeting inflammation.
By targeting both hepatic inflammation and fibrosis, CVC was regarded as a promising candidate for clinical trials in NASH patients. CVC was first investigated in clinical trials of patients with human immunodeficiency virus (HIV), a disease characterized by acquired lipodystrophy with NAFLD and higher rates of NASH than those observed in the general population [45]. In HIV patients, CVC was initially displayed to improve aspartate aminotransferase-to-platelet ratio index (APRI) and fibrosis-4 index (FIB-4), two noninvasive indices of hepatic fibrosis [46].
In terms of pharmacodynamics, CVC in vitro occupies > 95% of CCR2 receptors (at 6 nM) on human monocytes and > 90% of CCR5 of human CD4+ T cells (at 3.1 nM); contrariwise, CVC does not inhibit CCR1 [23]. In a phase 1 clinical trial (NCT02120547), CVC (150 mg/d for 14 days) was reported to be safe and well tolerated [47]. In terms of pharmacokinetics, the mean half-life of CVC in healthy individuals was about 22 h. The half-life was higher in patients with mild (about 30 h) or moderate hepatic impairment (38 h) [47]. This half-life probably favors the administration of CVC once daily. CVC circulates bound to plasma proteins at > 98%, it is metabolized in the liver and is mainly excreted via the bile. CVC is substrate of the cytochromes P450 (CYP)2C8 and CYP3A4, acting as a weak inhibitor of the latter [47]. Another phase 1 clinical trial of CVC in patients with severe hepatic impairment (NCT03376841; Table 1) has been reportedly completed, but the results have not been announced yet.
Α phase 2a clinical trial, entitled ORION (“Effects of Cenicriviroc on Insulin Sensitivity in Subjects With Prediabetes or T2DM and Suspected NAFLD”; NCT02330549; Table 1) has been completed, the results have been announced in clinicaltrials.gov, but an article has not been published yet. In ORION, 45 patients were recruited and were randomized in CVC (150 mg/d) or placebo for six months. The primary outcome of ORION was the change in IR, measured by the Matsuda index and Adipose Tissue IR. CVC did not improve IR as compared with placebo in ORION. Furthermore, CVC seemed to be safe and well tolerated, since adverse events were mild to moderate in severity and balanced between the CVC and placebo group (https://clinicaltrials.gov/ct2/show/results/NCT02330549).
A phase 2b clinical trial, entitled CENTAUR (“Efficacy and Safety Study of Cenicriviroc for the Treatment of NASH in Adult Subjects With Liver Fibrosis”; NCT02217475), has been completed and published [11, 12]. CENTAUR was a 2-year, multicenter study, focusing on the safety and efficacy of CVC in patients with NASH (NAS ≥ 4) and liver fibrosis, but not cirrhosis (F1-F3). The patients (n = 289) were randomized to receive CVC 150 mg or placebo once daily. At year 1, half of the patients initially on placebo switched to CVC for one year. Repeat liver biopsy was performed at the end of year 1 and 2. The primary outcome was the improvement in NAS ≥ 2 points (consisting of ≥ 1-point reduction in either lobular inflammation or hepatocellular ballooning) without worsening of fibrosis [11, 12]. CVC did not meet the primary endpoint at the 1-year interim analysis [11]. However, the endpoint was met in a subgroup of patients with more severe NASH (defined as NAS ≥ 5) [11]. It is of note that the improvement in fibrosis ≥ 1 stage (secondary endpoint) was observed in more patients on cenicriviroc than on placebo (20% vs. 10%, respectively). Furthermore, complete resolution of fibrosis was observed in more patients on CVC than placebo (7.0% vs. 3.5%, respectively). Hepatocellular ballooning, being the hallmark of hepatic inflammation in NASH, was ameliorated only in a subgroup of patients with severe hepatocellular ballooning at baseline (grade 2). Liver function tests and hepatic steatosis were not improved by CVC [11].
At the beginning of year 2, 242 (84%) patients continued the study [12]. Again, the primary endpoint was not met at the end of year 2. Furthermore, the overall rates of patients on CVC and placebo that achieved improvement in fibrosis ≥ 1 stage without worsening of NASH (secondary endpoint) were similar (14% vs. 13%, respectively). Moreover, treatment with CVC for a second year did not provide additional antifibrotic benefit [12]. However, when the patients with F1 fibrosis stage were excluded from the analysis, improvement in fibrosis ≥ 1 stage without worsening of NASH was observed in more patients on CVC than placebo (11% vs. 3%, respectively). Notably, higher rates of patients on CVC than placebo preserved the improvement in fibrosis achieved at year 1 at the end of year 2 (60% vs. 30%, respectively) [12]. CVC had no effect on weight, liver function tests, and metabolic parameters related to NAFLD, including IR. Enhanced liver fibrosis score (ELF), APRI and FIB-4, selected as noninvasive indices of fibrosis, reduced in responders to treatment compared to non-responders, as expected [12].
Safety and tolerability of CVC were similar to placebo in CENTAUR study [11, 12]. Overall, CVC has been well tolerated and no major safety issues were noticed. No deaths were reported. All serious adverse events (11% vs. 15% in patients receiving CVC or placebo, respectively, for two years) except for one (grade 2 arrhythmia; the patient remained on blinded treatment) were reportedly not associated with CVC. Ten patients on CVC (7.0%) and twelve patients on placebo (8.3%) discontinued the study at year 1 due to side effects [11]. The most frequently reported adverse events, e.g., fatigue, diarrhea, nausea, nasopharyngitis, headache, abdominal pain, arthralgia, were mild or moderate in severity. Notably, alanine aminotransferase concentration similarly increased in some patients in CVC and placebo group (year 1: 11.8% vs. 11.8%; year 2: 4.4% vs. 3.3%, respectively) [12].
A phase 3 clinical trial, entitled AURORA (“Cenicriviroc for the treatment of liver fibrosis in adults with NASH”; NCT03028740; Table 1) is ongoing. AURORA was designed to evaluate the efficacy and safety of CVC therapy on NASH patients with fibrosis stage F2 or F3 [13]. This seems to be rational, since patients with higher fibrosis stage were shown to benefit more in CENTAUR study; therefore, the primary endpoint of this study was the improvement in fibrosis ≥ 1 stage without worsening of NASH at month 12 [12]. AURORA has also a second, longer-term (5 years) primary endpoint, being the time to the first occurrence of a composite outcome, which contains harder endpoints, such as death, cirrhosis, HCC, and liver transplantation [13]. It is of note that the selection of patients subjected to liver biopsy at baseline was based on the “AURORA toolbox”, an algorithm consisting of transient elastography of the liver and other noninvasive indices of hepatic fibrosis (e.g., APRI, FIB-4). This strategy aimed to limit high screening failure rate observed in previous trials, including the CENTAUR, which may be up to 80% of screened patients [13].
There are some more ongoing studies on CVC in NASH patients, which are summarized in Table 1; some of them have been reportedly completed, but their results have not been published yet. Apart from the above mentioned ORION trial (NCT02330549) and NCT03376841, another phase 1 trial evaluated the comparative pharmacokinetics of CVC vs. pioglitazone vs. their combination (NCT02342067). Furthermore, the long-term safety of treatment with CVC in NASH patients is evaluated in an open-label, phase 2 trial (NCT03059446), in which patients that have completed the CENTAUR trial or discontinued the AURORA study are recruited; this study is expected to complete in 2030 (Table 1).
CVC is also under evaluation in combination with other agents targeting NASH via other pathways. This is interesting, since NASH is a multifactorial disease, therefore, we believe that it needs multiple-targeted treatment, since hardly one medication may be suitable in all NASH patients [48]. In this regard, CVC is under investigation in combination with tropifexor, a farnesoid X receptor (FXR) agonist in a phase 2 trial (Table 1). The combination of CVC and tropifexor had been previously evaluated in a study of drug-drug interaction, which showed that the combination therapy was well tolerated with a similar profile to that observed with the monotherapies, and that co-administration with tropifexor had no marked impact on pharmacokinetics of CVC [49]. FXR agonists have gained increasing interest in the treatment of NASH after the promising results of obeticholic acid, the first FXR agonist having been investigated in NASH patients [50, 51]. TANDEM (NCT03517540) is a multicenter, phase 2b trial, primarily targeting to evaluate the 1-year safety and tolerability of the combination of CVC and tropifexor in NASH patients with fibrosis stage F2 or F3 over a 48-week period [52]. The patients of the TANDEM trial are expected to undergo paired liver biopsy to evaluate the efficacy of the combination of CVC and tropifexor vs. either monotherapies on the improvement in hepatic fibrosis by ≥ 1-point and the resolution of NASH (secondary endpoints) [52].
Apart from CVC that targets both CCR2 and CCR5, other molecules targeting CCR2 or CCR5 are in the pipeline for the treatment of NAFLD/NASH. The therapeutic efficacy of CCR5 mono-antagonists was firstly evaluated in HIV infection, since they were demonstrated to prevent the CCR5-mediated entry of HIV strains into the cells. Nonetheless, some of the first developed CCR5 antagonists, such as aplaviroc and vicriviroc, initially provided some favorable results, but the relevant clinical trials were discontinued due to safety considerations (hepatotoxicity and carcinogenesis, respectively) [53]. It should be highlighted that CCR5 antagonism may combine the anti-HIV activity with a beneficial effect on NAFLD/NASH [54], which is very common in HIV infected patients [45].
Preclinical data on CCR5 antagonists in NAFLD are promising. Treatment with a CCR5 antagonist, named Met-CCL5, inhibited the migration and proliferation of HSC, as well as collagen secretion in cultures [31]. Likewise, treatment with Met-CCL5 in mice led to the regression of hepatic fibrosis [31]. Another CCR5 antagonist, MVC, inhibited HSCs in cultures [54]. In animal models of NAFLD, MVC was shown to reduce IR and steatosis [55, 56]. However, despite the initial speculations, hepatic inflammation was not attenuated by MCV in a high-fat diet mouse model [56].
Based on these preclinical data, there are currently three ongoing clinical trials, performed in Europe, aiming to evaluate the potential benefit of MVC to NAFLD/NASH in HIV infected patients (Table 2). The MAVMET study (“A Multicentre, 48 Week Randomized Controlled Factorial Trial of Adding Maraviroc and/or Metformin for Hepatic Steatosis in HIV-1-infected Adults on Combination Antiretroviral Therapy”; NCT03129113) primarily aims to evaluate the 12-month effect of MVC, metformin or their combination, as add-on to standard antiretroviral treatment (ART), on hepatic fat accumulation. The MASH study (“Maraviroc Add-On Therapy for Steatohepatitis in HIV”; EUCTR2017-003172-32) primarily aims to evaluate the 12-month effect of MVC on changes in immune cells infiltrating the hepatic parenchyma. Since patients will be subjected to paired liver biopsies in the MASH study, the effect of MVC on hepatic histology is also expected as secondary endpoint. The third study (ISRCTN31461655) primarily aims to investigate the safety of MVC and adherence to treatment. Noninvasive indices and liver imaging will be also performed as secondary outcomes.
Registered clinical trials on CCR antagonists (other than CVC) in patients with NAFLD
| ID, registration date (DD/MM/YYYY) | Title | Design | Arms | Patients (n) | Duration | Primary outcome(s) |
|---|---|---|---|---|---|---|
| ISRCTN31461655, 23/02/2018 | A phase IV, open-label pilot study investigating non-invasive markers of hepatic fibrosis in people living with HIV-1 and non-alcoholic fatty liver disease randomized to receiving optimized background therapy (OBT) plus maraviroc or OBT | Multicenter, randomized, open-label, phase 4 | MVC (NA mg) vs. No treatment (ART only) | 60 | 24 months | 1) Number of participants with adverse events 2) Adherence to MVC treatment (questionnaire) 3) Parameters related to the participation and missing values |
| EUC-TR2017-003172-32, 06/02/2018 | Maraviroc Add-On Therapy for Steatohepatitis in HIV (MASH study) | Multicenter, non-randomized, open-label, phase 2 | MVC (150 mg) vs. MVC (300 mg) | 30 | 12 months | Change in hepatic immune cells identified on liver biopsy (immuno-histochemistry) |
| NCT03129113, 26/04/2017 EUC-TR2016-003575-21, 09/05/2018 | A Multicentre, 48 Week Randomized Controlled Factorial Trial of Adding Maraviroc and/or Metformin for Hepatic Steatosis in HIV-1-infected Adults on Combination Antiretroviral Therapy (MAVMET) | Multicenter, randomized, open label, phase 2 | MVC (NA mg) vs. Metformin (500 mg) vs. MVC (NA mg) + Metformin 500 mg) vs. No treatment (ART only) | 90 | 12 months | Change in percentage of liver fat, measured by MR-PDFF |
References are presented in date of registration order (newer to older); ART: antiretroviral treatment; HIV: human immunodeficiency virus; MR-PDFFl: magnetic resonance spectroscopy measuring the hepatic proton density fat fraction; MVC: Maraviroc; NA: not available; OBT: optimized background therapy
Moreover, a humanized anti-CCR5 IgG4 antibody, named leronlimab (PRO140), is under evaluation, as monotherapy or in combination with ART, against HIV infection. Leronlimab is anticipated to be evaluated in NASH patients in a phase 2, multicentre, double-blind, placebo controlled trial (https://www.globenewswire.com/news-release/2019/10/02/1923792/0/en/CytoDyn-Announces-FDA-Clearance-to-Proceed-with-Phase-2-Clinical-Trial-of-Leronlimab-PRO-140-for-Treatment-of-NASH.html).
Regarding CCR2 antagonists, there are preclinical data favoring their potential efficacy in NASH. Early treatment of a choline-deficient amino acid diet mouse model with a CCR2 inhibitor, named RS102895, reduced hepatic steatosis, decreased the infiltration of immune cells into the liver, and suppressed the expression of fibrogenic genes. Later treatment of the same model with RS102895, when NASH was established, ameliorated hepatic inflammation and fibrosis, but not steatosis [37]. Treatment of a lipoatrophic mouse model with RS504393, another CCR2 antagonist, also reduced hepatic inflammation and IR [57]. Treatment of two murine models with mNOX-E36, a structured L-enantiomeric RNA oligonucleotide inhibiting CCL2, improved hepatic steatosis and immune cell infiltration into the liver, but not fibrosis [38]. As aforementioned, propagermanium, acting as CCR2 inhibitor, had beneficial effects on IR, hepatic steatosis, and inflammation [9]. Despite favorable preclinical data, to the best of our knowledge, there are no clinical trials with CCR2 antagonists in NASH.
Interestingly, aspirin has been also proposed to ameliorate atherosclerosis and NAFLD by downregulating CCR2 and inducing anti-inflammatory phenotype in macrophages [58].
It should be underlined that there are not currently studies on antagonists of other CCR relevant to NAFLD in the literature.
Although NAFLD is highly prevalent and its prevalence is expected to increase in parallel with the epidemics of obesity and T2DM, there is currently no licensed pharmacological treatment [8]. Guidelines recommend pioglitazone or vitamin E (tocopherol) in selected patients with NASH and significant fibrosis [59, 60], but the use of both medications remains off-label [8]. These render NASH pharmaceutical treatment attractive for both the investigators and pharmaceutical industry [61]. Therefore, there is a paroxysm in the drug development pipeline, with more than 300 molecules in clinical trials for NASH in 2018; however, only a limited number of molecules have reached phase 3 clinical trials, including CVC [8].
Most preclinical data supported a beneficial effect of CVC on both hepatic inflammation and fibrosis [10, 21], which were close to the initial expectations, based on the speculated inhibitory effect of CVC on Kupffer cells and infiltrating macrophages, which target mainly hepatic inflammation, and HSCs, which target mainly hepatic fibrosis [20, 43]. Nonetheless, since extrapolation of animal studies to human often fails, CVC did not improve hepatic inflammation in CENTAUR study (primary endpoint), exhibiting, however, promising results in hepatic fibrosis (secondary endpoint), especially in patients with more severe disease at baseline [11, 12]. These results led CVC to a phase 3 trial (AURORA) with NASH patients with more severe fibrosis (F2–F3) that those of CENTAUR trial (F1–F3) and improvement in fibrosis as its primary endpoint [13]. The one-year results of AURORA are expected to be announced at the end of 2021, whereas its long-term results at the end of 2028.
Although some preclinical data also showed a favorable effect of CVC [21] or MVC [55, 56] on IR and hepatic steatosis, the results of both ORION and CENTAUR [11, 12] studies did not reveal any special effect of CVC on human IR or hepatic steatosis. This seems rational, since the response to any treatment depends on the underlying pathophysiological mechanism of action of the medication. By targeting CCR2 and CCR5, CVC has a potential to affect hepatic inflammation and fibrosis, but not IR and steatosis. Contrariwise, other medications that primarily target the metabolic contributors (e.g., IR, lipid metabolism) to NAFLD (e.g., pioglitazone, glucagon-like peptide-1, statins) are expected to be effective on IR and hepatic steatosis, but have a lower or null effect on hepatic inflammation and fibrosis. The above considering, combing medications that target different pathogenetic contributors (“hits”) of NASH seems a rational approach [6, 48]. For example, CVC combined with pioglitazone and/or a statin might probably target all main NASH components, i.e. IR, steatosis, inflammation, and fibrosis. Nonetheless, whether this multifactorial approach will provide fruitful benefits in NASH patients remains to be shown in future clinical trials [8, 48].
Regarding the effect of CCR antagonists on extrahepatic diseases related to NAFLD (obesity, T2DM, dyslipidemia, kidney disease), there are no studies to-date, specifically designed for this aim. Limited data retrieved mainly from the CENTAUR trial showed that CCR2/CCR5 antagonism does not essentially affect body weight, parameters of glucose metabolism (glucose, HbA1c), lipid metabolism (total cholesterol, high-density and low-density lipoprotein cholesterol, triglycerides), and renal function [11, 12]. However, since these are secondary endpoints, specifically focused studies are needed to definitely show that CCR2/CCR5 antagonism has a neutral effect on extrahepatic diseases related to NAFLD.
In summary, CVC provided favorable effects on hepatic fibrosis in NASH patients and is under evaluation in a large multicenter phase 3 clinical trial (AURORA), which is expected to provide a definite answer on its effect on hepatic fibrosis. A positive result in AURORA may open a window towards the approval of CVC for NASH patients with significant or advanced fibrosis, which is the main histological prognostic factor of the disease, in the near future.
APRI: aspartate aminotransferase-to-platelet ratio index
ART: antiretroviral treatment
CCL: CC-chemokine ligands
CCR: C-C chemokine receptor
CVC: cenicriviroc
CYP: cytochrome P450
DCs: dendritic cells
ELF: enhanced liver fibrosis score
F: fibrosis stage
FIB-4: fibrosis-4 index
FXR: farnesoid X receptor
HCC: hepatocellular carcinoma
HIV: human immunodeficiency virus
HSC: hepatic stellate cell
IL: interleukin
IR: insulin resistance
MCP: monocyte chemoattractant protein
MetS: metabolic syndrome
MVC: maraviroc
NAFL: nonalcoholic fatty liver
NAFLD: nonalcoholic fatty liver disease
NAS: NAFLD activity score
NASH: nonalcoholic steatohepatitis
NK: natural killer
PDGF: platelet derived growth factor
RANTES: regulated on activation, normal T expressed and secreted
T2DM: type 2 diabetes mellitus
TGF: transforming growth factor
TLR: Toll-like receptor
MD, KP, AP: design of the review, drafting the manuscript; JK: design of the review, critical revision of the manuscript; SAP: conception and design of the review, drafting and critical revision of the manuscript; all the authors: final approval of the version to publish, agreeing to be accountable for all aspects of the work.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Noel C. Salvoza ... Natalia Rosso
Amedeo Lonardo, Stefano Ballestri
Marvin Ryou ... Gyorgy Baffy
Marica Meroni ... Paola Dongiovanni
Ivana Mikolasevic ... Sandra Milic
Valerio Rosato ... Marcello Persico
Carlo Acierno ... Ferdinando Carlo Sasso
Rosa Lombardi ... Anna Ludovica Fracanzani
Angelo Di Vincenzo ... Marco Rossato
Angelo Colucci ... Claudia Mandato
