 Review
Review

Affiliation:
Department of Medicine, Division of Gastroenterology and Hepatology, University of Missouri, Columbia, MO 65212, USA
Email: nassirf@health.missouri.edu
ORCID: https://orcid.org/0000-0002-3653-3621
Explor Med. 2020;1:248–258 DOI: https://doi.org/10.37349/emed.2020.00017
Received: May 31, 2020 Accepted: July 24, 2020 Published: August 31, 2020
Academic Editor: Amedeo Lonardo, Ospedale Civile di Baggiovara, Azienda Ospedaliero-Universitaria di Modena, Italy
The article belongs to the special issue Exploring NAFLD/NASH
Nonalcoholic fatty liver disease (NAFLD) has become the most prevalent liver chronic disease worldwide. The pathogenesis of NAFLD is complex and involves many metabolic enzymes and multiple pathways. Posttranslational modifications of proteins (PMPs) added another layer of complexity to the pathogenesis of NAFLD. PMPs change protein properties and regulate many biological functions, including cellular localization, stability, intracellular signaling, and protein function. Lysine acetylation is a common reversible PMP that consists of the transfer of an acetyl group from acetyl-coenzyme A (CoA) to a lysine residue on targeted proteins. The deacetylation reaction is catalyzed by deacetylases called sirtuins. This review summarizes the role of acetylation in NAFLD with a focus on sirtuins 1 and 3.
Nonalcoholic fatty liver disease (NAFLD) has become the most prevalent liver chronic disease worldwide [1]. NAFLD consists of the accumulation of fat in the liver (steatosis) that might progress to nonalcoholic steatohepatitis (NASH), fibrosis, cirrhosis, hepatocellular carcinoma. NAFLD affects 25% of the global population; 1.5–6.5% is estimated to have NASH [2–5]. The pathogenesis of NAFLD is complex and implicates multiple factors, acting together in the development and aggravation of the disease [6, 7]. NAFLD has become a silent epidemic rising with the increase in obesity and insulin resistance [6, 8]. The mechanisms involved in NAFLD include the excessive accumulation of fat in hepatocytes, impaired mitochondrial function, and mitochondrial damage, increased intracellular fat, oxidative stress, inflammation, cellular damage, apoptosis, and activation of fibrosis [9, 10]. Apart from lifestyle modifications, no pharmacotherapy is currently approved to treat NAFLD despite the alarming health concerns. NAFLD therapies, currently being evaluated, target some aspects of metabolic enzymes and metabolic dysfunction. The discovery of posttranslational modifications (PTMs) of proteins (PMPs) added a new layer to the complexity of the mechanisms involved in NAFLD. PMPs represents a major protein diversification mechanism by which the cell increases the diversity of its proteome [11, 12]. PMPs involve modifications that change protein properties and regulate many biological functions, including cellular localization, stability, intracellular signaling, protein-protein interactions, and protein function [11–13]. Several PMPs have been identified, including acetylation, glycosylation, phosphorylation, palmitoylation, ubiquitination, succinylation, and prenylation. Here, we summarize the role of the deacetylases sirtuin (SIRT)1, and SIRT3 in NAFLD.
The liver plays an essential role in lipid metabolism [14]. The pathogenesis of NAFLD includes the accumulation of hepatic triglyceride as a result of increased flux of fatty acids (FAs) into hepatocytes, increased de novo lipogenesis (DNL), reduced export of lipids into lipoproteins, mitochondrial dysfunction, mitochondrial damage, oxidative stress and inflammation which lead to cellular damage, apoptosis, and fibrosis [9, 10]. Insulin resistance increases hepatic DNL, reduces adipose tissue insulin sensitivity, and increases the flux of FAs to the liver [15]. Lipid export capacity and the oxidation of fat in the mitochondria increase to compensate for the increased FAs flux to the liver [15–17]. However, mitochondrial dysfunction eventually occurs, likely due to increased lipids and impaired electron chain activity [10, 18, 19].
Acetylation of proteins is a key PTM, initially known to regulate transcription through modification of nuclear proteins [20, 21]. The role of lysine acetylation has been extended to thousands of non-nuclear proteins, including metabolic enzymes [20, 22–24]. Lysine acetylation regulates a wide range of cellular processes and metabolism [22]. Lysine acetylation consists of the addition of an acetyl group from acetyl-coenzyme A (acetyl-CoA) to a lysine residue within a protein. Large-scale proteomics studies identified over 3,000 acetylation sites [24–27]. More than 20% of mitochondrial protein is acetylated in mouse liver [26]. Using high-resolution mass spectrometry, studies by Choudhary et al. [25] identified acetylation as a major PMP comparable to phosphorylation. Acetylation regulates metabolic pathways, including the tricarboxylic acid (TCA) cycle and FA metabolism [24]. The acetylation status of metabolic enzymes appears to be responsive to environmental, dietary, lifestyle factors providing mechanisms for rapid changes in protein properties in response to the cellular environment [24, 28]. Acetylation of cellular proteins regulates their localization, stability, interaction with other proteins, and function [26, 29]. In addition to acetylation, a lysine residue in a protein can be subject to other PTMs, such as methylation, phosphorylation, ubiquitination, succinylation, and sumoylation. Modification of lysine residues by PTM may occur in a mutually exclusive manner suggesting an interplay between the different lysine modifications in the function of the regulatory protein [13, 30, 31].
Although lysine acetylation was discovered about 50 years ago, it has regained more attention in recent years. Not much is known about the specific changes in the acetylation profile with NAFLD. However, protein acetylation pattern in fatty livers is significantly different from healthy livers [32]. Studies on the role of acetylation and NAFLD are emerging. The transcription factor, cyclic AMP-responsive element-binding protein 3-like 3-hepatocyte-specific (CREBH), is one example of the modulation of NAFLD by acetylation [33]. CREBH regulates many metabolic pathways, including lipolysis, fatty acid oxidation (FAO), and ketogenesis [34]. Defects in CREBH might directly lead to NASH and hyperlipidemia under an atherogenic high-fat diet (HFD) or fasting conditions [34, 35]. The P300/CBP-associating factor (PCAF) and SIRT1 are the acetyltransferase and deacetylase, respectively, that regulate CREBH [33]. The acetylation of CREBH by PCAF at lysine 294 is required for CREBH transcriptional activity and for maintaining hepatic lipid homeostasis in fasting states [33]. The carbohydrate-response element-binding protein (ChREBP) is another example of the modulation of NAFLD by acetylation. In a high-glucose state, ChREBP is acetylated by p300 at lysine 672 [36, 37]. Acetylation of ChREBP induces hepatic lipid accumulation by increasing the expression of lipogenic genes (Figure 1). These studies suggest that lysine acetylation is associated with hepatic steatosis. Since most studies on acetylation and NAFLD focused on the role of the deacetylases SIRT1 and SIRT3, this review will mainly summarize the role of SIRT1 and SIRT3 in NAFLD.
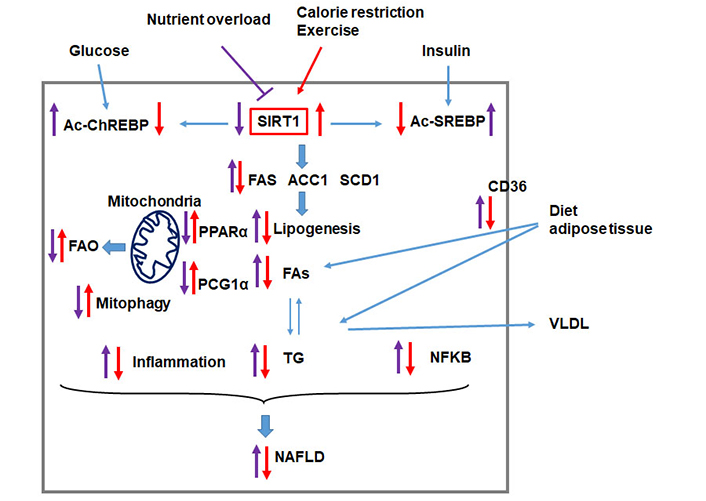
SIRT1 protects the liver from nutrient overload-induced NAFLD. Nutrient overload triggers metabolic reprogramming that leads to hepatic lipid accumulation and liver injury. Nutrient overload induces the hyper-acetylation of the transcription factors ChREBP (Ac-ChREBP) and SREBP (Ac-SREBP) leading to induction of lipogenic enzymes [FA synthase (FAS), acetyl-CoA carboxylase 1 (ACC1), and stearoyl-CoA desaturase-1 (SCD1)] and the peroxisome proliferator-activated receptors (PPARα)/peroxisome proliferator-activated receptor γ coactivator-1α (PGC1α) pathway leading to increased FAs in the liver reduced FAO. Nutrient overload also upregulates the FA transport protein CD36 and increases the uptake of FAs. Altogether, the reprogramming of these pathways leads to the accumulation of hepatic triglyceride (TG). On the other hand, lipid overload is associated with increased inflammation, reduced mitophagy, and induction of the NF-kappa B (NFKB) pathway, altogether leading to liver injury and the progression of NAFLD. Factors that upregulate SIRT1, such as calorie restriction and exercise, counteract the development and the progression of NAFLD. VLDL: very-low-density lipoprotein
An increasing number of studies have shown the regulation of metabolic enzymes by the SIRTs. Mammalian SIRTs (SIRT 1–7) are seven members belonging to the silent information regulator 2 family with different subcellular functions. Sirtuins regulate cellular proteins through various PTMs. SIRT1 and SIRT3 are protein deacetylases that act as a critical metabolic/energy sensor, which directly links the product of metabolism to cellular activities involved in metabolic homeostasis [38–41]. SIRT1 and SIRT3 regulate hepatic carbohydrate and lipid metabolism, insulin signaling, and inflammation [20, 42, 43]. SIRT1 and SIRT3 use the product of cellular nicotinamide adenine dinucleotide (NAD) as a cofactor to post-translationally deacetylate cellular proteins and consequently link the metabolic status of the cell to protein function. Changes in sirtuin expression are critical in several diseases, including metabolic syndrome, diabetes, and NAFLD. Here we focus on the role of the most commonly studied deacetylases SIRT1 and SIRT3 in the regulation of NAFLD.
SIRT1 regulates the acetylation of both histones and other cellular proteins. SIRT1 is expressed in the liver, pancreas, heart, muscle, and adipose tissue [44], and the protein is localized both in the nucleus and cytoplasm [44, 45]. SIRT1 plays an essential role in the pathophysiology of many metabolic diseases, including NAFLD [38, 46–49]. SIRT1 is downregulated in patients with NAFLD [43, 50]. SIRT1 has been shown to improve NAFLD in part through its effect on improving insulin sensitivity, its antihyperlipidemic, and its anti-inflammatory activities [47, 51]. Nutrition overload impairs SIRT1 function by reducing cellular NAD levels [52], while calorie restriction increases NAD levels and activates SIRT1 [53, 54] (Figure 1). Interestingly, late gestation and early postnatal life exposure to excess dietary fat increase the susceptibility to develop NASH in adulthood, and this was associated with reduced SIRT levels and altered expression of genes involved in NAFLD [55–57]. Treatment of mice fed HFD with the polyphenol resveratrol that activates SIRT1, improves lipid metabolism, and decreases NAFLD and inflammation in the liver [58]. In HFD-induced hepatic steatosis, SIRT1 improves NAFLD by reducing DNL and increasing β-oxidation [59] (Figure 1). SIRT1 activates lipogenic enzymes through SIRT1-mediated deacetylation of transcription factors in the promoter of downstream genes. Two major transcriptional factors, sterol regulatory element-binding transcription factor 1c (SREBP-1c) and ChREBP, control triglyceride synthesis [59, 60]. SREBP-1c and ChREBP activate lipogenic enzymes ACC1, FAS, and SCD1 (Figure 1). SIRT1 deacetylates and inhibits SREBP-1c activity [61]. siRNA-mediated depletion of SIRT1 increases the acetylation of SREBP-1c and activates lipogenic enzymes [61]. However, adenoviral overexpression of SIRT1 in the liver decreases acetylated SREBP-1c levels and lipogenic gene expression [61]. PPARα/PGC-1α signaling pathway plays a significant role in the oxidation of fat in the mitochondria [62]. PGC-1α is a transcriptional coactivator that induces the expression of PPARα target genes [62]. SIRT1 increases the transcription of PPARα and promote β-oxidation in the liver, mainly through its ability to deacetylate PGC-1α (Figure 1) [63–65].
Damaged mitochondria are degraded by selective autophagy, called mitophagy. SIRT1 plays an important role in mitochondria by regulating mitophagy [49, 66]. SIRT1 also mitigates HFD-induced fatty liver and liver injury by inhibiting the expression of the FA transport protein CD36 and the NF-KappaB signaling pathway (Figure 1) [67, 68]. The available findings so far demonstrate that SIRT1 plays a vital role in negatively regulating the development and the progression of NAFLD by acting on multiple cellular pathways.
SIRT3 is a NAD-dependent deacetylase localized in the mitochondrial matrix [44, 45, 69–71]. SIRT3 is a global regulator of mitochondrial protein acetylation with the capability to coordinate cellular responses to nutrient status and energy homeostasis [72–77]. SIRT3 is expressed in the liver, adipose tissue, heart, brain, and kidney [71, 73] and regulates β-oxidation, ketogenesis, mitophagy, and stress-related pathways (Figure 2) [74, 78–83]. The expression of SIRT3 is activated during fasting and calorie restriction [82, 84, 85], while chronic HFD and obesity reduce SIRT3 activity [86] (Figure 2). In the absence of SIRT3, mitochondrial proteins become hyperacetylated, mitochondrial function impaired, leading to NAFLD [76, 82, 87] (Figure 2). At least 65% of all mitochondrial proteins from the liver tissue of SIRT3 knockout (KO) mice have at least one lysine-acetylated [85]. SIRT3 regulates proteins of the mitochondrial electron transport chain such as complex I [87], succinate dehydrogenase A of complex II [88, 89], and ATP synthase (complex V) [90] and increase ATP production (Figure 2) [87]. Basal levels of ATP in the liver is reduced by 50% in mice lacking SIRT3 [87]. SIRT3 promotes β-oxidation by activating long-chain acyl CoA dehydrogenase (LCAD) activity and ketone body generation by promoting the deacetylation of 3-hydroxy-3-methylglutaryl CoA synthase 2 (AceCS2) [79, 82, 84, 85] (Figure 2). Fasting upregulates SIRT3, induces LCAD and increases its activity [82]. LCAD deficiency is also associated with reduced β-oxidation, increased lipid accumulation, and the development of insulin resistance and steatohepatitis in mice [91, 92]. Mitochondrial trifunctional protein (MTP) catalyzes the last three steps in β-oxidation. Heterozygous MTP KO (MTP+/–) mice have reduced SIRT3 levels, reduced β-oxidation, and develop fatty liver [74, 93] (Figure 2). Overexpression of SIRT3 in MTP+/– mice deacetylates MTP increases its hepatic levels and increased mitochondrial function [74] (Figure 2). Interestingly, the hydroxy acyl-CoA dehydrogenase (HADHA), the α-subunit of the MTP complex, was found to be highly regulated by SIRT3 [73].
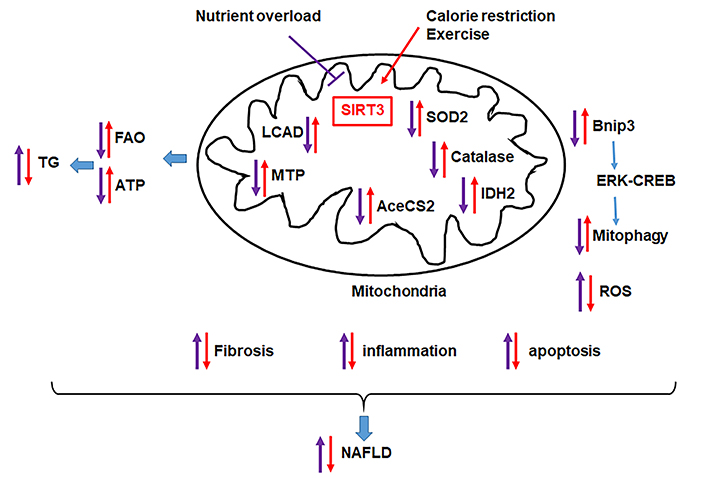
SIRT3 protects the liver from nutrient overload-induced NAFLD. Nutrient overload triggers metabolic reprogramming that leads to hepatic lipid accumulation and liver injury due to reduced mitochondrial function (FAO, ATP production), increased inflammation, reduced mitophagy. Factors that increase hepatic SIRT3, such as dietary restriction and exercise, counteract the effect of nutrient overload and improve NAFLD
SIRT3 also modulates oxygen consumption and reactive oxygen species (ROS) levels in hepatocytes [86, 94]. SIRT3 deacetylates superoxide dismutase (SOD2), which converts O–2 to H2O2 and enhances its ability to scavenge ROS [77, 95] (Figure 2). Consistently, SIRT3 KO mice exhibit decreased SOD2 activity and increased oxidative stress, suggesting a role of SIRT3 in NASH [95]. Moreover, SIRT3 activates isocitrate dehydrogenase 2 (IDH2), a crucial enzyme that promotes the restoration of antioxidant capability [96, 97] (Figure 2). SIRT3 KO mice subjected to a methionine and choline-deficient diet, a classical dietary model of NASH, exhibit increased hepatic lipid content associated with increased serum ALT levels, reduced SOD2 activity, and increased expression of inflammatory [TNFα, interleukin β1, inducible nitric oxide synthase (iNOS)] and fibrogenic [collagen 1 and alpha-smooth muscle actin (αSMA)] genes [98]. However, overexpression of SIRT3 results in opposite effects supporting an essential role of SIRT3 in NAFLD and NASH [99]. In addition, chronic HFD is associated with hepatocellular damage, and this is associated with reduced hepatic SIRT3 [94]. Interestingly, SIRT3 overexpression in mice restored hepatic function and reversed liver fibrosis, inflammation, and hepatocyte apoptosis [83].
Recently Cheng et al. [100] identified SIRT3 as a target for the differentially expressed microRNA-421 (miRNA-421) in HFD-fed mice compared with controls. miRNA-421 decreases SIRT3 and FOXO3 protein levels, as well as the downstream antioxidant targets SOD2 and catalase [100]. The progression of NAFLD in SIRT3 KO mice is associated with diet-induced mitochondrial damage [101]. SIRT3 overexpression protected hepatocytes against mitochondrial apoptosis via promoting Bnip3-required mitophagy [83] (Figure 2). Gut microbiota imbalance also contributes to the pathogenesis of NAFLD [8, 65]. Lack of SIRT3 results in an impaired intestinal permeability in HFD-fed mice through gut microbiota dysbiosis and inflammation [102]. Together the available finding indicates a protective role of SIRT3 in NAFLD and NASH.
NAFLD is a silent epidemic increasing with the increase in obesity. Apart from lifestyle modifications such as diet and physical activity, no approved pharmacological options currently exist to treat NAFLD. Regulation of proteins by acetylation mimics the effect of exercise and caloric restriction, the current recommended method for the management of NAFLD and NASH, with SIRT1 and SIRT3 having a protective role against the development and the progression of the disease. Proteins with differential acetylation levels could be a potential diagnostic marker and target for the treatment of NAFLD. Further studies are needed to determine the interaction between the different sirtuins and how this affects the metabolic pathways in the liver. The nature of the crosstalk between the different PMPs and their role in the modulation NAFLD is an area that needs more investigation. The discovery of selective and potent SIRTs activators and inhibitors is still in its early stages. More studies are needed to develop specific activators and inhibitors of SIRTs activity as well as agonists that target a particular cellular compartment.
acetyl-CoA: acetyl-coenzyme A
ChREBP: carbohydrate-responsive element-binding protein
CREBH: cyclic AMP-responsive element-binding protein 3-like 3-hepatocyte-specific
DNL: de novo lipogenesis
FA: fatty acid
FAO: fatty acid oxidation
HFD: high-fat diet
KO: knockout
LCAD: long-chain acyl CoA dehydrogenase
MTP: mitochondrial trifunctional protein
NAD: nicotinamide adenine dinucleotide
NAFLD: nonalcoholic fatty liver disease
NASH: nonalcoholic steatohepatitis
PMPs: posttranslational modifications of proteins
PPARα: peroxisome proliferator-activated receptors
PTM: posttranslational modification
ROS: reactive oxygen species
SIRT: sirtuin
SOD2: superoxide dismutase 2
SREBP-1c: sterol regulatory element-binding transcription factor 1c
TG: triglyceride
The author contributed solely to the work.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The University of Missouri Research Board grant to Fatiha Nassir; the funder had no role in the design and preparation of the manuscript, or decision to publish the manuscript.
© The Author(s) 2020.
Copyright: © The Author(s) 2020. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Noel C. Salvoza ... Natalia Rosso
Amedeo Lonardo, Stefano Ballestri
Marvin Ryou ... Gyorgy Baffy
Michael Doulberis ... Stergios A. Polyzos
Marica Meroni ... Paola Dongiovanni
Ivana Mikolasevic ... Sandra Milic
Valerio Rosato ... Marcello Persico
Carlo Acierno ... Ferdinando Carlo Sasso
Rosa Lombardi ... Anna Ludovica Fracanzani
Angelo Di Vincenzo ... Marco Rossato
Angelo Colucci ... Claudia Mandato
