 Review
Review

Affiliation:
1General Medicine and Metabolic Diseases, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milano, Italy
2Department of Pathophysiology and Transplantation, Università degli Studi di Milano, 20122 Milano, Italy
†These authors contributed equally to the work.
Affiliation:
1General Medicine and Metabolic Diseases, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milano, Italy
3Department of Clinical Sciences and Community Health, Università degli Studi di Milano, 20122 Milano, Italy
†These authors contributed equally to the work.
Affiliation:
1General Medicine and Metabolic Diseases, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milano, Italy
Email: paola.dongiovanni@policlinico.mi.it
Explor Med. 2020;1:218–243 DOI: https://doi.org/10.37349/emed.2020.00015
Received: May 28, 2020 Accepted: June 20, 2020 Published: August 31, 2020
Academic Editor: Lindsay A. Farrer, Boston University School of Medicine, USA
The article belongs to the special issue Exploring NAFLD/NASH
The prevalence of nonalcoholic or more recently re-defined metabolic associated fatty liver disease (MAFLD) is rapidly growing worldwide. It is characterized by hepatic fat accumulation exceeding 5% of liver weight not attributable to alcohol consumption. MAFLD refers to an umbrella of conditions ranging from simple steatosis to nonalcoholic steatohepatitis which may finally progress to cirrhosis and hepatocellular carcinoma. MAFLD is closely related to components of the metabolic syndrome and to environmental factors. In addition to the latter, genetic predisposition plays a key role in MAFLD pathogenesis and strictly contributes to its progressive forms. The candidate genes which have been related to MAFLD hereditability are mainly involved in lipids remodeling, lipid droplets assembly, lipoprotein packaging and secretion, de novo lipogenesis, and mitochondrial redox status. In the recent years, it has emerged the opportunity to translate the genetics into clinics by aggregating the genetic variants mostly associated with MAFLD in polygenic risk scores. These scores might be used in combination with metabolic factors to identify those patients at higher risk to develop more severe liver disease and to schedule an individual therapeutic approach.
Nonalcoholic or more recently re-defined metabolic associated fatty liver disease (MAFLD) is regarded as the most relevant liver disease of the twenty-first century, affecting at least one third of the general population [1–3] and it is predicted to become the leading cause of liver transplantation by 2030 [4]. Thus, given its pandemic proportion, MAFLD constitutes a tremendous socio-economic and health burden [5]. MAFLD is characterized by excessive hepatic fat deposition that exceeds 5% of liver weight, in the absence of alcohol intake. It embraces a variable phenotypic spectrum of hepatic disorders, spanning from isolated steatosis to its inflammatory form, nonalcoholic steatohepatitis (NASH), defined by lobular inflammation, hepatocyte ballooning degeneration and fibrotic processes activation. NASH may then progress towards end-stage liver injuries, such as cirrhosis and hepatocellular carcinoma (HCC) [6, 7].
At epidemiological level, MAFLD is tightly correlated with excessive adiposity, type 2 diabetes (T2D) and metabolic syndrome (MetS) hallmarks [8] and its pathophysiology is closely intertwined with obesity, insulin resistance (IR) and atherogenic dyslipidemia [9]. Hence, it is referred to as the hepatic manifestation of MetS and unhealthy dietary habits, fructose over-consumption and sedentary lifestyle may represent pivotal environmental risk factors for its pathogenesis [10, 11].
However, as a complex disease, MAFLD has a multi-factorial pathogenesis and the individual susceptibility to develop it may be partly attributable to inherited risk factors [12]. Hitherto, the best-known MAFLD inherited components are represented by single nucleotide polymorphisms in genes regulating hepatic lipid remodeling, turn-over and dismissal, among which patatin-like phospholipase domain-containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), membrane bound o-acyltransferase domain-containing 7 (MBOAT7), and Glucokinase regulator (GCKR) [12]. Nonetheless, only a minor percentage of inter-individual variability is explained by these common mutations and the majority of the phenotypic differences in hepatic manifestations may also derive from gene-environment interaction, due to hereditable epigenetic modifications [13, 14]. Furthermore, among the plethora of multiple parallel hits, a new perspective in the pathophysiology of fatty liver is represented by the crucial role of intestinal dysbiosis, microbial metabolites and enhanced intestinal permeability [15, 16].
Nowadays, liver biopsy remains the gold standard procedure for diagnosis of MAFLD and no therapeutic consensus exists for the treatment of MAFLD patients [10, 11]. However, the deep-knowledge of the genetic landscape responsible for the individual susceptibility towards MAFLD and its progressive forms may open the possibility to identify score-based strategies of combined genetic risk factors for liver damage prediction and pave the way to personalize therapeutic approaches. In addition, the study of the interaction between environment and inherited factors may be also useful to address specific clinical tailored advices, health practices for disease prevention and genome-based dietary guidelines [10, 11].
For this reason, this review aimed to focus on the relevance of the assessment of the individual genetic make-up in order to ameliorate disease diagnosis, by using non-invasive strategies and to individualize the clinical management of MAFLD patients, avoiding its progression towards end-stage conditions.
In the past years, it has been widely demonstrated that IR and obesity are the most prevalent risk factors of MAFLD pathogenesis. However, among subjects with the same grade of adiposity, there is a substantial variability in hepatic lipid deposition, raising the possibility that several other risk factors may participate to steatosis development. Familial, twin and epidemiological studies indicate that hepatic fat accumulation has a strong inherited component [17, 18]. In particular, Schwimmer et al. [19], revealed that parents and siblings of overweight children with MAFLD display an enhanced susceptibility to develop fatty liver compared to obese children without MAFLD. Even more, a familial NASH clustering is common, reaching 18% of patients having a similarly affected first degree relative [20].
Twin studies demonstrated that approximately 60% of the variation in serum alanine aminotransferase (ALT) as well as in circulating insulin concentrations, are genetically determined in absence of alcohol abuse or viral hepatitis and that are both significantly correlated with liver fat content [21]. Moreover, Loomba et al. [19], demonstrated that hepatic steatosis, assessed by using magnetic resonance imaging proton-density fat fraction (MRI-PDFF) and hepatic fibrosis, based on stiffness determined by MR elastography, are both strictly correlated in monozygotic twins compared to dizygotic twins. In a multivariable model, adjusting for age, gender and ethnicity, the percentages of hereditability of hepatic steatosis and fibrosis were attested at 52% and 50%, respectively [22]. In addition, in the same cohort of twins, Cui et al. [23], revealed a high level of shared genetic components between hepatic steatosis and fibrosis.
Even the ethnicity can influence the predisposition towards MAFLD [17, 18]. In two large multi-ethnic population studies conducted in the United States, it has been demonstrated that Hispanic individuals have an higher risk to develop MAFLD than those of European descent [24, 25]. More specifically, intra-ethnic differences exist and amongst Hispanics, Mexicans have much higher prevalence of MAFLD compared to Dominican or those from Puerto Rico [26]. On the contrary, African-Americans are protected irrespectively of diabetes, excess in body weight and socioeconomic factors, supporting the notion that heritability may exert a crucial role in MAFLD pathophysiology [12]. In particular, African-Americans differed in the metabolic response to obesity and IR when compared to either Hispanics or Caucasians, appearing more resistant to triglyceride (TG) accumulation both in the adipose tissue and in the liver [27].
Several inherited risk factors have been identified as modifier of genetic predisposition to develop MAFLD and its progressive forms [12]. In particular, Dongiovanni et al. [28], demonstrated that hepatic fat accumulation represents the main driver of the progression to end-stage liver damages in genetically predisposed individuals. Therefore, the effect of each genetic variation on the spectrum of MAFLD is closely intertwined with their ability to induce fat accumulation [28]. Nowadays, the major common predictors of the inherited predisposition to severe MAFLD are the variants in PNPLA3, TM6SF2, MBOAT7 and GCKR genes. However, given the complex genetic architecture of MAFLD, a huge number of other genetic risk factors has been identified, and score-based strategies which evaluate polygenic determinants of MAFLD seems to be the most highly predictive and they can be exploited to improve diagnostic accuracy and to guide treatment options [29, 30].
To date, the major genetic predictor of the inter-individual and ethnicity-related differences in hepatic fat content and the main risk factor of increased susceptibility to develop progressive MAFLD is the rs738409 C > G variant in PNPLA3 gene (also referred to as adiponutrin), encoding the amino acid substitution isoleucine to methionine at the position 148 (p.I148M) [31]. It was firstly identified by a genome-wide association study (GWAS) in 2008 which evaluated North American population of diverse ethnicity and revealed that the prevalence of the G at-risk allele is higher in Hispanics (49%) than in Europeans (23%) and less frequent in African Americans (17%), thereby explaining the inter-ethnic susceptibility to MAFLD [31]. PNPLA3 encodes a 481-amino acid membrane lipase, mainly localized in the endothelial reticulum (ER) and at the lipid droplet surface in hepatocytes, adipocytes and in hepatic stellate cells (HSCs) [32, 33]. At transcriptional level, PNPLA3 expression is controlled by the activation of the sterol regulatory element-binding protein 1 (SREBP1c)/liver X receptor and by Carbohydrate response element binding protein pathways, induced by post-prandial or pathological hyperinsulinemia. Moreover, PNPLA3 protein is post-transcriptionally modified by the presence of fatty acids, hindering its degradation [34, 35]. Thus, an interplay between PNPLA3 and environmental factors exists. In particular, high-sucrose and high-fructose consumption, sedentary lifestyle habits and excessive adiposity trigger the detrimental effect of p.I148M both in adults and children [36–38]. Indeed, the associations between the rs738409 variant and MAFLD may be unmasked by the increased adiposity, enhancing the genetic risk [36]. Nonetheless, it has been demonstrated that this variation constitutes a risk factor for MAFLD development and progression towards NASH and fibrosis, even in lean subjects (BMI < 25 kg/m3) [39].
In vitro studies demonstrated that PNPLA3 hydrolyzes lysophosphatidic acid to phosphatidic acid, and the p.I148M inherited variation strongly affects its function [40, 41]. As a consequence of the loss of PNPLA3 enzymatic activity, patients carrying G alleles have elevated plasma liver enzymes and a strong predisposition towards the entire spectrum of liver damage related to fatty liver, encompassing NASH, severe fibrosis and HCC, even influencing the response to therapeutic approaches [42].
However, the physiological implication of PNPLA3 in lipid metabolism remains to be fully elucidated. In particular, neither Pnpla3 genetic deficiency nor Pnpla3 wild-type over-expression in mice induces hepatic steatosis development [41, 43, 44], whereas Pnpla3 I148M knock-in mice are characterized by fatty-laden hepatocytes upon a steatogenic high-sucrose diet challenge [45]. In these mice, Pnpla3 silencing through antisense oligonucleotides ameliorated all histological features of MAFLD, including liver fibrosis [46]. More specifically, it has been recently proposed that the pathogenic effect of PNPLA3 variant is mediated by its interfering with the activity of the adipose triglyceride lipase (ATGL)/patatin-like phospholipase domain-containing 2 (PNPLA2), which is the main TG lipase in liver and adipose tissue, directly by interacting with its cofactor, namely comparative gene identification-58 (CGI-58) [47]. Indeed, overexpression of Pnpla3 I148M increased hepatic TG levels in wild-type, but not in Cgi-58 knock-out (KO) mice, suggesting that the PNPLA3 mutation may promote steatosis through a CGI-58-dependent inhibition of ATGL/PNPLA2 on lipid droplets. Superimposable results have been obtained in brown adipocytes [48]. In addition, it has been demonstrated that the p.I148M variant is able to abolish PNPLA3 ubiquitylation and proteasomal degradation resulting in the accumulation of PNPLA3 I148M, without altering lipid synthesis [49]. Therefore, the likely mechanisms through which the p.I148M variant induces intracellular fat deposition seems to be due to the accumulation of the PNPLA3 mutated protein on the lipid droplet surface due its reduced ubiquitination, thus impairing TG mobilization even by other lipases (i.e. ATGL/PNPLA2) and hampering TG turnover and dismissal [49, 50]. The proof of this concept is widely explained by BasuRay et al. [51], which engineered a synthetic isoform of PNPLA3 that uncouples protein accumulation from loss of enzymatic activity. These authors demonstrated that the expression of an ubiquitylation-resistant form of Pnpla3 in mice is responsible for the accumulation of Pnpla3 mutated protein on hepatic lipid droplets. Furthermore, the reduction of Pnpla3 expression by shRNA or by or proteolysis-targeting chimera (PROTAC)-mediated degradation dampened TG storage formation in mice overexpressing Pnpla3 I148M [51]. In keeping with these findings, PNPLA3 E434K variant ameliorates the impact of the p.I148M on steatosis development in MAFLD patients, whereby down-modulating PNPLA3 expression on the lipid droplets [50]. Another potential mechanism that explain TG accumulation in presence of the p.I148M variant is related to the impairment of lipophagy in hepatocytes, thus hampering autophagic fluxes and lipid droplet degradation [52].
A lipidomic analysis, provided by Luukkonen et al. [53], demonstrated that TG in very-low density lipoproteins (VLDL) are depleted of polyunsaturated fatty acids (PUFAs) in I148M homozygous subjects both under fasting and postprandial conditions. As a consequence, in I148M hepatic cells, PUFA incorporation into TG is increased and PUFA-containing diacylglycerols (DAGs) are accumulated, at the expense of phosphatidylcholines (PCs) [53]. The hepatic lipid composition of DAG species may, in turn, strongly affect insulin sensitivity. However, the alteration in hepatic DAG levels has not confirmed by Franko et al. [54], which supported the idea that PNPLA3 variation is tightly associated with fatty liver, but not with IR, whereby dissociating these two hallmarks of MAFLD. Indeed, IR-related MAFLD is characterized by high levels of metabolically harmful saturated and mono-unsaturated TG, ceramides and free fatty acids (FFAs), whereas PNPLA3-related MAFLD by hepatic PUFA-containing TG. These observations may possibly explain why metabolic MAFLD and not PNPLA3-related MAFLD, is tightly correlated with increased risk of T2D and cardiovascular events [55]. Likewise, the excess of PUFA-containing lipids has been observed even in the adipose tissue of PNPLA3 I148M carriers, where this variant did not alter the rate of lipolysis or the composition of fasting serum FFAs [56].
Intriguingly, PNPLA3 mutated protein in HSCs exerts a detrimental effect on retinol secretion [57], directly participating to fibrogenesis and carcinogenesis, therefore, enhancing cirrhosis and HCC risks, irrespective of the predisposition to steatosis [58–61]. Indeed, MAFLD patients carrying the G allele display a distinctive histological pattern, characterized by enhanced steatosis and portal inflammation accompanied by a prominent proliferation of hepatic progenitor cell, extensive ductular reaction, HSCs and myofibroblasts activation, inducing even portal fibrosis generation and severe systemic oxidative stress [62]. Therefore, the size effect of the I148M variant on the risk of MAFLD is the strongest ever reported for a common variant [10].
Notably, this variation has been also associated with hepatic decompensation and liver-related death in a recent Italian prospective study [63], with increased risk of fibrosis progression and HCC even in patients affected by viral hepatitis or alcoholic liver disease (ALD) and with poor prognosis in patients affected by autoimmune hepatitis, independently of steatosis [64–66].
In 2014, an exome-wide association study conducted by Kozlitina et al. [67], in more than 80, 000 participants of three independent cohorts identified the missense rs58542926 C > T variant in the TM6SF2 gene encoding the lysine to glutamate substitution at residue 167 (E167K). Ever-increasing evidence has strengthened the role of TM6SF2 as a key regulator of cholesterol biosynthesis and lipid composition in the liver of both humans and experimental models [68–75]. TM6SF2 localizes in the ER and ER-Golgi compartments [76] and it participates to hepatic VLDL lipidation and assembly in the ER cisternae [67, 69]. In Tm6sf2−/− mice, Smagris et al. [69], firstly reported that TM6SF2 is required to mobilize neutral lipids for VLDL assembly but it did not affect lipoproteins’ trafficking. A lipidomic analysis in both human livers and in HuH7 human hepatoma cell line revealed that TM6SF2 silencing impairs hepatic synthesis of PC-containing PUFAs. The reduction of the unsaturated PCs pool causes an excess of free PUFAs which, in turn, inhibits VLDL assembly and favors the formation of TGs and cholesterol-esters clusters thus disturbing membrane composition and dynamics [71, 73]. The impact of TM6SF2 deficiency has been also investigated in a fine-tuned research by O’Hare and colleagues who explored the role of TM6SF2 in small intestine of zebrafish and in Caco-2 enterocytes. They demonstrated that intestinal TM6SF2-deficiency induces lipid droplets accumulation, reduces lipid clearance, and promotes ER stress [77]. Interestingly, Fan and coworkers hypothesized that TM6SF2 might also exert an enzymatic activity by converting zymosterol into 5-α-cholesta-7,24-dien-3β-ol, a critical step in cholesterol biosynthesis, although further studies will be crucial to corroborate this finding [70].
In presence of the 167Lys substitution, TM6SF2 expression is 50% reduced in the HuH7 hepatocytes and generated a TM6SF2 misfolded protein which may run into rapid intracellular turnover and degradation further causing its hepatic down-regulation [67]. In addition, Tm6sf2 knock-down in the livers of mice resulted in 3-fold enrichment of intrahepatic TGs, probably due to VLDL retention, paralleled with decreased plasma cholesterol [67]. The E167K genetic variant was independently associated with higher circulating levels of ALT, a marker of liver damage, hepatic TG content, and MAFLD stages in both children and adults [67, 77–82]. The minor T allele was also correlated with lower serum cholesterol and TGs levels in several cohorts of MAFLD patients [77, 81] and in large population studies including the Dallas Heart Study, the Dallas Biobank and the Copenhagen Study [67]. In another exome-wide association study encompassing 300, 000 participants, the authors identified 444 coding and noncoding genetic variants associated with plasma lipids and, among them, the E167K TM6SF2 variant resulted as one of the causal gene that strongly influenced metabolic traits as it mitigated circulating TG levels and increased risk of fatty liver and T2D [83]. Intriguingly, in a large cohort of 1, 201 biopsy-proven MAFLD individuals who underwent liver biopsy, carriers of the rs58542926 variant showed a higher degree of steatosis, necroinflammation, ballooning and fibrosis but were protected against cardiovascular events [81]. In keeping with these results, numerous studies revealed that the non-synonymous rs58542926 polymorphism reduces serum concentration of “bad” cholesterol enriched in low density lipoproteins (LDL) [67, 77, 79–81, 84–86]. O’Hare et al. [77], evaluated the effects of the rs58542926 single nucleotide polymorphism (SNP) in two independent cohorts entailing 983 patients who underwent bariatric surgery and 3, 556 participants who were enrolled in the Amish Complex Disease Research Program (ACDRP). Despite the metabolic biochemical differences, carriers of E167K variant showed an improved fasting lipid profile in both case studies and they exhibited lower postprandial serum TGs after high fat challenge [77]. Therefore, it has been suggested that the T risk allele may disentangle MAFLD from cardiovascular disorders but it increases liver disease severity [81, 87]. Even Sookoian et al. [88], found a significant correlation between the E167K variant and histological steatosis in 361 subjects and such findings were consistent with those reported by Mancina et al. [78], and Dongiovanni et al. [81]. In a multiethnic pediatric cohort consisting of 957 obese individuals, of whom 459 underwent MRI for MAFLD diagnosis, the minor T allele positively associated with serum ALT levels, fatty liver and with improved lipid profile only in Caucasian and Afro-American children, thus highlighting how the solely ethnical branch may confer a different genetic predisposition to MAFLD [79]. In addition, in 60 non-obese normo-lipemic MAFLD patients, whose liver biopsies were available, the TM6SF2 at-risk genotype increased the susceptibility to both hepatic and adipose tissues IR as well as it impaired the post-prandial incretin secretion and, consequently, its effects on pancreatic β-cells [89]. Furthermore, at multivariable models, the TM6SF2 rs58542926 variant is associated with MAFLD even in lean individuals, who showed a higher prevalence of carriage of the T allele compared to obese patients [90].
Overwhelming data supported that TM6SF2 loss-of-function affects lipid metabolism and predisposed to hepatic steatosis development and progressive liver injury [79, 81, 82, 91, 92]. However, the association between the minor T allele with both fibrosis and HCC is still controversial, probably due different diagnostic tools exploited for MAFLD diagnosis (MRI, ultrasound, and liver biopsy) [74, 88, 92]. For instance, Sookoian et al. [88], did not find any associations between the TM6SF2 mutation and circulating transaminases, necroinflammation and fibrosis possibly due to the low frequency of the rs58542926 polymorphism and lack of statistical power. Conversely, a meta-analysis of 24, 147 individuals affected by heterogeneous chronic liver disorders associated the TM6SF2 rs58542926 genotype with higher susceptibility to develop MAFLD, ALD, cirrhosis, and HCC than viral hepatitis [93]. In alcohol abusers affected by alcohol-related cirrhosis, the presence of the rs58542926 C > T variant represented an additional risk factor to develop HCC [94]. According to these results, the E167K TM6SF2 did not influence fibrosis severity in 694 consecutively biopsied Caucasians with chronic hepatitis C [95] whereas it correlated with alcohol-related HCC in 511 cirrhotic patients [96] and in a prospective cohort of 249 ALD patients [97]. Liu et al. [91], reported that carriers of the T risk allele conferred an increased predisposition to develop MAFLD-related advanced fibrosis in two independent cohort regardless of other confounders as gender, sex, BMI, T2D and PNPLA3 rs738409 genotype. In 11 biopsy-proven MAFLD pediatric subjects, carriers of the TM6SF2 C > T variant displayed a higher grade of fibrosis compared to CC genotype [79]. Finally, the association among the T risk allele with hepatic fibrosis [92] and HCC risk [98] was further observed in a cross-sectional and in a small cohort study including 502 and 129 MAFLD patients, respectively.
In 2015, a GWAS revealed that the common rs641738 C > T variant in the MBOAT7-transmembrane channel like 4 (TMC4) locus on chromosome 19, increased the susceptibility to cirrhosis in alcoholics [99, 100]. Then, Mancina and Dongiovanni et al. [101], demonstrated that the rs641738 variation associates with a strong predisposition towards hepatic fat accumulation and to the entire phenotypic spectrum of liver injuries related to MAFLD, including HCC [101–103]. This evidence has been further corroborated in pediatric individuals in which MBOAT7 variation is strongly correlated with enhanced ALT levels, C-reactive protein concentrations, and with increased total body fat percentage [104]. Moreover, it has been demonstrated that in pediatric MAFLD patients, T risk allele carriers were characterized by increased ALT, and more advanced steatosis and fibrosis, revealing a combined effect of the MBOAT7 rs641738, PNPLA3 I148M, and TM6SF2 E167K variants on pediatric MAFLD risk [105], even confirmed in adult patients [29, 30, 106]. Notably, the rs641738 has been also identified as a risk factor for fibrosis development even in viral hepatitis B and C, possibly representing a common modifier of liver damage [107, 108] and it has been shown to be involved in primary biliary cholangitis, exerting a positive effect on transplant free survival [109, 110]. However, the association between the rs641738 variant and liver injuries remains still controversial and not fully replicated, mainly due to the different sample size and ethnicity of the cohorts enrolled in the studies or to the diverse assessment of hepatic steatosis [111–115]. Notwithstanding, a very recent meta-analysis across 42 studies, confirmed the associations between the rs641738 variant and liver fat, ALT, histological severity of MAFLD, fibrosis and HCC in individuals of European descent [116].
MBOAT7, also known as lyso-phosphatidylinositol (lyso-PI) acyl-transferase1 (LPIAT1), encodes for an enzyme member of the “Lands’ Cycle” of phospholipid acyl-chain remodeling of the membranes. It is mainly localized in the membrane bridging ER and mitochondria in which the fat biosynthesis and lipid droplets formation occurs. It conjugates an acyl-CoA to the second acyl-chain of lyso-phospholipids, using arachidonoyl-CoA. Thus, it modulates the desaturation of phospholipids and the amount of free arachidonic acid, precursor of proinflammatory eicosanoids [117]. Mancina and Dongiovanni et al. [101], have deeply demonstrated that the mechanisms underlying the association between the rs641738 variant and liver damage is related to the hampered hepatic gene and protein expression of MBOAT7, altering in turn phosphatidylinositol species [118], as confirmed by Luukkonen et al. [102]. This evidence is supported by an our very recent paper, that pointed out that hepatic MBOAT7 down-regulation is a dysfunctional response to hyperinsulinemia and that its reduced expression induces intracellular fat accumulation in clinical samples, in in vivo models of MAFLD and in genetically edited HepG2 cells (MBOAT7−/−) [118]. In particular, MBOAT7 is hampered in presence of hyperinsulinemia and severe liver damage in obese patients, irrespectively of the individual genetic make-up, as well as in experimental models of obesity-related or IR-related MAFLD. An impairment in MBOAT7 function contributes to accumulate saturated phospholipids, mainly, phosphatidylinositol species that may be shunted to the synthesis of saturated and mono-unsaturated TG, further corroborating fatty-laden hepatocyte formation [118].
Alongside PNPLA3, TM6SF2 and MBOAT7 variations, even the common loss-of-function rs1260326 C > T variant in the GCKR gene, encoding the p.P446L substitution has been widely associated with increased fasting TG concentrations, large VLDL, steatosis and liver damage [115, 119–121]. GCKR gene codifies for the glucokinase regulatory protein, that exerts a crucial role in glucose homeostasis, whereby regulating glucose influx into the hepatocytes and the activation of de novo lipogenesis. In particular, the alteration of GCKR affects glucokinase partitioning between the cytosol and nucleus, thus impairing its negative modulation in response to fructose-6-phosphate and in turn, it constitutively activates the hepatic glucose uptake [122]. On one hand, this effect may ameliorate circulating fasting glucose and insulin levels, on the other it may provide malonyl-CoA as substrate for de novo lipogenesis and it may avoid fatty acid oxidation through the inhibition of carnitine-palmitoyltransferase, favoring glycolysis and steatosis development [123, 124]. Santoro et al. [125], reported for the first time the rate of de novo lipogenesis through incorporation of deuterium into the palmitate contained in the VLDL after the administration of a carbohydrate drink (75 g glucose and 25 g fructose) in obese adolescents. These authors demonstrated that the GCKR rs1260326 variation in homozygosity increased hepatic lipid synthesis in obese adolescents, as a result of the enhanced glycolytic carbon flux to TG formation [125]. This study was the first that revealed in pediatric subjects that a common variant might favor steatosis onset by enhancing the ability of the liver to convert carbohydrate into TG [125]. Moreover, it has been demonstrated that the combination of PNPLA3 and GCKR minor alleles may explain up to 32% of the liver fat content in Caucasian obese children, 39% in African-Americans and 15% in Hispanics [120]. The joint effect of GCKR variant with PNPLA3 has been indicated even by the strong increased risk of MAFLD in histologically confirmed MAFLD patients [126]. Furthermore, this variant has been also associated with increased susceptibility to NASH and NASH-derived HCC [126, 127] and enhanced fibrosis in adult MAFLD patients along with elevated serum TG levels, without altering LDL and HDL cholesterol levels and cardiovascular risk [119, 128].
In 2018, an exome-wide sequencing identified the rs72613567 variant, which has been shown to protect against histological steatohepatitis and against clinically significant fibrosis and cirrhosis, in both MAFLD and ALD [129]. The rs72613567 is an insertion of an adenine adjacent to the donor splice site of the last exon (TA allele), resulting in a truncated transcript, reduced expression and impaired enzymatic activity of the HSD17B13, which is widely expressed in the hepatocytes, at the lipid droplets surface [129–131]. The precise function of HSD17B13 is currently under definition. However, it has been demonstrated that HSD17B13 is up-regulated in MAFLD-affected patients, at the lipid droplets surface and in experimental models of NASH [131]. In particular, HSD17B13 over-expression is enabled to exacerbate the number and the size of the lipid droplets in cultured hepatocytes [131]. Conversely, HSD17B13 KO mice had an impairment in hepatic-lipid metabolism, resulting in an increased TG content into the liver and steatosis onset [132], directly inducing de novo lipogenesis through SREBP1 and fatty acid synthase over-expression [131], without altering reproductive performance and serum steroid concentrations [132]. Thus, the effect of HSD17B13 on hepatic steatosis remains controversial. The rs72613567 variant has never been related to a reduced risk of hepatic steatosis development [129]. Moreover, Kozlitina et al. [133], revealed also the presence of another loss of function variant in HSD17B13 (c.573delC, rs143404524) more common in African-Americans than in Hispanics or Caucasians, that seems to confer a protection against chronic liver disease, but the association between this variant and hepatic TG content has not been found. In a case-control study, Pirola et al. [134], reported that TA allele protects against histological steatohepatitis, ballooning degeneration, lobular inflammation and fibrosis in MAFLD patients. This data is even more confirmed in a very recent GWAS that demonstrated that the HSD17B13 protective effect is more relevant to development of steatohepatitis than progression of fibrosis [115]. Furthermore, the rs72613567 variant has been related to a reduced risk of elevated transaminases and HCC in 111, 612 individuals from the Danish general population and in 3, 315 European patients, respectively [135, 136]. The likely mechanism behind these genetic associations seems to be due to an increased concentration of phospholipids in the liver of carriers compared to noncarriers, that is coupled to a down-regulation of pro-inflammatory genes [137].
An interaction between HSD17B13 rs72613567 and PNPLA3 I148M has been reported. Indeed, the HSD17B13 TA allele seems to be able to mitigate the effect of PNPLA3 I148M variant on liver injuries, without affecting hepatic fat accumulation [129]. Furthermore, the lowering effect of HSD17B13 variant on transaminases is amplified in patients with high risk of fatty liver, such as in carriers of the PNPLA3 variant [135]. All these notions introduce the concept that the modulation of HSD17B13 in patients carrying PNPLA3 G allele may represent a potential therapeutic strategy against chronic liver diseases. However, the precise mechanism linking the HSD17B13 variant with inflammation and fibrosis remains uncharacterized.
Another variant that has been shown to be protective against liver damage is the rs4841132 G > A, which enhances the expression of PPP1R3B, involved in glycogen synthesis [121, 138]. Indeed, it has been reported to reduce the risk of MAFLD but at the same time may favor liver disease by facilitating glycogen accumulation [138, 139]. In addition, the minor A allele associated with increased aminotransferases and liver disease diagnosis [138]. Conversely, in a rodent model, hepatic genetic deletion of PPP1R3B significantly reduce glycogen synthase protein abundance, glucose incorporation into hepatic glycogen, total hepatic glycogen content and fasting plasma glucose compared to wild-type littermates [140]. Therefore, the overall impact of PPP1R3B variation on hepatic fat and progressive liver damage remains disputed. However, Dongiovanni et al. [139], demonstrated that PPP1R3B variant was associated with protection from steatosis and fibrosis, resulting into protection from HCC development in individuals at high risk of MAFLD, but not in the general population.
Several other less frequent variants may influence MAFLD pathophysiology and progression, by exerting an effect on a particular aspect of the disease. Overwhelming data demonstrated that IR is a key player in the development of MAFLD and progressive forms [141, 142]. Indeed, IR correlates with the severity of liver fibrosis [141], the main determinant of MAFLD prognosis [143], and fibrosis progression is observed in T2D patients with MAFLD, irrespective of NASH [144, 145]. Thus, genetic variants that impair insulin receptor (InsR) signaling activation may favor fibrosis development in MAFLD [146]. For instance, the loss-of-function rs1801278 (G972R) mutation in insulin receptor substrate (IRS1) and the gain-of-function mutation in the ectonucleotide pyrophosphatase/phosphodiesterase1 (ENPP1) 121Q genes are both associated with a marker reduction of insulin sensitivity, increased body weight, dyslipidemia and liver disease severity, in particular in terms of fibrosis [146]. Conversely, the rs2954021 polymorphism in the tribbles homolog1 (TRIB1), involved in glycogenesis, impacts on MAFLD onset and circulating TG [147].
Several mutations in genes governing hepatic lipid secretion and utilization have also been shown to be causative of inherited fatty liver. Indeed, variants within Apolipoprotein B (APOB), involved in VLDL assembly and release, have been associated with low circulating lipoproteins and in turn, severe hepatic fat deposition, that may favor the progression of liver injury until HCC [148, 149]. Similar findings have been observed even in presence of microsomal triglyceride transfer protein (MTTP) variations that cause VLDL retention [150]. Another component of VLDL particles, chylomicrons, and HDL cholesterol is the apolipoprotein C3 (APOC3). Two common promoter variants (APOC3 T-455C and C-482T) predispose to liver fat accumulation in Indian individuals, but not in other ethnic groups, suggesting that genetic factors influencing TG metabolism outside of the liver are less involved in the pathogenesis of progressive MAFLD [151–153].
More recently, Dongiovanni et al. [154], demonstrated that the proprotein convertase subtilisin/kexin type 7 (PCSK7) rs236918 G > C variant impacts on circulating lipids and liver damage in a large cohort of MAFLD patients, bridging atherogenic dyslipidemia with hepatic inflammation and fibrosis. In this cohort, the variant did not impact on hepatic fat accumulation overall, however stratifying patients for the presence of the PNPLA3 I148M allele, the rs236918 polymorphism seems to be also associated with more severe steatosis. In particular, in HepG2 cells carrying the PNPLA3 I148M allele in homozygosity, PCSK7 genetic deletion reduced lipogenesis, fat accumulation, inflammation and fibrogenesis, even after the exposure to a FFA challenge [154]. In addition, Huang et al. [155], identified a significant correlation between the minor C allele and a strong increase in fasting insulin levels and homeostatic model assessment for IR in response to high-carbohydrate diet consumption.
Furthermore, alterations in PCSK9, another member of the proprotein convertase subtilisin/kexin family, have been broadly associated with familial hypercholesterolemia [156], severe steatosis [157] and cardiovascular risk [158], as a consequence of its role in the modulation of low-density lipoprotein LDL uptake. PCSK9 expression is strongly influenced by nutritional status and its expression impressively decreases in mice after 24 h of fasting. On the contrary, PCSK9 mRNA levels are restored trough SREBP1c activation upon high carbohydrate refeeding or insulin stimulation [159]. Loss-of-function mutations in PCSK9 have been reported to reduce plasma LDL cholesterol, without altering hepatic fat content [160].
Another rare familiar cause of hepatic disorders is represented by the alterations of lysosomal acid lipase (LIPA) gene, that cause a lysosomal acid lipase (LAL) deficiency. LAL is responsible for the hydrolysis of cholesteryl esters, TG, and LDL particles into free cholesterol and fatty acids. Thus, its functional impairment induces the accumulation of these un-hydrolyzed compounds in hepatocytes, whereby favoring hypercholesterolemia, cardiovascular disease, hepatic steatosis and cirrhosis [161, 162].
Furthermore, even variants implicated in fatty acid uptake and metabolism, such as the rs56225452 in fatty acid transport proteins (FATP5) or the rs13412852 in Lipin1 (LPIN1), may influence IR, steatosis and ALT levels, by regulating the flux of FFAs to the liver [163, 164].
In the contest of fatty liver, a broad number of adverse events may precipitate liver injury towards end-stage conditions. Indeed, oxidative stress, ER and mitochondrial dysfunctions, innate immune inflammation [165], intestinal high permeability, dysbiosis and enhanced circulating gut-derived by-products [166], and HSCs activation [167] participate to the switching from simple steatosis to NASH and fibrosis.
The stronger association reported for genetic variants involved in the regulation of inflammatory response is provided by tumor necrosis factor α (TNF-α) and interleukin 28 (IL28) polymorphisms. The latter gene codifies for the interferon λ3/λ4 (IFNL3/4), and the rs12979860 CC variant has been widely related to elevated production of interferon λ3, but not to interferon λ4 [168]. Thus, interferon λ3 is responsible for the increased clearance of hepatitis C virus, and more severe NASH and fibrosis in MAFLD patients [169, 170]. In particular, it has been proposed, by Eslam et al. [171], a gene model, referred to as FibroGENE, based on the rs12979860 genotype, age, gender and the routinely assessed clinical and laboratory variables to predict significant fibrosis. The rs12979860 is in high linkage disequilibrium with the rs368234815 TT > δG in IFNL4. Carriers of the rs368234815 TT allele are characterized by more severe necroinflammation and fibrosis compared to non-carriers [172]. Opposite results have been observed in presence of the rs3480 A > G polymorphism in the fibronectin type III domain-containing protein 5 (FNDC5) gene, that encode irisin, a novel myokine, that mediated fibrogenic activation and collagen synthesis in HSCs [173, 174]. On the contrary, it has been described that the minor G allele is associated with more severe steatosis, likely by modulating irisin expression in MAFLD patients [174]. As well as, the rs2228603 variant in Neurocan, the rs12137855 SNP in lysophospholipase-like 1 (LYPLAL1), and the rs10883437 near the carboxypeptidase n subunit 1 have related to advanced liver damage in MAFLD subjects [175, 176].
Even more, several line of evidence indicates that the gut-derived fibroblast growth factor (FGF) 19 signal, mainly involved in the regulation of lipid and carbohydrate metabolism in response to nutritional status, is associated with metabolic diseases and MAFLD pathogenesis [177]. Dongiovanni et al. [178], recently reported that the rs17618244 G > A variant in the β-Klotho (KLB) gene, encoding the hepatic co-receptor of the FGF19 receptor (fibroblast growth factor receptor 4, FGFR4), leads to reduced KLB plasma levels, severe lobular inflammation, ballooning and fibrosis in obese MAFLD pediatric patients. Indeed, KLB reduced expression causes the over-expression of genes involved in lipotoxicity and inflammation [178]. Thus, therapeutic strategies aimed to ameliorate KLB levels may represent an attractive approach to MAFLD management.
Furthermore, the rs4374383 non-coding variant in the macrophage c-mer tyrosine kinase (MERTK) has been associated with a protection against fibrosis in both MAFLD and in viral hepatitis C, thereby down-modulating the expression of MERTK, a tyrosine kinase that initiates the removal of dying cells by phagocytes and that is involved in the activation of HSCs [179, 180]. According to these notions, it has been recently reported that MerTK cleavage in hepatic macrophages is reduced during the transition from simple steatosis to NASH, promoting in turn transforming growth factor β (TGF-β) release and HSCs activation [181].
During their proliferation and activation, the HSCs express Krueppel-like factor 6 (KLF6). Alternative splicing of the KLF6 gene associates with mild MAFLD and reduced fibrosis [182]. In addition, variants that predispose to hepatic iron depot formation, such as alterations in HFE and TMPRSS6 genes, are also correlated with more advance fibrosis in MAFLD patients [183].
Finally, the susceptibility to fibrogenesis and carcinogenesis is also influenced by cellular senescence and cell cycle arrest. Therefore, the rs762623 in cyclin dependent kinase inhibitor 1A (CDKI1A) which encodes the cellular senescence marker p21, was significantly associated with disease progression in MAFLD [184]. Likewise, telomerase reverse transcriptase (TERT) gene loss-of-function mutations associate with familial cirrhosis and accelerate HCC development [185].
Mitochondrial dysfunction is closely related to the pathogenesis of MAFLD so much so that in the past years the latter has been defined as a mitochondrial disease [186]. In the early stages of MAFLD, mitochondrial activity increases in response to hepatic IR and FFA accumulation. Sustained mitochondrial oxidative flux results in increased reactive oxygen species production associated with mitochondrial DNA damage, ER stress, tissue inflammation and cell death which may contribute to the progression to NASH and more advanced liver damage [187].
Common polymorphisms in genes regulating mitochondrial function have been associated with MAFLD development and to its progressive forms. The superoxide dismutase 2 (SOD2) gene encodes the antioxidant enzyme manganese superoxide dismutase. The rs4880 C47T polymorphism in the SOD2 gene results in a valine to alanine amino acid substitution in the signal region targeting the protein to the mitochondrial matrix, where it exerts its function and the T allele has been related to higher enzyme activity. Al-Serri et al. [188], by exploiting both case-control and intra-familial association studies found a consistent association between the rs4880 and advanced fibrosis in MAFLD patients providing a convincing evidence that mitochondria-derived oxidative stress is crucial for the development of fibrosing NASH. In keeping with these findings, Namikawa et al. [189], reported that 63 patients with biopsy-proven NASH had a higher incidence of the SOD2 T/T genotype compared to 150 healthy controls
The uncoupling protein 2 (UCP2) is involved in the regulation of mitochondrial lipid efflux and oxidative metabolism and its hepatic expression has been found to be increased in NASH patients leading to a proton leak and to a reduced redox pressure on the mitochondrial respiratory chain, thus exerting a protective role on liver damage progression [190]. Fares et al. [191], performed a case-control study in which 688 Italian patients with biopsy-proven NASH and 232 healthy controls were enrolled to investigate the association between the -866 G > A UCP2 promoter region polymorphism and the susceptibility to develop NASH. The authors found that homozygosity for the -866A UCP2 allele was protective towards NASH and was associated with higher hepatic UCP2 expression.
The uncoupling protein 3 encoded by the UCP3 gene is a mitochondrial proton carrier which uncouples the oxidative phosphorylation by increasing the proton leak of the inner mitochondrial membrane. The UCP3 transporter is considered to be protective against oxidative stress derived from-oxidation of fatty acids. Aller et al. [192], demonstrated that a non-coding variant in the promoter (-55C > T, rs1800849) of UCP3 was associated with IR, low adiponectin levels, moderate-severe steatosis and inflammation in obese patients with MAFLD.
Sirtuins (SIRTs) which are a family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases involved in cellular metabolism and UCPs may modify oxidative stress thereby affecting the risk of atherosclerosis and cardiovascular diseases. In a group of 1, 018 stroke-free subjects from the Northern Manhattan Study (NOMAS) who had high-definition carotid ultrasonography and a GWAS data available, the SIRT6 rs107251 and the SIRT5 rs12216101 were associated with an increased risk for carotid plaque, whereas T-carriers of the UCP5 SNP rs5977238 had a decreased risk [193]. However, the impact of genetic variations in SIRTs genes has not been deeply investigated in MAFLD patients who are highly susceptible to cardiovascular complication.
It’s only recently been described a novel common missense variant (rs2642438 A165T) in the mitochondrial amidoxime-reducing component 1 (MARC1) gene which encodes for the mitochondrial amidoxime reducing component 1, a molybdenum-containing enzyme. The A165T variant is located at the N-terminal domain which anchors the protein to the outer membrane of the mitochondria. The threonine to alanine amino acid substitution results in a truncating protein making the rs2642438 variant a loss of function mutation. The A165T variant has been associated with protection against all-cause cirrhosis, reduced hepatic fat content and lower levels of liver enzymes pointing to MARC1 as a potential therapeutic target for liver diseases although further studies are required to clarify its function and its role in oxidative stress regulation [194]. A schematic over-view of the main genetic risk factors involved in MAFLD onset and progression is represented in Figure 1 and in Table 1.
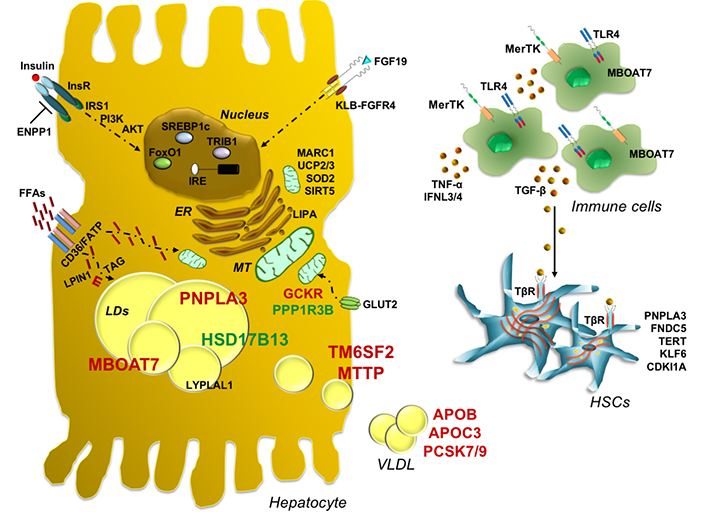
Genetic contribution to MAFLD pathogenesis and progression towards worsen stages. Schematic representation of the main genetic risk variants implicated in MAFLD onset and progression, which highlights their possible functional effects. PNPLA3 is an intracellular membrane lipase, localized on the surface of lipid droplets in hepatocytes where catalyzes TG hydrolysis. The PNPLA3 148M variant increases hepatic TG content upon accumulation of the mutant protein on the surface of lipid droplets, reducing TG turnover and dismissal. The I148M variant impairs the amount of released VLDL, exacerbating fat deposition. TM6SF2 is directly involved in VLDL secretion, whereas MBOAT7 catalyzes the transfers of PUFA, such as arachidonoyl-CoA to lysophospholipids, thus maintaining the fluidity of membranes. Thus, the TM6SF2 E167K variant impairs physiological VLDL secretion and affects cholesterol metabolism and TG synthesis, while the rs641738 variant in MBOAT7 reduces membrane fluidity and dynamism by altering phospholipid acyl-chains remodeling and lipogenic program in hepatocytes, while in immune cells it enhances the amount of free arachidonic acid, thus triggering inflammation. On the contrary genetic variants such as HSD17B13 and PPP1R3B may exert a protective effect against liver injuries. Furthermore, inherited variations that influence glucose and insulin signaling, FFA uptake, fat deposition and VLDL assembly and release may contribute to fatty liver development. Viceversa, hyperinsulinemia and elevated FFAs derived from adipose tissue exacerbate fat deposition induced by genetic modifiers, even activating de novo lipogenesis, through SREBP-1c. Common polymorphisms in genes regulating mitochondrial (MT) function have been associated with MAFLD development and to its progressive forms, further corroborating the switching from simple steatosis to NASH and fibrosis. In addition, it has been reported that genetic variants involved in the regulation of inflammatory response in immune cells and in the activation of HSCs to myofibroblasts phenotype may precipitate fatty liver to worsen conditions. Finally, the combined effect of PNPLA3, TM6SF2, and MBOAT7 genetic variants may predispose to an increased risk of progressive liver damage. PI3K: Phosphoinositide 3-kinase; FOXO1: Forkhead box protein O1; CD36: cluster of differentiation 36; TLR4: toll like receptor 4
List of the main genetic variants associated with the histological spectrum of MAFLD
| Variant | Gene | Function | Effect | Impact | Phenotype |
|---|---|---|---|---|---|
| rs738409 C > G | PNPLA3 | Lipid remodeling | p.I148M | Loss-of-function | ↑ MAFLD, NASH, fibrosis, HCC |
| rs58542926 C > T | TM6SF2 | VLDL secretion | p.E167K | Loss-of-function | ↑ MAFLD, NASH, fibrosis |
| rs641738 C > T | TMC4/MBOAT7 | Lipid remodeling | p.G17E | Loss-of-function | ↑ MAFLD, NASH, fibrosis, HCC |
| rs1260326 C > T | GCKR | Regulation of de novo lipogenesis | p.P446L | Loss-of-function | ↑ MAFLD, NASH, fibrosis |
| rs72613567 T > TA | HSD17B13 | Lipid remodeling | Truncated protein | Loss-of-function | ↓ NASH, fibrosis, HCC |
| rs4841132 G > A | PPP1R3B | Glycogen synthesis | Non-coding | Gain-of-function | ↓ MAFLD, fibrosis, HCC |
| rs1801278 A > C | IRS1 | Insulin signaling | p.G972R | Loss-of-function | ↑ Fibrosis |
| rs1044498 A > C | ENPP1 | Insulin signaling | p.K121Q | Gain-of-function | ↑ Fibrosis |
| rs2954021 G > A | TRIB1 | Regulation of de novo lipogenesis | Non-coding | Gain-of-function | ↑ MAFLD |
| rs12137855 C > T | LYPLAL1 | Lipid metabolism | Intronic | Loss-of-function | ↑ MAFLD |
| Several | APOB | VLDL secretion | Protein change | Loss-of-function | ↑ MAFLD, NASH, fibrosis, HCC |
| Several | MTTP | VLDL secretion | Protein change | Loss-of-function | ↑ MAFLD |
| rs236918 G > C | PCSK7 | Membrane transferrin receptor shedding and regulation of circulating lipids | Intronic | Gain-of-function | ↑ NASH, fibrosis |
| Several | PCSK9 | LDL uptake | Protein change | Loss-of-function | No evidence of association with steatosis |
| Several | LIPA | Lipid remodeling | Protein change | LAL deficiency | ↑ MAFLD, NASH, fibrosis |
| rs56225452 G > A | FATP5 | FFAs uptake | Non-coding | Gain-of-function | ↑ NASH, fibrosis |
| rs13412852 C > T | LPIN1 | Lipid metabolism | Intronic | NA | ↓ NASH, fibrosis |
| rs17618244 G > A | KLB | FGF19/FGFR4 pathway | R728Q | Loss-of-function | ↓ NASH, fibrosis |
| rs4374383 G > A | MERTK | Innate immunity | Intronic | Loss-of-function | ↓ Fibrosis |
| rs3750861 G > A | KLF6 | HSCs activation | Splice variant IVS1-27G | Loss-of-function | ↓ Fibrosis |
| Several | TERT | Telomere maintenance | Protein change | Loss-of-function | ↑ Fibrosis, HCC |
| rs12979860 C > T | IL28B | Innate immunity | Alternative IFNL3/4 transcription | Loss-of-function | ↓ NASH, Fibrosis |
| rs3480 A > G | FNDC5 | HSCs activation | Non-coding | Loss-of-function | ↓ Fibrosis |
| rs4880 C > T | SOD2 | Mitochondrial antioxidant | p.A16V | Loss-of-function | ↑ Fibrosis |
| rs695366 G > A | UCP2 | Mitochondrial lipid metabolism Oxphos | -866 promoter variant | Gain-of-function | ↓ NASH, fibrosis |
| rs2642438 G > A | MARC1 | Mitochondrial detoxification | A165T | Loss-of-function | ↓ MAFLD, NASH, fibrosis |
NA: not applicable
In the past years, single candidate gene studies and GWAS have contributed to pinpoint the role of genetic variants in MAFLD pathogenesis and progression. Similarly, to what has been done for other complex disorders, a recent approach is to combine individual loci into PRSs to convert genetic data into a measure of the risk of developing MAFLD. The aggregation of these scores with the most common risk factors may be helpful in identifying those patients at higher risk to develop more severe liver damage and to decide the more effective therapeutic strategy [195].
We firstly applied a mendelian randomization analysis and a PRS to demonstrate that hepatic steatosis promotes the full spectrum of liver disease and the impact of risk alleles in PNPLA3, TM6SF2, MBOAT7 and GCKR on liver damage is proportional to their effect on hepatic fat accumulation [28]. In keeping with these findings, Di Costanzo et al. [196], evaluated the impact of genetic and metabolic variables on liver fat accumulation in a cohort of obese children. They found that genetic variants contribute more strongly to fatty liver compared to IR which is a known driver of steatosis and fibrosis. Moreover, hepatic fat content variation was explained for 8.7% by metabolic factors and for 16.1% by the weighted effect of PNPLA3, TM6SF2, and GCKR variants [196]. Similarly, in a cohort of 2, 042 children Suomela et al. [197], demonstrated that a combined score including BMI, insulin levels and genetic variants in PNPLA3 and TM6SF2 genes was more accurate to predict fatty liver compared to the one used in adulthood based only on BMI and insulin.
Krawczyk et al. [106], analyzed the joint effect of PNPLA3 I148M, TM6SF2 E167K, and MBOAT7 rs641738 variants on liver damage in a large cohort of patients with fatty liver and demonstrated that increasing number of risk alleles was associated with higher aminotransferase, which may reflect hepatic injury in MAFLD. The additive effect of PNPLA3 and TM6SF2 gene variation seems to affect lipid metabolism and MAFLD possibly by upregulating the expression of genes involved in de novo lipogenesis [198]. Risk estimation of progressive MAFLD by PRS was also calculated in a Japanese study which demonstrated that the effect of the risk alleles, namely PNPLA3, GATAD2A and GCKR was cumulative and increased the risk of NASH [127].
Genetic variants predisposing to hepatic fat accumulation promote HCC development and may be useful biomarkers for patients’ stratification. We previously demonstrated that the number of genetic risk variants in PNPLA3, TM6SF2 and MBOAT7 was strongly associated with HCC, with a 13.4-fold higher risk in MAFLD patients carrying five risk alleles as compared to none [103]. Gellert-Kristensen et al. [199], have recently assessed that a PRS including the genetic variant in PNPLA3, TM6SF2 and HSD17B13 genes is associated with a 12-fold and 29-fold higher risk to develop cirrhosis and HCC respectively, in the general population.
All these studies raise the important question about the possibility to use these scores in the clinical setting to predict the development of MAFLD and its progression to more severe forms and how to combine them with the metabolic risk factors to apply the appropriate therapeutic intervention. The new challenge is to understand how to use the genetic architecture of MAFLD, which has been explored in the past years, to identify new druggable targets and to accelerate the MAFLD drug discovery pipeline [200].
Genetics plays a crucial role in the development of MAFLD and in its progression towards NASH and HCC. Inherited variant in PNPLA3, TM6SF2 and MBOAT7 genes provide the strongest evidence for association with MAFLD but it remains to be determined how genetic data may be translated in the clinical setting. Both PNPLA3 and TM6SF2 have been related with “metabolically silent” MAFLD thus contributing to its development and progression even in the absence of metabolic factors. Moreover, genetically determined fat accumulation is associated with biochemical markers of liver damage and higher risk of fibrosis. The opportunity to aggregate genetic variants in polygenic risk scores and to combine them with metabolic information represents an appealing therapeutic choice to design preventive intervention in higher risk individuals with MAFLD.
ALD: alcoholic liver disease
ALT: alanine aminotransferase
APOB: Apolipoprotein B
APOC3: apolipoprotein C3
ATGL: adipose triglyceride lipase
CGI-58: comparative gene identification-58
DAGs: diacylglycerols
ENPP1: ectonucleotide pyrophosphatase/phosphodiesterase1
ER: endothelial reticulum
FATP5: fatty acid transport proteins
FFAs: free fatty acids
FGF: fibroblast growth factor
FGFR4: fibroblast growth factor receptor 4
FNDC5: fibronectin type III domain-containing protein 5
GCKR: Glucokinase regulator
GWAS: genome-wide association study
HCC: hepatocellular carcinoma
HSCs: hepatic stellate cells
HSD17B13: hydroxysteroid 17-beta dehydrogenase 13
IFNL3/4: interferon λ3/λ4
IL28: interleukin 28
InsR: impair insulin receptor
IR: insulin resistance
IRS1: insulin receptor substrate
KLB: β-Klotho
KLF6: Krueppel-like factor 6
KO: knock-out
LAL: lysosomal acid lipase
LDL: low-density lipoprotein
LIPA: lysosomal acid lipase
LYPLAL1: lysophospholipase-like 1
MAFLD: metabolic associated fatty liver disease
MARC1: amidoxime-reducing component 1
MBOAT7: membrane bound o-acyltransferase domain-containing 7
MERTK: macrophage c-mer tyrosine kinase
MetS: metabolic syndrome
MRI-PDFF: magnetic resonance imaging proton-density fat fraction
MTTP: microsomal triglyceride transfer protein
NASH: nonalcoholic steatohepatitis
PCs: phosphatidylcholines
PCSK7: proprotein convertase subtilisin/kexin type 7
PNPLA2: patatin-like phospholipase domain-containing 2
PNPLA3: patatin-like phospholipase domain-containing 3
PPP1R3B: protein phosphatase 1 regulatory subunit 3B
PRSs: polygenic risk scores
PUFAs: polyunsaturated fatty acids
SIRTs: sirtuins
SNP: single nucleotide polymorphism
SOD2: superoxide dismutase 2
SREBP1c: sterol regulatory element-binding protein 1
T2D: type 2 diabetes
TERT: telomerase reverse transcriptase
TG: triglyceride
TGF-β: transforming growth factor β
TM6SF2: transmembrane 6 superfamily member 2
TMC4: transmembrane channel like 4
TNF-α: tumor necrosis factor α
TRIB: tribbles homolog1
UCP2: uncoupling protein 2
VLDL: very-low density lipoproteins
MM, ML and PD all took part in writing the manuscript, preparing figures and reading and approving the final draft. All authors have read and agreed to the published version of the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The study was supported by the Ricerca Corrente Fondazione IRCCS Cà Granda and Ricerca Finalizzata Ministero della Salute RF-2013-02358319. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Noel C. Salvoza ... Natalia Rosso
Amedeo Lonardo, Stefano Ballestri
Marvin Ryou ... Gyorgy Baffy
Michael Doulberis ... Stergios A. Polyzos
Ivana Mikolasevic ... Sandra Milic
Valerio Rosato ... Marcello Persico
Carlo Acierno ... Ferdinando Carlo Sasso
Rosa Lombardi ... Anna Ludovica Fracanzani
Angelo Di Vincenzo ... Marco Rossato
Angelo Colucci ... Claudia Mandato
