Review
Review

Affiliation:
School of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, Jiangsu, People’s Republic of China
ORCID: https://orcid.org/0009-0004-9191-2247
Affiliation:
School of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, Jiangsu, People’s Republic of China
Affiliation:
School of Life Science and Technology, China Pharmaceutical University, Nanjing 211198, Jiangsu, People’s Republic of China
Email: zhengh18@hotmail.com
ORCID: https://orcid.org/0000-0002-1810-5842
Explor Drug Sci. 2026;4:1008161 DOI: https://doi.org/10.37349/eds.2026.1008161
Received: February 08, 2026 Accepted: April 01, 2026 Published: June 01, 2026
Academic Editor: Kamal Kumar, Smartbax GmbH, Germany
The article belongs to the special issue Discovery and development of new antibacterial compounds
The global rise of antimicrobial resistance (AMR) has emerged as one of the most pressing threats to public health. This crisis calls for the urgent development of alternative therapeutic agents against antibiotic-resistant pathogens. Antimicrobial peptides (AMPs) are widely present in nature, with a broad range of effects and a low risk of causing drug resistance. Therefore, they are an ideal choice for the development of the next generation of antimicrobial drugs. To overcome the inefficiencies of traditional AMP discovery, artificial intelligence (AI) and machine learning (ML) technologies have been increasingly used to predict and design AMPs. Multiple AMP databases were used to train ML models for predicting the activity of AMPs or generating AMP sequences. This review briefly provides a comprehensive overview of AMP databases and computational tools, highlighting their capabilities and challenges. Future work should integrate larger datasets and experimental validation to accelerate clinical translation.
Antimicrobial resistance (AMR) has escalated to a global public health emergency. The World Health Organization reports that antibiotic resistance directly caused 1.27 million deaths in 2021 and indirectly contributed to another 4.95 million deaths. Predictions suggest that without decisive intervention measures, the number of deaths due to drug-resistant infections could reach 10 million annually by 2050, surpassing cancer as the leading cause of death worldwide [1].
The development of traditional antibiotics has focused primarily on several major classes, such as β-lactams, macrolides, tetracyclines, aminoglycosides, and glycopeptides [2]. These constraints in structural diversity confine antibiotic mechanisms of action to a narrow spectrum, enabling bacteria to readily develop multidrug resistance through point mutations, enzymatic inactivation, or efflux pump activation [3].
Antimicrobial peptides (AMPs) are a class of small-molecule proteins naturally produced by organisms. They are an important part of the innate immune system and play a crucial role in the host defense mechanisms of all living forms, including microorganisms, plants, and animals [4]. The natural AMPs, due to their diverse mechanisms of action, high sequence diversity, and relatively low tendency to induce drug resistance, have provided a new solution path for the structural homogeneity problem of traditional antibiotics [5]. As of now, more than 60 kinds of AMPs have entered the clinical trial stage worldwide, among which 10 such substances, such as daptomycin and polymyxin B, have been approved for market release [6]. However, it is extremely difficult to conduct exhaustive screening for AMPs. Theoretically, a vast array of peptide chains with different sequences and spatial structures can be formed using no more than 25 natural amino acids [7]. As of 2024, the Database of Antimicrobial Activity and Structure of Peptides (DBAASP) v3 database only contains approximately 3.2 × 104 experimentally verified AMP sequences [8]. This significant disparity in quantity makes comprehensive experimental verification nearly impossible.
The development of artificial intelligence (AI) provides a potential way to solve this problem. Driven by breakthroughs in image recognition, natural-language processing, and computer vision, contemporary AI systems are uniquely positioned to resolve long-standing bottlenecks in pharmaceutical research, converting terabyte-scale datasets into actionable insight and compressing discovery timelines while elevating success rates across the entire drug-development pipeline [9]. AI has also delivered unprecedented efficiency in AMP discovery. For example, generative models jointly design peptide sequences and self-assembling water-rich hydrogel matrices under multi-objective optimization, yielding composites that rapidly eradicate drug-resistant pathogens while simultaneously orchestrating tissue regeneration [10]. By analyzing large amounts of natural AMP data, these models automatically learn design patterns and successfully guide the synthesis of novel AMPs that have been verified to be effective in experiments and animal models [11]. These breakthroughs demonstrate that AI can reliably design and optimize antibacterial molecules, thus providing a promising solution for combating drug-resistant bacteria.
This review systematically summarizes the application of AI and machine learning (ML) technologies in the discovery and design of AMPs, covering the latest advancements in related databases and computational tools, as well as their current challenges. The aim is to address the increasingly severe AMR crisis through efficient and intelligent computational methods, providing technical paths and theoretical support for the development of new generations of antimicrobial drugs.
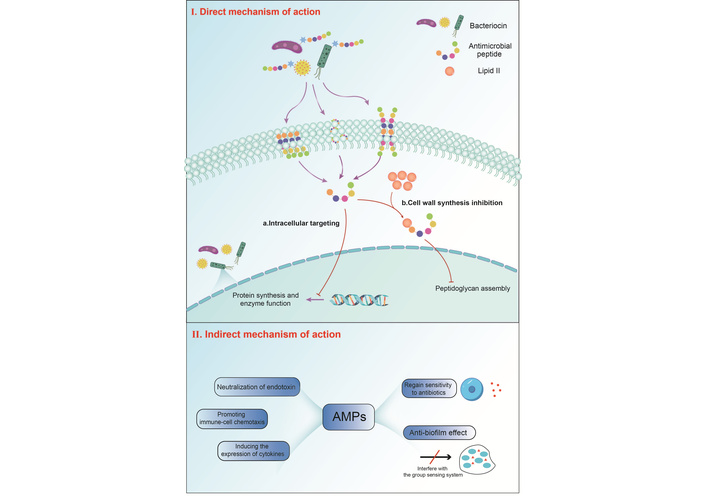
The mechanisms underlying bacterial resistance to traditional antibiotics encompass multiple strategies, such as the production of inactivating enzymes, modification of drug targets, reduced membrane permeability, enhanced efflux pump activity, biofilm formation, and entry into a dormant state (Figure 1) [12]. The distinct mechanisms of action between AMPs and traditional antibiotics position AMPs as a promising solution for the therapy of multidrug-resistant (MDR) pathogens [13], especially in the treatment of MDR pathogens such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE), and MDR tuberculosis (MDR-TB). The potential mechanisms of action of AMPs include disrupting bacterial cell membranes, inhibiting critical cellular processes, and modulating the host’s immune response. These mechanisms can be categorized into two main types: direct killing (encompassing both membrane-targeted and non-membrane-targeted actions) and immune modulation [14–16]. Cationic amphiphilic peptides (such as magainin, AMP from Xenopus laevis [PGLa], and LL-37) adopt a random coiling conformation in aqueous solution. When they bind to negatively charged bacterial membranes, they transform into α-helices parallel to the membrane surface. This “interface parallel” conformation causes the peptide’s polarity to face the aqueous phase and its hydrophobic side to insert into the lipid layer, resulting in local loosening of lipid chains and a thinning of the membrane, eventually forming transient pores or complete rupture [17].
AMPs exert their effects through multi-target mechanisms. They can disrupt bacterial cell wall synthesis (such as targeting lipid II) or fungal cell wall components, directly lysing microbial cell membranes through barrel-shaped pores, carpet models, or toroidal-pore model [18, 19]. The barrel-stave model shows that AMPs alter the conformation of bacterial membranes through electrostatic interactions and binding to the cell membrane, and insert into the phospholipid bilayer of the cell membrane in the form of polymers from a direction perpendicular to the cell membrane. Their hydrophobic sides face the acyl chains of the membrane outward, while the hydrophilic sides face inward to form pores or channels, thereby creating ion channels in the cell membrane. The carpet model was used to describe the action of AMPs such as dermaseptin [20]. The mechanism of action of this model: First, the AMP monomers combine with the phospholipid groups. Then, the AMP monomers are arranged successively on the bacterial membrane surface. Due to the combination of phospholipid groups with the peptide and the action of water, the orientation of the hydrophilic surface of the membrane changes, and a hydrophobic core is formed on the membrane. Finally, the AMP breaks the bacterial membrane [21]. The toroidal-pore model was first proposed by Yoneyama et al. [22]. The positively charged AMP was electrostatically captured by the negatively charged bacterial membrane. Its hydrophobic end inserted into the lipid bilayer, causing a breach in the hydrophobic center of the cell membrane, resulting in the positive curvature and stretching of the bacterial membrane, and further stretching of the breach further disrupted the integrity of the bacterial membrane.
For drug-resistant bacteria and biofilms, AMPs utilize positive charge adsorption and hydrophobic insertion to disrupt membrane structure [23], increase membrane permeability to restore antibiotic sensitivity [24], and target intracellular metabolic processes to achieve “internal and external synergy” killing [25, 26]. In addition, AMPs can also enter the interior of cells, inhibiting nucleic acid replication, protein synthesis, or interfering with key enzyme functions [27].
For example, LL-37, a cationic amphiphilic α-helical peptide, exerts its antimicrobial effect through multiple synergistic mechanisms, mainly including: membrane disruption and pore formation; its inhibitory effect on the palmitoyl transferase that maintains membrane integrity results in enhanced AMPs [28]; it has lipopolysaccharide (LPS) neutralization, biofilm inhibition by outer membrane penetration, and intracellular inhibition by non-membrane targeting [29]. Plectasin is a fungal peptide that achieves antimicrobial effects by inhibiting cell wall biosynthesis. Through in vitro experiments, it can be confirmed that plectasin exerts antimicrobial activity by directly binding to lipid II. Nuclear magnetic resonance spectroscopy and computational models further determined the key residues involved in this interaction [30]. Apidaecins I and II are derived from bees and are also a type of gram-negative bacterial AMP that does not damage cell membranes. Studies have shown that apidaecins competitively bind to the A site of the ribosome, specifically inhibiting translation termination related to the UAG stop codon and suppressing the release of newly synthesized proteins [31, 32]; P5 exerts antimicrobial effects on carbapenem-resistant Pseudomonas aeruginosa (CRPA) by disrupting the biofilm and promoting their death [24].
AMPs have significant immunomodulatory functions: They possess the functions of neutralizing endotoxins and regulating the local immune microenvironment [24]. They can also attract immune cells such as neutrophils and macrophages to the infection site, upregulate the expression of pro-inflammatory factors such as TNF-α and IL-6, and act as vaccine adjuvants to enhance adaptive immune responses [14–16]. Moreover, AMPs can effectively inhibit biofilm formation by interfering with the quorum-sensing system, inhibiting extracellular polymer synthesis, changing the morphology and adhesion ability of free bacteria, and significantly reducing the drug resistance of biofilms [33]. The multi-modal and low-resistance characteristics of these mechanisms make AMPs ideal candidate drugs for combating drug-resistant infections. For instance, cathelicidin BF has immunomodulatory activity against Pseudomonas aeruginosa and can significantly alleviate the symptoms of pneumonia caused by it [34]; human α-lactalbumin made lethal to tumor cells (HAMLET) causes MRSA to lose resistance to methicillin and VRSA to lose resistance to vancomycin by depolarizing the bacterial membrane and dissipating proton dynamics [35].
The core technologies used in the research on the AMP mechanism: Experimental methods such as ARGO assay [36] and NMR [37] provide high-resolution complex structure and affinity data, and reverse-verify the computational predictions, forming a closed research system from atomic simulation to in vivo validation; molecular dynamics (MD) simulations achieve the revelation of the interaction between AMPs and cellular components, especially the interaction with cell membranes, by simulating the movements of atoms and molecules. This enables the understanding of the transition states during pore formation and the mechanism of AMPs’ action [38]. MD simulations through software such as GROMACS track the dynamic trajectories of AMP insertion into lipids, binding to DNA, or filamenting temperature-sensitive mutant Z (FtsZ) at the atomic scale, enabling real-time observation of pore formation or enzyme inhibition details. For instance, in the 2025 study by Tian et al. [39], MD simulation was used as the core validation step in the screening of AMPs. Firstly, nine candidate peptides selected by AI reached an equilibrium state in the solution (MD1). Subsequently, they were placed outside the bacterial membrane model composed of DMPC/DMPG (3:1) and observed for their adsorption behavior through a 2-microsecond simulation (MD2). Additionally, a two-microsecond simulation of a four-fold system (MD3) revealed that only AMP-12 and AMP-48 could spontaneously insert into the membrane and induce the formation of stable water channels (with over 500 water molecules present in the membrane for more than 50% of the time), while the other seven peptides only adsorbed on the membrane surface. The reliability of the screening strategy was verified using positive control (PGLa) and negative control (nAMP1) [39]. DTBind, proposed by Li et al. [40] in 2025, is a successful example of the systematic integration of AI with computational chemistry methods. AI automatically learn the nonlinear relationships between features to capture the deep patterns in sequence and structure, while computational chemistry provides physical priors such as geometric invariance description, surface morphology analysis, and interaction type identification to achieve unified and accurate prediction of drug-target binding occurrence, site, and affinity [40]. Quantitative structure-activity relationship (QSAR) and deep learning [DL; random forest (RF), AntiBERTy, etc.] convert peptide sequences into thousands of dimensional descriptors such as hydrophobicity, charge, and secondary structure, establish activity-to-toxicity prediction models, and achieve millions of virtual screenings; bioinformatics tools and platforms such as APD3, AutoDock, etc., complete sequence alignment, homology modeling, and molecular docking, quickly identifying potential targets [16]; through homology modeling, the three-dimensional structure of AMP is constructed using tools such as SWISS-MODEL or MODELLER [41].
The global spread of bacterial resistance to conventional antibiotics has spurred the research and development (R&D) of novel anti-infective agents, in turn accelerating the discovery of AMPs. Concurrently, the integration of ML and DL into pharmaceutical R&D workflows has further expedited the expansion of curated information within AMP databases. The AMP database is divided into two main categories: the general database and the specific database. The general database includes all types of AMPs, without considering peptide families with specific properties. The specific database covers information related to a certain type of AMP (such as only anti-parasitic peptides or bacteriocins) or includes a certain supergroup of AMPs (such as only anti-parasitic peptides or only bacteriocin peptides) [42].
Among these databases, tools for predicting the three-dimensional structure of peptides are often included (such as AlphaFold or ESMFold). They often play a role in accurately predicting the three-dimensional structures of various biomolecular complexes, such as proteins, nucleic acids, small-molecule ligands, ions, and modified residues, within a unified DL framework [43]. Protein structure prediction models based on DL use deep neural networks (DNN, especially architectures such as Transformer) to automatically learn complex evolutionary patterns and physicochemical constraints from the amino acid sequence of proteins, so as to achieve high-precision prediction of their three-dimensional spatial structure [44]. However, these tools for dealing with AMPs are limited. Due to the flexible C-terminal tail that can freely swing to insert and penetrate the cell membrane, the diffusion model of short peptide structures with antimicrobial activity lacks the necessary physical confinement mechanism when working with engineered alternately folded systems with intrinsic conformational ambiguity, resulting in frequent unphysical atomic overlap in the prediction results [45, 46]. At the same time, it also has limitations in producing “illusion” structures in disordered regions, failing to capture the dynamic behavior of molecules, and requiring a large number of samples to obtain high-precision predictions for some complex targets [43]. Thus, dynamic simulation needs to be taken into consideration.
In this article, we list several comprehensive AMP databases and specialized AMP databases (Tables 1 and 2).
| Name | Sequence | Update the year of the current version | Remarks | Reference |
|---|---|---|---|---|
| AMPDB v1 | 59,122 | 2023 | Basic information, physical and chemical properties, activity information | [49] |
| dbAMP 3.0 | 35,518 | 2025 | Sequence information, functional activity data, physicochemical properties, and structural annotations | [50, 51] |
| DRAMP 4.0 | 30,260 | 2025 | Antimicrobial activity data, structure information, hemolytic and cytotoxic information | [52–55] |
| DBAASP v3 | > 15,700 | 2021 | Sequences, chemical modifications, 3D structures, bioactivities, and toxicities of peptides | [8, 56] |
| LAMP2 | 23,253 | 2020 | Primary structure, collection, composition, source, and function | [57] |
| CAMPR4 | 24,243 | 2023 | Sequences, source organisms, target organisms, minimum inhibitory, hemolytic concentrations, N- and C-terminal modifications, and presence of unusual amino acids | [58, 59] |
| APD | 5,188 | 2026 | Structure, activity, stability, synergistic effect, toxicity, animal models, and recombinant expression systems | [60–62] |
| YADAMP | 2,133 | 2012 | Explicit presence of antimicrobial activity against the most common bacterial strains | [63] |
| InverPep | 702 | 2017 | Including InverPep code, phylum and species source, peptide name, sequence, peptide length, secondary structure, molar mass, charge, isoelectric point, hydrophobicity, Boman index, aliphatic index, and percentage of hydrophobic amino acids | [64] |
Specialized antimicrobial peptide (AMP) databases.
| Name | Sequence | Description | Update year of the current version | Remarks | Reference |
|---|---|---|---|---|---|
| PlantPepDB | 3,848 | AMPs derived from plants | 2020 | Activity, hemolytic activity, cytotoxicity | [65] |
| ACovPepDB | 518 | Anti-coronavirus peptides | 2022 | Organism, activity, mechanism | [66] |
| AVPdb | 2,683 | Antiviral peptide | 2014 | Organism, activity, mechanism | [67] |
| HIPdb | 981 | HIV inhibiting peptides | 2013 | Organism, activity, mechanism | [68] |
| DRAVP | 5,688 | Antiviral peptide | 2023 | Antiviral activity, structure information, physicochemical information, and literature information | [69] |
| DCTPep | 6,214 | Cancer therapy peptides | 2024 | Tumor active peptide, cancer targeted peptides, targeted peptide conjugates, and collection of targeted peptides | [70] |
Our laboratory has constructed DRAMP, a comprehensive open-access database of AMPs dedicated to accelerating the clinical translation of AMPs. The version 4.0 contains 30,260 entries (8,001 new additions compared to the previous version), with core innovations and data integration including: the first introduction of a stability dataset, 110 newly added verified serum/protease stability and half-life data; the expansion of clinical entries to 96, integrating repetitive sequences and associating clinical trial numbers; 2,891 new hemolytic activity and 2,674 new cytotoxicity experimental data, systematically marking toxicity bottlenecks; the addition of an “Expanded AMPs” subset (179 entries) including experiments verified by Chinese journals not indexed in international databases; the number of experimentally verified structures (PDB) increased to 419, and the number of predicted structures reached 1,570. The database provides categorized downloads, covering 70.47% of unique sequences (18,580 entries not found in APD/DBAASP/CAMP). The database can be accessed at: http://dramp.cpu-bioinfor.org/ [52–55].
dbAMP is dedicated to addressing the increasingly severe AMR challenges in the post-pandemic era. This database has significantly expanded its data scale and functional depth through continuous updates. The latest version (dbAMP 3.0, 2025) integrates data from over 5,200 scientific papers, including 33,065 AMPs and 2,453 antimicrobial proteins, covering 3,534 source organisms, representing a nearly threefold increase from the initial version in 2019 (12,389 entries). The core breakthrough lies in the use of ESMFold to predict the three-dimensional structures of over 30,000 AMPs and to provide contact maps and secondary structure information, which greatly facilitates structural-function research. To accelerate clinical translation, the platform has added an AMP rational design pipeline, using multi-objective optimization algorithms (balancing antimicrobial activity, low toxicity, and stability) to assist in the development of new antibacterial peptides. At the same time, dbAMP integrates over 20 prediction tools and benchmark data set portals, becoming the core hub for AMP discovery, mechanism research, and clinical design. The database can be accessed at: https://awi.cuhk.edu.cn/dbAMP/ [50, 51].
DBAASP is currently the most comprehensive resource library for antimicrobial and cytotoxic peptides, containing over 15,700 entries, including > 14,500 monomers and nearly 400 homo- and hetero-multimers. Its core advantage lies in integrating multi-dimensional data, covering more than 2,700 ribosomal synthesis peptides, over 170 non-ribosomal peptides, and more than 12,000 synthetic peptides, including various modifications such as linear, cyclic, and disulfide bonds; providing experimental activity data for 4,200+ targets [such as minimum inhibitory concentration (MIC)/IC50], as well as hemolysis/cytotoxicity indicators; generating 3,200+ peptide conformational trajectories through an automated MD process and correlating with 400+ PDB experimental structures, supporting online visualization and analysis; the newly added PAASS tool can predict the activity of peptides against specific pathogens, and expands the calculation of physicochemical properties. As a key platform for antibacterial peptide design, DBAASP v3 promotes the rational development of new anti-infective peptides through high-precision experimental data, dynamic structure simulation, and ML tools. The access address is: http://dbaasp.org [8, 56].
AVPdb is a specialized database dedicated to experimental verification of antiviral peptides (AVPs), containing 2,683 peptide sequences targeting over 60 medically significant viruses (such as influenza, HCV, HSV, etc.), including 624 chemically modified peptide segments. This database provides detailed experimental information for each peptide, including sequence, source, target virus, inhibitory activity, mechanism of action, cell line, detection method, and complete PubMed literature reference. Additionally, AVPdb integrates various analysis tools, such as BLAST alignment, physicochemical property calculation, and structure prediction, enabling users to browse, search, and download data. This resource aims to promote AVP research and drug development, and is publicly accessible at: http://crdd.osdd.net/servers/avpdb [67].
PlantPepDB is a comprehensive database focusing on plant-derived active peptides, which includes 3,848 peptide sequences from 443 plant species, covering 9 major functional categories, such as antimicrobial, anti-cancer, antiviral, anti-hypertension, immune regulation, and many other biological activities. This database integrates data from 11 existing databases and 835 research papers. Each peptide entry provides detailed sequence, source plant, functional description, experimental verification level (protein level, transcriptional level, prediction or homology inference), physicochemical properties, tertiary structure prediction, and dictionary of protein secondary structure (DSSP) secondary structure report. The database offers multiple search methods and integrates analysis tools such as BLAST, Smith-Waterman alignment, and peptide mapping, enabling users to perform function prediction, structure analysis, and target screening. The database also places particular emphasis on data deduplication and information integration, merging duplicate peptide entries from different sources into a single record with more comprehensive information. The database is publicly accessible at: http://www.nipgr.ac.in/PlantPepDB/ [65].
DCTPep is an open-source comprehensive database specifically designed for cancer therapeutic peptides. It contains a total of 6,214 entries, including 6,106 peptide sequences and 108 peptide drugs. This database not only encompasses traditional anti-cancer peptides (ACPs) but also integrates information on cell-penetrating peptides with targeting functions, cancer-targeting peptides, and peptide-drug conjugates. It provides detailed annotations such as activity, targets, physicochemical properties, and three-dimensional structures predicted by AlphaFold. The database covers data on over 60 targets and more than 400 cancer cell lines. Its purpose is to offer valuable resources for the design, screening, and mechanism-of-action research of ACPs. The database is publicly accessible at: http://dctpep.cpu-bioinfor.org/ [70].
In evaluating AMP databases, it is typically necessary to establish comparative benchmarks across multiple dimensions. First, data scale and source diversity refer to the total number of peptides included in the database and whether it encompasses various types, such as natural and synthetic peptides. Second, data uniqueness and functional richness are crucial, as low-redundancy, independent datasets are essential for preventing model overfitting. Third, annotation depth and comprehensiveness extend beyond basic sequence information to include details such as source species, three-dimensional structures, post-translational modifications, MIC data, toxicity profiles, and mechanisms of action, which directly determine the research value of the database. Additionally, data update frequency and maintenance status are significant indicators, with databases that undergo continuous updates and data corrections, such as APD, being more reliable. Finally, the integration of functional tools and their utility—including retrieval, design capabilities, and support for emerging technologies such as ML predictions—forms a critical dimension for assessing the applicability of a database. Key aspects include whether it offers tools for BLAST alignment, sequence search, physicochemical property calculation, and even ML-based predictive capabilities [71].
Take dbAMP and DBAASP mentioned above as example. At the basic data level, dbAMP leads significantly over DBAASP with its scale advantage of 35,518 peptide sequences (including 33,065 AMPs) and 3,534 source organisms, compared to DBAASP’s over 15,700 records. In terms of annotation depth, the former categorizes 58 types of functional activities and fully annotates MIC efficacy data, while the latter provides a unique negative example resource for ML by including experimentally verified “inactive” peptides. In the aspect of structural information processing, dbAMP adopts the cutting-edge protein language model (pLM) ESMFold to complete high-throughput 3D structure prediction for over 30,000 peptides, with a prediction speed approximately 50 times faster than AlphaFold2, while DBAASP uses a classic computational path to provide high-precision 400 ns MD simulation trajectories for over 3,200 peptides. In terms of technical adaptability: dbAMP comprehensively utilizes generative AI, not only introducing GPT-4 to assist in literature mining but also integrating AMPActiPred based on deep forests, HemoFinder for hemolytic toxicity prediction, and a multi-objective peptide optimization platform based on genetic algorithms; DBAASP, on the other hand, focuses on specific strain activity prediction models (PAASS) and DiSAAP statistical analysis tools, and ensures the biophysical authenticity of simulations through the CHARMM36m force field. In summary, dbAMP is more suitable for large-scale structure-function mining and AI-driven peptide design optimization, while DBAASP has advantages in targeted strain prediction and sequence pattern statistics [8, 50].
Genome mining refers to the analysis of sequenced microbial genomes to identify biosynthetic gene clusters (BGCs) that may encode novel AMPs or secondary metabolites. Most of the data in the database are derived from genome mining, and the main research subjects are actinomycetes and fungi. Mining strategies can be divided into two categories: one is to identify BGCs based on conserved biosynthetic enzymes such as non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS); the second is to combine algorithms to predict precursor peptides and their post-translational modifications. To achieve these strategies, researchers have developed a variety of specialized tools, such as BAGEL, the earliest tool for mining bacteriocins; antiSMASH, the most comprehensive tool that supports the identification of 81 BGCs; ThioFinder, which specializes in identifying thiopeptides; RODEO, which combines ML to identify lasso peptides; Ripp-prism, which can predict 21 classes of ribosomally synthesized and post-translationally modified peptides (RiPP) structures and BGCs, and RiPPMiner, which can predict RiPP classes and cleavage sites based on ML. The successful application of these tools has led to the discovery of a large number of new AMPs, such as stambomycins from Streptomyces ambofaciens and medipeptin A from Pseudomonas mediterranea. The strategies of genome mining have evolved from the early reliance on BLAST homology search to the identification based on conserved modifying enzymes, and now to the intelligent development process that combines ML and DL to achieve automated and high-throughput predictions, enabling the discovery of high-throughput and culture-independent AMPs [72–75].
The establishment of AMP databases has significantly facilitated the development of computational prediction methods for AMPs. Certain AMP databases are equipped with predictive functionalities. For instance, the CAMPR3 database provides a suite of prediction tools that allow users to submit any peptide sequence for evaluation. Using ML models such as support vector machine (SVM) and RF, the system predicts whether the peptide exhibits antimicrobial activity. It can also identify localized antimicrobial regions (e.g., functional domains) within longer peptide sequences, or predict potential AMPs based on family-specific characteristics using publicly available data [58]. Similarly, DBAASP supports predictive analysis based on its database content. Its PAASS tool enables the prediction of antimicrobial activity of user-submitted peptide sequences against specific microbial strains (including Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 25923, and seven pathogenic organisms in total), allowing for strain-level targeting. Another tool, PGA, predicts whether a peptide sequence possesses broad-spectrum antimicrobial activity against a comprehensive set of microbial targets [8]
The computer-based prediction of AMPs using established known peptides was first introduced by the APD database in 2004 [61]. Later, in 2007, methods such as artificial neural network (ANN), quantitative matrices (QM), and SVM were developed based on the APD dataset [76]. Recently, DL methods and large language models (LLMs) have also been applied to the discovery of AMPs.
Since 2007, a wide range of different ML algorithms have been applied in AMP classification and prediction models. The ML methods successfully applied to the modeling of AMPs include decision tree (DT) [77–79], K-nearest neighbor (K-NN) [79, 80], naive bayes (NB) [81, 82], eXtreme Gradient Boosting (XGBoost) [82], SVM [79, 82], RF [79, 82, 83], neural network (including its DL variants) [84], and many other methods. Many AMP prediction models, such as CalcAMP [85], AMPpred-EL [86], PTPAMP [87], etc., have utilized these ML models. Next, we will introduce several of them.
The utility of DTs in AMP discovery primarily stems from their high interpretability and efficient processing of structured features. Through the three-step learning rules of feature selection, tree generation, and pruning, they are used in the detection of AMPs for classifying active peptides and non-active peptides. Their advantages include strong interpretability, which can intuitively display the key discriminative features and corresponding hypotheses. For example, the ADTree model, by identifying key discriminative features such as specific dipeptide composition, cationic amino acid content, and alpha-helical structure, can accurately pinpoint potential bacteriocins. The clear rules it generates directly inform the rational design of novel AMPs with desired properties [77].
K-NN is a distance-based classification method that relies on the similarity between the feature vectors of peptide sequences in AMP prediction. Since K-NN does not require explicit training, researchers can quickly assess which category of peptides a new sequence is closer to in the database. For example, iAMP-2L uses fuzzy K-NN (FKNN) for multi-label functional prediction, by calculating the similarity between the input sequence and the known AMPs in the training set to determine whether it has antimicrobial activity and other functions [80]. The advantage of K-NN is that it does not require a training process; however, when the feature dimension increases, the “dimension curse” causes the distance measurement to lose its discriminatory power, and the computational cost also grows linearly with the data size.
DL in AMP prediction mainly falls into four categories: basic models, language models, graph neural networks (GNN), and mixed and multimodal models [88].
The basic models mainly include DNN, convolutional neural networks (CNN), recurrent neural networks (RNN), their variants (LSTM, BiLSTM, GRU, etc.), and other hybrid models. CNN is adept at extracting local conserved motifs using multi-scale convolution and can effectively capture local sequence patterns, making it suitable for sequence encoding; DNN, which is a multi-layer fully connected network, is suitable for the fusion and classification of various manual features (such as amino acid composition, dipeptide composition, physical and chemical properties), and achieves this by globally optimizing the network parameters through minimizing the selected loss function. This is similar to CNN. Many works have implemented AMP prediction. For instance, Ma et al. [89] employed DL tools, including DNN, to mine and predict novel AMPs from human gut metagenomic data on a large scale. Subsequent experimental validation confirmed the antimicrobial activity of over a hundred of these predicted peptides, demonstrating the powerful application of DNN models in the field of AMP discovery [89]. RNN captures long-range dependencies between amino acids through a recurrent mechanism, but has a slower training speed and requires a large amount of data. It is often cascaded with CNN to form the CNN-BiLSTM architecture, and is supplemented by transfer learning, multi-task learning, or stacking integration to address the issues of insufficient data and model robustness [90]. DL variants (such as CNN, LSTM, DNN) simulate the processing of complex patterns by human brain neurons. In 2022, the research integrated LSTM, Attention, BERT, and other natural language processing (NLP) models. The prediction accuracy of AMP exceeded 91%, and the AUPRC reached 0.9244. Moreover, it does not require sequence homology or amino acid composition information and can discover new peptides with a homology lower than 40%, significantly reducing false positive (FP) [89]. TriNet further integrates sequence fingerprints, position-specific scoring matrix (PSSM) evolutionary information, and physicochemical properties with a three-layer network. It introduces the training-validation interaction (TVI) mechanism and supports hyperparameter tuning to enhance the generalization ability of ACP/AMP predictions [84].
Currently, such prediction models generally encounter the following common bottlenecks: Firstly, the models are highly dependent on known databases for training, which limits their generalization ability and makes it difficult for them to identify sequences with low homology and novel categories. Moreover, the training data is mostly concentrated on short peptides, resulting in poor prediction performance for long sequences. Secondly, most models only learn based on amino acid sequences and lack the integration of key physical and chemical information, such as protein spatial conformation, dynamic behavior, and interactions. Finally, the selected candidate molecules often have toxicity issues, and the experimental verification cost is high, which limits their efficient transformation from computational prediction to practical application [84, 89].
The role of the language model is to use large-scale unsupervised pre-training to transform amino acid sequences into semantic-rich context vectors, and then perform lightweight fine-tuning to achieve performance even surpassing that of traditional feature engineering. This includes BERT and its variants, pLMs, and peptide-specific lightweight models, etc. BERT and its variants obtain context-sensitive sequence embeddings through large-scale masked language modeling and encode and extract features for AMPs [49, 91], significantly reducing the need for labeled data and improving prediction performance, and can further fine-tune the input for other models to adapt to AMP classification, functional annotation, and even design tasks. pLMs are inspired by BERT and capture complex dependencies in protein sequences through self-supervised learning and multi-task pre-training, such as Diff-AMP [92] and AMP-BERT [93], etc. In 2023, Lobo et al. [94] used a concise and transferable “pre-training and fine-tuning” pipeline for rapid prediction of the antifungal activity of peptide segments, and ultimately found that the combination of SeqVec + k-Best feature selector + support vector classification (SVC) had the best effect. Peptide language models draw on the idea of pre-trained language models in NLP, are specifically designed for protein or peptide sequences, but need to be combined with structural biology and efficient algorithms to solve the key bottlenecks in the design of AMPs [88].
In recent years, the Transformer neural network architecture has demonstrated significant application value in the field of bioinformatics. Originally proposed by Vaswani et al. [95], its core self-attention mechanism effectively captures long-range dependencies in sequences, providing new technical approaches for protein and peptide sequence analysis.
In protein sequence representation learning, the evolutionary scale modeling (ESM)-2 model developed by Lin et al. [96] employs self-supervised pre-training strategies to learn semantically rich embeddings from large-scale protein sequence data. Similarly, the ProtT5 model proposed by Elnaggar et al. [97] utilizes advanced pre-training methods and has achieved excellent performance in various protein-related tasks.
In the field of drug discovery, the Transformer architecture also plays an important role. The PepMLM model developed by Chen et al. [98] employs masked language modeling and fine-tuning techniques to enable effective prediction and design of peptide sequence functions. This model can adapt to specific biomedical application scenarios with minimal labeled data, demonstrating good practicality and generalization value. However, such models often encounter the problem of a limited experimental verification scope during the experiment. The selection process may still have a relatively high FP rate. Leveraging features from larger-scale sequence datasets can improve AMP classification performance, but when it comes to model size, bigger isn't always better. Moreover, incorporating structural information can greatly enhance screening efficiency [98, 99].
The core idea of GNN in the prediction of AMPs is: treating the atoms/residues/fragments of the peptide as nodes, and establishing edges using chemical bonds, field atomic information, or spatial adjacency relationships to construct a graph structure, converting the sequence into a structural graph. GNN aggregates neighbor information through message passing to simultaneously capture sequence order and three-dimensional/topological features, featuring direct peptide graph representation, hierarchical graph representation, and alternative representations leveraging DL. It can incorporate physical-chemical properties (charge, hydrophobicity) as node or edge features, but the structural prediction error will gradually increase with each level or due to reasons such as over-smoothing [88, 100, 101]. For example, graph neural network models such as PNAbind can be used for high-throughput annotation of nucleic acid binding functions of unknown proteins, which can complete local prediction at the level of a single residue and global prediction at the level of the whole protein complex. As a structural and functional characterization tool, PNAbind is suitable for functional analysis of various proteins [102].
Although graph neural networks can effectively model the structural features and spatial relationships of peptide sequences in AMP prediction, their application still faces certain limitations: Firstly, the model performance is highly dependent on the prediction accuracy of the input structure, which requires a large number of high-quality experimental resolved structures. However, the AMP peptide sequences often provide enough information to construct complex graph structures, resulting in unstable performance and low accuracy. Secondly, it is difficult for graph models to integrate non-graphical information (such as evolutionary spectrum, chemical modification, etc.), which limits their ability to model multi-dimensional features. In addition, although its inference speed is fast, the training process has certain requirements on computational resources, which may limit its wide application in resource-limited environments. Therefore, the advantages of graph models in AMP prediction have not been fully exploited, and it is still necessary to combine multimodal information (such as language models) for optimization and fusion [88, 102].
The mixed and multimodal model achieves information complementation within a unified framework by jointly employing sequence embeddings, predictive structural graphs, evolutionary spectra, and physical-chemical properties, and aligning multi-source features through attention or contrastive learning. For example, APEX screened out 37,176 broad-spectrum active peptides from extinct species (11,035 of which only appeared in extinct species), verified through 69 synthetic experiments, 59% of which were active, and the in vivo efficacy was comparable to polymyxin B, proving that multimodal DL combined with molecular de-extinction is an efficient path for discovering new antibiotics [103]; UniproLcad is an AMP predictor that integrates a multi-pLM. It concatenates the sequence representations of three pre-trained models, ESM-2, ProtBert, and UniRep, and then uses BiLSTM, 1D-CNN, and Attention to refine the features, and finally uses multi-layer perceptron (MLP) for classification. It significantly outperforms existing methods [104].
The wide application of such models still faces a series of common problems. First of all, such models usually rely on large-scale labeled datasets for training. When the size of the dataset is insufficient and the source is single, it is difficult to truly reflect the robustness of the model in diverse scenarios. However, the heterogeneity, unbalanced length distribution, and potential redundancy of biological data may lead to the model learning the specific deviation of the dataset instead of the universal biological law. Secondly, multimodal fusion often introduces multiple deep subnetworks and complex interaction modules to form a highly nonlinear “black box” system. Although the prediction accuracy is improved, it is difficult to explain the decision basis from the biological perspective. Even with the introduction of attention mechanisms, most studies still stay at the visualization level and lack deep integration with experimental verification, which leads to doubts about the biological significance of attention weight. Thirdly, it is common practice to send the outputs of each modal encoder to the prediction layer after simple splicing or weighted summation. This late fusion method fails to fully model the complex intersection between modes, which limits the ability of the model to describe the internal mechanism of the biological system. In addition, most hybrid multimodal models do not take into account the dynamics and physicochemical details of biological systems, such as protein conformational changes, water molecule-mediated interactions, ligand flexibility, and other key factors, resulting in a gap between the prediction results and the real physiological environment [44, 103, 104].
Overall, these four models have their own focuses: Basic models are lightweight and efficient, language models are semantically rich, graph models have precise structures, and multimodal fusion balances performance and generalization, collectively forming the current technical panorama of AMP prediction. DL has four main technical routes in AMP prediction: CNN/RNN basic models, language models, graph neural networks, and multimodal models.
LLMs, particularly those based on the Transformer architecture, have revolutionized NLP and are now making significant impacts in bioinformatics and cheminformatics. Their core advantage lies in the ability to learn complex, context-dependent representations of sequential data through self-supervised learning on vast unlabeled corpora. In the context of AMPs, amino acid sequences can be treated as a biological language, allowing LLMs to capture long-range dependencies, evolutionary constraints, and structural semantics without the need for manual feature engineering [95, 105].
The foundation of modern LLMs is the Transformer architecture, introduced by Vaswani et al. [95], which utilizes a self-attention mechanism to weigh the importance of different parts of the input sequence, effectively capturing global dependencies. This architecture has been adapted for proteins, leading to the development of specialized “pLMs”. These models are pre-trained on massive datasets of protein sequences (e.g., from UniRef or PDB) to learn the underlying “grammar” of protein sequences.
Several prominent pLMs have been developed and applied to AMP discovery. For instance, the ESM family of models, such as ESM-1b and ESM-2, developed by Lin et al. [96], uses self-supervised learning to generate rich embeddings that capture biological and structural properties from sequence alone [106]. Similarly, the ProtTrans family, including ProtT5 proposed by Elnaggar et al. [97], leverages Transformer architectures and has achieved state-of-the-art performance in various protein prediction tasks by providing powerful feature representations.
pLMs serve as powerful feature extractors for downstream tasks like AMP prediction. Their pre-trained knowledge can be leveraged through fine-tuning or by using their embeddings as input to simpler classifiers. This approach has proven highly effective, especially in data-sparse scenarios common in peptide research [105].
For example, HMD-AMP, developed by Yu et al. [106], utilizes the ESM-1b pLM combined with a deep forest classifier to accurately identify AMP functions across 11 target types, demonstrating robustness even with small sample sizes. Another model, AMP-BERT, proposed by Lee et al. [93], fine-tunes a BERT-based model specifically for AMP function prediction, showcasing the adaptability of the Transformer architecture. PeptideBERT, introduced by Guntuboina et al. [107], is a lightweight Transformer-based language model specifically fine-tuned for peptide property prediction, including antimicrobial activity. It demonstrates that even smaller, specialized models can achieve high performance by leveraging the power of the Transformer architecture [107]. Furthermore, Lobo et al. [94] successfully used a “pre-training and fine-tuning” pipeline with the SeqVec pLMs to rapidly and accurately predict antifungal peptides, outperforming traditional methods.
A more recent advancement is the integration of LLMs with other architectures to create hybrid models. PepGraphormer, developed by Lin et al. [108], fuses the semantic power of the ESM-2 language model with a graph attention network (GAT). This hybrid framework constructs a heterogeneous graph containing peptide and amino acid nodes, allowing it to capture both global semantic understanding and local structural relationships for high-precision AMP identification without requiring explicit 3D structures [108].
Beyond prediction, LLMs and deep generative models are increasingly used for the de novo design of novel AMPs. These models learn the distribution of functional peptide sequences and can generate new ones with desired properties. For instance, deepAMP, introduced by Li et al. [109], is a deep generation framework based on peptide language models. It uses a pre-training and fine-tuning strategy to optimize peptides from low-activity to high-activity and broad-spectrum candidates, with subsequent experimental validation confirming its efficiency [109]. Similarly, BroadAMP-GPT, proposed by Li et al. [110], combines CNN and LSTM architectures for AI-driven design and multi-model screening, achieving high experimental hit rates. In the same type, BatGPT-Chem proposed by Yang et al. [111], based on the learning of massive chemical data, establishes the mapping relationship between molecular properties and molecular structures represented by SMILES, and the SMILES code is used as a “special language” to carry out unified sequence modeling with natural language. The peptide molecular codes that meet the requirements of these properties are directly output, so as to complete the conversion from “property description” to “peptide structure generation” [111].
Other generative models, while not strictly LLMs, also play a crucial role. HydrAMP, a conditional variational autoencoder (cVAE) developed by Szymczak et al. [112], creates a continuous latent space for peptide sequences, enabling both the optimization of existing peptides and the generation of entirely new, highly potent AMPs. Multi-CGAN, introduced by Yu et al. [113], uses a conditional generative adversarial network to design peptides with multiple desired attributes, such as high activity, low toxicity, and specific secondary structures.
The application of LLMs represents a paradigm shift in AMP discovery, moving towards more intelligent and data-efficient design [5]. By treating sequences as a language, these models can uncover hidden patterns and design principles that are difficult to capture with traditional methods. However, challenges remain. As highlighted by several studies, the quality and scale of training data are paramount [47, 114]. Models trained on biased or limited datasets may have poor generalization. Furthermore, while powerful, LLMs are often “black boxes”, and enhancing their interpretability to reveal the key structural motifs driving activity is a critical area of future research [115]. The integration of LLMs with structural modeling and experimental validation will be key to realizing their full potential in developing next-generation antimicrobial therapeutics [6].
In the model evaluation criteria, a series of performance indicators are calculated based on four basic parameters [true positive (TP), true negative (TN), FP, false negative (FN)], including sensitivity, specificity, accuracy, precision, F-measure, and Matthews correlation coefficient (MCC). Additionally, two evaluation methods that are independent of threshold values, namely ROC curve and AUC value, are also employed to comprehensively assess the classification performance of the AMP prediction model (Tables 3 and 4) [42, 116].
Definition of prediction model evaluation basic parameters [42].
| Acronym | Definition description |
|---|---|
| TP | These are the experimentally validated AMPs that have been correctly predicted as AMPs by the prediction method. |
| TN | These are the non-AMP sites that have been correctly predicted as non-AMPs. |
| FP | These are the non-AMPs that have been incorrectly predicted as AMPs. |
| FN | These are the experimentally validated AMPs that have been incorrectly predicted as non-AMPs. |
AMPs: antimicrobial peptides; FN: false negative; FP: false positive; TN: true negative; TP: true positive.
Definition of prediction model evaluation indicators [42].
| Name | Definition description |
|---|---|
| Sensitivity | The proportion of samples that are truly positive and are correctly predicted as positive by the model. |
| Specificity | Among all the samples that are truly negative cases, the proportion of those that were correctly predicted as negative by the model. |
| Accuracy | The proportion of samples that were correctly predicted (whether positive or negative) in all the samples. |
| Precision | The proportion of samples that are truly positive among all those predicted as positive by the model. |
| F-measure | The harmonic mean of precision and recall. |
| MCC | A balanced metric used to measure the performance of binary classification, with its value ranging from –1 to +1. +1 indicates perfect prediction, 0 indicates random prediction, and –1 indicates completely incorrect prediction. |
FN: false negative; FP: false positive; MCC: Matthews correlation coefficient; TN: true negative; TP: true positive.
The typical AMP prediction process consists of the following steps: a. data collection, obtaining positive and negative samples from the database; b. data cleaning and redundancy removal, using tools such as cluster database at high identity with tolerance (CD-HIT) to eliminate similar sequences and reduce bias; c. feature extraction and selection, converting peptide sequences into numerical feature vectors; d. model training and optimization, evaluating model performance using cross-validation or independent test sets; e. performing performance evaluation using indicators such as sensitivity, specificity, accuracy, F1-score, MCC, and AUC [42].
CD-HIT is a tool used for clustering biological sequences. The new version has significantly improved its speed through parallelization and multiple optimizations, and supports multi-core acceleration, making it suitable for processing large-scale sequencing data [117]. Its tools are integrated into the CDSnake process for processing 16S sequencing data. It clusters paired-end read lengths separately, clustering read 1 (R1) and read 2 (R2) respectively to avoid information loss caused by merging errors or poor quality, thereby retaining more effective data. It is suitable for large-scale microbiota research and performs exceptionally well, especially in high-redundancy data [118].
Feature extraction is a crucial step in ML, referring to the process of extracting numerical information from raw data that can effectively represent the characteristics of samples and facilitate model learning. For instance, in the Ensemble-AMPPred study, in order to predict AMPs, 517 features were extracted from the sequences, including amino acid composition, pseudo-amino acid composition (PseAAC), physicochemical properties, secondary structure propensity, antimicrobial index (IC50), etc., to convert variable-length peptide sequences into fixed-length numerical vectors. To further enhance the model performance, the researchers fused four key features through logistic regression and created a new “mixed feature”. This mixed feature ranked highly in terms of feature importance and effectively improved the sensitivity and overall performance of the prediction model [119]. ftrCOOL, developed by Amerifar et al. [120], is an R-language tool designed for biological sequence feature extraction. Its functions encompass seven categories of feature extraction methods: k-mer frequency calculation, grouped amino acid composition, physicochemical index substitution, mathematical transformations such as autocorrelation, K-NN similarity based on sequence alignment, position-specific k-mer frequency, and open reading frame feature analysis. This tool covers all the functions of iLearnPlus and adds 30 new features, such as the codon adaptation index and Fickett score [120].
To enhance the efficiency of feature extraction and reduce storage space, representation learning has become the fundamental and crucial first step in the computational research of AMPs. Its core objective is to convert the symbolic data of amino acid sequences into numerical features that can retain their biological and functional information, so that subsequent ML or DL models can process them [121]. Among them, pLMs, such as ESM-2 and ProtBert, encode amino acid sequences into context-dependent dense vectors through self-supervised pre-training, directly replacing traditional one-hot or Word2Vec, significantly reducing the dimensionality problem and improving prediction accuracy [122].
Graph contrastive learning (GCL) is a graph representation learning method based on self-supervised learning. The core idea is to first generate dual views of the original features using two different Laplacian smoothing techniques. Then, positive and negative sample pairs are constructed at the structural level. Specifically, the embeddings of the same node in the two views form a positive pair, while those of different nodes form a negative pair. Through the joint optimization of the structural alignment loss and the contrastive loss, the distance between positive pairs is reduced, and the distance between negative pairs is increased [123]. In 2024, Yang et al. [124] proposed a GCL framework called Local Structure-aware GCL (LS-GCL), which consists of three key steps: Firstly, multiple related views are generated from the original graph through data augmentation techniques such as node feature masking, edge dropout, or subgraph sampling; secondly, the node or graph embedding representations are extracted using graph neural network encoders (such as GCN, GAT); finally, the contrastive loss function (such as InfoNCE loss) is used to maximize the consistency of the representation of the same node in different views or contexts, and minimize the similarity between different nodes [124].
Dynamic Network Contrastive representation Learning (DNCL) is a contrastive learning framework specifically designed for dynamic networks. Its core idea is to learn node representations through a dual contrast mechanism involving inter-snapshot and intra-snapshot within snapshots. This framework generates multi-view network sequences using enhancement techniques, extracts spatio-temporal features through a GCN + GRU encoder, and utilizes an improved contrastive loss (introducing time decay) to maximize the consistency of positive samples and minimize the similarity of negative samples, thereby enabling the learning of robust and discriminative dynamic node representations without the need for manual annotation [125].
As of now, many online tools and software for predicting AMPs and their functions have been put into use, which has facilitated the discovery and design of more effective antimicrobial agents (Tables 5 and 6). With the development of computer science and computational methods, these tools have also been continuously optimized (Figure 2).
AMP prediction tool.
| Predictor | Core methodology | Key features | Reference |
|---|---|---|---|
| PyAMPA | random forest | Multifunctional platform for prediction, validation, and optimization | [126] |
| AMPlify | BiLSTM + attention | Deep learning model with balanced/imbalanced training modes | [127] |
| TriNet | Triple fusion network | Simultaneously predicts AMPs and anti-cancer peptides (ACPs) | [84, 128] |
| Deep-AmPEP30 | CNN | Using pseudo-amino acids combined with CNN for AMP prediction | [129] |
| AI4AMP | LSTM | An AMP prediction tool based on physical-chemical coding | [130] |
| PTPAMP | SVM | SVM performed the best and was specifically used for identifying and classifying plant-derived AMPs | [87] |
| PeptideBERT | Protein language model | A language model for fine-tuning peptides, for AMP prediction | [107] |
| AutoPeptideML | Automated modeling platform | Supports multiple machine learning algorithms and automatically builds peptide prediction models | [131] |
| AmPEP | RF | Predicting AMP based on amino acid distribution patterns | [132] |
| HMD-AMP | ESM-1b; DF | Accurately identify the functions of AMP and its 11 types of targets, with both robustness in small sample sizes and interpretability | [106] |
| iAMP-CA2L | Multi-scale convolution and attention mechanism | Extract key features of the sequence, accurately identify and conduct interpretable analysis | [26] |
| iAMPCN | CNN | A large-scale and high-quality training and testing dataset was constructed, and it demonstrated higher accuracy in most functional activity predictions | [133] |
| PAMPred | k-means | Hierarchical heterogeneous integration architecture, improved particle swarm optimization (PSO) weight optimization, superior experimental performance | [134] |
| PepGraphormer | ESM2; GAT | An AMP prediction framework that does not require a three-dimensional structure and integrates language models with graph neural networks | [108] |
AMP: antimicrobial peptide; BiLSTM: bidirectional long short-term memory; CNN: convolutional neural networks; DF: decision forest; ESM: evolutionary scale modeling; GAT: graph attention network; PSO: particle swarm optimization; RF: random forest; SVM: support vector machine.
Examples of peptide generation tools.
| Predictor | Core methodology | Key features | Reference |
|---|---|---|---|
| HydrAMP | cVAE | By constructing a low-dimensional continuous latent space representation of peptide sequences, analogue generation and unconstrained generation can be achieved | [112] |
| deepAMP | Protein language model | Data augmentation, multi-round iterative optimization, and experimental verification are highly efficient | [109] |
| Multi-CGAN | CGAN | Adopt a three-stage generation strategy, and introduce the multi-label embedding mechanism and the automatic integration module | [113] |
| LSTM_Pep | Long short-term memory | After pre-training with large-scale general peptide data, then fine-tuning with specific active peptides | [135] |
| BroadAMP-GPT | CNN + LSTM | AI-driven design, multi-model fusion screening, high experimental hit rate | [110] |
AI: artificial intelligence; CNN: convolutional neural network; cVAE: conditional variational autoencoder.
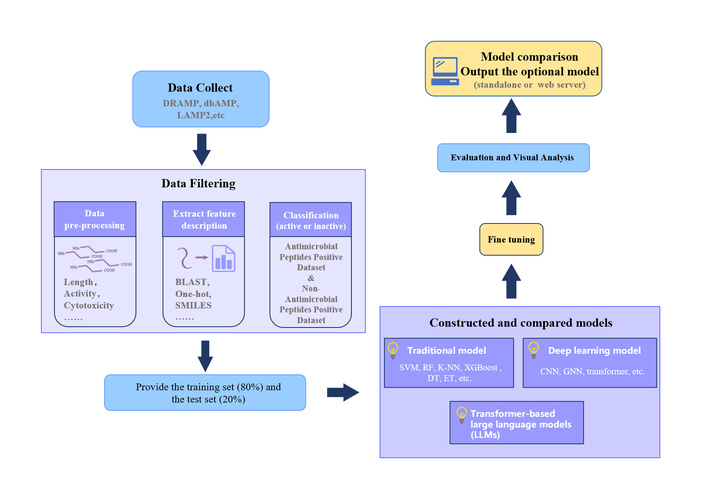
General workflow for developing a prediction model for AMPs. This figure outlines the basic steps for building an AMP prediction model. Starting from the collection of AMP sequences from the database, a dataset containing active and inactive peptides was created and then divided into a training set (80%) and a test set (20%). Feature extraction can be achieved by BLAST, motif search, and computational approaches. After building and training the model with algorithms, the resulting model can be deployed as a standalone tool or a web server. AMPs: antimicrobial peptides; CNN: convolutional neural networks; DT: decision tree; ET: extra trees; K-NN: K-nearest neighbor; RF: random forest; SVM: support vector machine; XGBoost: eXtreme Gradient Boosting.
PyAMPA is a comprehensive AMP discovery and optimization platform developed in Python. It uses RF classifiers for predicting antimicrobial activity, cell-penetrating capacity, hemolytic activity, and toxicity. The AMPScreen module uses a sliding window method based on a propensity scale to rapidly scan entire proteomes. It consists of five core modules: AMPScreen is used for high-throughput screening of potential antimicrobial sequences; AMPValidate uses ML models to verify antimicrobial activity; AMPSolve predicts the hemolysis, toxicity, half-life, and antimicrobial spectrum of peptides; AMPMutate assesses the impact of single-point mutations on function; and AMPOptimize uses genetic algorithms to optimize the comprehensive performance of peptides. PyAMPA integrates multiple databases and prediction models, featuring efficiency, accuracy, and scalability, and provides a complete solution from initial screening to optimization for the development of antibacterial peptide drugs [126].
iAMPCN is a DL-based computing framework designed for efficient identification of AMPs and their 22 functional activities (such as antibacterial, antifungal, antiviral, anti-cancer, anti-parasitic, etc.). This model integrates four sequence encoding methods (one-hot, BLOSUM62, AAIndex, and PAAC) and employs CNN for feature extraction and prediction. The research team constructed a large-scale and high-quality training and testing dataset, covering over 49,000 experimentally verified AMP sequences, and systematically evaluated the impact of different sequence similarity thresholds on the model’s performance. Benchmark experiments demonstrated that iAMPCN significantly outperformed existing mainstream tools on independent test sets, showing higher accuracy, AUC, and MCC values in most functional activity predictions. Additionally, this study conducted model interpretability analysis, revealing that positively charged amino acids such as lysine (K) and arginine (R) play a crucial role in AMP activity. The code of iAMPCN is open-source: https://github.com/joy50706/iAMPCN, providing powerful support for the discovery and functional annotation of AMPs and promoting their application in drug development [133].
PepGraphormer is a DL framework that integrates the pLM ESM2 with the GAT, which is designed to directly predict AMPs from amino acid sequences. First, it utilizes the pre-trained ESM2 model to extract the deep semantic features of peptide sequences, which serve as the initial prediction and the initial embedding of peptide nodes in the graph. Meanwhile, it constructs a heterogeneous graph containing peptide nodes and amino acid nodes. In this graph, the term frequency-inverse document frequency (TF-IDF) and mutual information are used to quantify the peptide-amino acid composition relationship and the global co-occurrence pattern among amino acids respectively, thus avoiding the reliance on time-consuming and error-prone three-dimensional structure prediction. Subsequently, the GAT learns the complex association features between peptides and amino acids on this graph. Finally, the model linearly fuses the global semantic understanding of ESM2 with the graph structural relationships learned by the GAT to jointly make the final classification decision, achieving high-precision and interpretable AMP identification [108].
HydrAMP is an AMP generation model based on cVAE. By constructing a low-dimensional continuous latent space representation of peptide sequences, it achieves prototype peptide-based optimization for analogue generation and unconstrained generation from scratch. The model introduces Jacobian decoupling regularization, decoupling the peptide’s latent representation from its antimicrobial activity conditions, thereby enhancing the generation control. HydrAMP supports parameterized adjustment of creativity (temperature τ), flexibly balancing diversity and conservation. Its core advantage lies in being able to generate highly active AMPs starting from both active and inactive peptides, significantly enhancing the diversity and activity of peptide classes; combined with consensus screening by an auxiliary classifier and pre-assessment through MD simulation, it improves the efficiency of experimental verification. Experimental results show that the peptides generated by HydrAMP exhibit excellent antimicrobial activity, low toxicity, and broad-spectrum properties, making it a powerful tool in combating AMR [112].
deepAMP is a deep generation framework based on peptide language models. It achieves targeted optimization from low-activity peptides to high-activity and broad-spectrum AMPs through a combination of pre-training and fine-tuning. Its core methods include pre-training a general generation model (deepAMP-general) on a large-scale peptide sequence dataset, and then using sequence degradation strategies to construct “low-activity→high-activity” peptide pairs, and fine-tuning the AMPs optimization model (deepAMP-AOM) and the penetration peptide optimization model (deepAMP-POM), respectively. This model has features such as data augmentation, multi-round iterative optimization, and efficient experimental verification. The designed peptides exhibit significantly superior activity compared to the template peptide penetration peptide, with some reaching antibiotic levels. They are also less likely to induce drug resistance and have good biocompatibility and in vivo anti-infection effects, providing an efficient and intelligent new strategy for the discovery of AMPs [109].
Multi-CGAN is a multi-attribute AMP generation model based on conditional generative adversarial networks. It adopts a three-stage generation strategy: single-label generation, multi-label integration, and multi-label generation, enabling the learning and generation of peptide sequences with multiple attributes such as antimicrobial properties, low toxicity, and specific secondary structures from single-attribute data. Its core method introduces a multi-label embedding mechanism and an automatic integration module, allowing the generator to generate peptide chains that meet the requirements of multiple attributes under specified conditions. The model has good attribute controllability and sequence diversity and can be used for data augmentation in downstream tasks, effectively improving the performance of the prediction model and supporting targeted attribute generation by regulating input noise, providing an efficient and interpretable generation tool for aAMP design [113].
After being pre-trained with large-scale general peptide data and then fine-tuned with specific active peptides, the research-generated model LSTM_Pep is based on the Long Short-Term Memory network. Through pre-training with large-scale general peptide data and fine-tuning with specific active peptides, it generates peptide sequences with potential therapeutic activity. Combined with the DeepPep DL model for protein-peptide interaction prediction, it enables rapid screening. The core process includes peptide generation, modeling, docking, MD simulation, and iterative optimization, significantly improving the efficiency of peptide drug discovery. The model supports the generation of multiple types of active peptides, has high diversity, low redundancy, and controllability, and is suitable for the screening of various targets such as antiviral, antimicrobial, and anti-cancer [110, 135].
Although AMPs have broad prospects, their clinical translation still faces some problems, such as high toxicity, low activity, easy degradation, and short retention time. For example, Na-SNK2-2a (C18W) is a cationic AMP obtained by bioinformatics mining and rational design of plant snakin peptides. Through a single point mutation in the key site (replacing the 18th cysteine with tryptophan), the toxicity and allergenicity of the peptide were reduced, while its hydrophobicity and membrane targeting selectivity were enhanced, and the ability to penetrate cells was retained [136].
PMAP-23 of the cathelicidin family exerts a broad spectrum of antimicrobial effects through membrane destruction mechanism, but its binding ability to LPS is limited, and it may face the challenge of balancing stability and toxicity in vivo. To this end, the researchers replaced the C-terminal or N-terminal of PMAP-23 with the LPS binding sequence GWKRKRFG (G8) with a β-boomerang structure, and used the PQKP itself as the linker region to construct a series of heterozygous peptides, which not only significantly enhanced the binding ability of PMAP-23 to LPS, but also significantly enhanced the binding ability of PMAP-23 to LPS. It further enhanced the antimicrobial activity and cell selectivity, and showed good salt ion and serum stability [137, 138].
Human host defense peptide LL-37 has broad-spectrum antibacterial and immunomodulatory functions, but its linear structure leads to prominent proteolytic degradability and poor stability, which limits its direct clinical application [139]. In order to break through this bottleneck, the minimal antibacterial core fragment KR-12 was used as a template, and the two KR-12 units were covalently linked to form a circular dimer through the main chain cyclizing and dimerization strategy. The best derivative, such as cd4(Q5K, D9K), had significantly increased antibacterial activity and could stably exist in serum for several hours. Simultaneous enhancement of activity and stability was achieved [140].
Despite significant progress in the application of AI to the discovery and design of AMPs, current research still faces many challenges that hinder experimental validation and clinical translation. The first issue concerns data quality and consistency. Although tens of thousands of AMP sequences are available in public databases, there is a severe scarcity of data on specific pathogens, toxicity profiles, structural stability, and ADMET properties, which are severely lacking, leading to biased model training and insufficient generalization capability [114].
Another critical barrier is the lack of model interpretability. Most mainstream DL approaches function as “black-box” models. While they often achieve high classification accuracy, they struggle to reveal the key residues or structural motifs in the peptide sequence that determine antimicrobial activity. This opacity restricts the practical utility of AI in guiding rational peptide design [115]. Additionally, the issue of insufficient utilization of structural information needs to be addressed. Although GNN and pLMs have been preliminarily applied to structure modeling, their performance in representing short peptides, cyclic peptides, and peptides with post-translational modifications remains limited and requires further improvement [89].
Despite the great theoretical potential of AMPs, the clinical translation of AMPs is difficult. A major challenge is that clinical validation of efficacy is difficult, with many drug candidates, such as pexiganan and iseganan, performing poorly in phase III trials. In addition, some drugs may not be able to replicate the efficacy results in the earlier phase of the trial. This indicates that AMP not only needs to overcome its own stability and toxicity problems, but also must pass rigorous clinical trials to prove its clear and safe therapeutic advantages in the complex human environment [141].
To address these challenges, several strategic improvements can be pursued. First, establishing a standardized, multimodal, and high-quality AMP database is essential. This can be initiated by standardizing experimental conditions and evaluation criteria, systematically collecting multi-dimensional data—including sequences, structures, functions, toxicity, MIC values, ADMET properties, etc.—and clearly defining negative samples to facilitate data sharing and open access [47].
Second, developing highly interpretable and flexible AI models is crucial. Integrating explainability tools such as SHAP, LIME, and attention visualization, along with combining biological physical features with sequence embeddings, can help transition models from mere statistical fitting to biologically meaningful prediction, thereby enhancing their translational value [115].
We can also promote multimodal fusion and structural perception modeling, strengthening the deep integration of multiple information sources such as sequences, structures, and functions, developing GNN and multimodal Transformer architectures suitable for short, cyclic, and modified peptides, and enhancing the model’s ability to understand and predict complex structural characteristics. Alternatively, we can utilize advanced generative technologies such as cVAE, cGAN, and diffusion models to achieve simultaneous optimization of multiple objectives, including antimicrobial activity, low toxicity, and target specificity, improving the synthesizability and functionality of generated peptides, and developing conditional generation models and multi-objective optimization strategies [6].
AI-driven AMP discovery is currently at a critical stage of transitioning from “tool-assisted” to “intelligent design”. Systematic breakthroughs in data quality, model interpretability, structural modeling, and experimental validation are essential to fully realize the potential of AI in antibacterial peptide drug development and to provide sustainable solutions to the global AMR crisis.
The escalating global crisis of AMR underscores the urgent need for innovative therapeutic alternatives. AMPs represent a promising class of agents with broad-spectrum activity, low resistance development, and diverse mechanisms of action. However, the vast and largely unexplored peptide sequence space makes traditional experimental screening methods impractical.
The integration of AI and big data models has revolutionized AMP discovery and characterization. ML and DL approaches—including discriminative models like RFs and LSTMs, and generative models such as variational autoencoders—have demonstrated remarkable efficacy in predicting, classifying, and even designing novel AMPs with desired properties. These models leverage large-scale AMP databases (e.g., dbAMP, DRAMP, DBAASP, LAMP2, CAMPR3) and diverse feature sets to achieve high predictive accuracy, sensitivity, and specificity.
Despite these advances, several challenges remain, including data scarcity, class imbalance, and inconsistent experimental annotations. The present study highlights the effectiveness of even simple models like linear kernel SVM in data-scarce settings, achieving high performance metrics and enabling the experimental validation of AI-predicted peptides.
ACP: anti-cancer peptide
AI: artificial intelligence
AMP: antimicrobial peptide
AMR: antimicrobial resistance
AVPs: antiviral peptides
BGCs: biosynthetic gene clusters
CD-HIT: cluster database at high identity with tolerance
CNN: convolutional neural networks
cVAE: conditional variational autoencoder
DBAASP: Database of Antimicrobial Activity and Structure of Peptides
DL: deep learning
DNN: deep neural networks
DT: decision tree
ESM: evolutionary scale modeling
FP: false positive
GAT: graph attention network
GCL: graph contrastive learning
GNN: graph neural networks
K-NN: K-nearest neighbor
LLMs: large language models
LPS: lipopolysaccharide
MCC: Matthews correlation coefficient
MD: molecular dynamics
MDR: multidrug-resistant
MIC: minimum inhibitory concentration
ML: machine learning
MRSA: methicillin-resistant Staphylococcus aureus
NLP: natural language processing
pLM: protein language model
R&D: research and development
RNN: recurrent neural networks
SVM: support vector machine
YZ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. ZW: Investigation, Writing—original draft, Writing—review & editing. HZ: Validation, Writing—review & editing, Supervision, Funding acquisition. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
National Natural Science Foundation of China [82073767]; Priority Academic Program Development of Jiangsu Higher Education Institutions [PAPD 2023007]; 2026 College Students’ Innovation and Entrepreneurship Training Program [2026303]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 645
Download: 11
Times Cited: 0
Irina Tiganova ... Yulia Romanova
Ebenezer Aborah ... Manuel F. Varela
Daniel Buldain ... Laura Marchetti
Ayuba Olanrewaju Mustapha ... Yusuf Oloruntoyin Ayipo
Monika I. Konaklieva ... Balbina J. Plotkin