 Review
Review

Affiliation:
1Food and Drug Investigation Laboratory, The Indonesian Food and Drug Authority, Jakarta 10560, Indonesia
Email: alfi.sophian@pom.go.id
ORCID: https://orcid.org/0000-0002-5206-2110
Affiliation:
2Biological Product Laboratory, The Indonesian Food and Drug Authority, Jakarta 10560, Indonesia
Affiliation:
2Biological Product Laboratory, The Indonesian Food and Drug Authority, Jakarta 10560, Indonesia
ORCID: https://orcid.org/0009-0003-2865-9659
Affiliation:
2Biological Product Laboratory, The Indonesian Food and Drug Authority, Jakarta 10560, Indonesia
ORCID: https://orcid.org/0009-0007-5488-8083
Explor Drug Sci. 2026;4:1008162 DOI: https://doi.org/10.37349/eds.2026.1008162
Received: January 23, 2026 Accepted: March 02, 2026 Published: June 03, 2026
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa, Universidad de Barcelona, Spain
Advanced Therapy Medicinal Products (ATMPs) represent a transformative class of innovative therapies based on genes, cells, or engineered tissues that aim to modify, repair, or replace biological functions at a fundamental level. This review provides a foundational overview of ATMPs, addressing their scientific basis, regulatory classification, clinical translation, and key challenges for future development. The article outlines the four principal categories of ATMPs: gene therapy medicinal products, somatic cell therapy medicinal products, tissue-engineered products, and combined ATMPs, and discusses their distinct mechanisms of action and therapeutic applications. Recent clinical successes, including chimeric antigen receptor T-cell therapies, gene replacement therapies for inherited disorders, and tissue-engineered constructs for regenerative medicine, demonstrate the paradigm shift from symptomatic management towards disease modification or potential cure. However, the clinical implementation of ATMPs presents substantial challenges related to safety monitoring, long-term efficacy, complex manufacturing processes, regulatory evaluation, high treatment costs, and equitable patient access. Ethical considerations, including informed consent, long-term follow-up obligations, and global disparities in availability, further complicate their integration into routine clinical practice. Regulatory agencies have introduced adaptive pathways and accelerated approval mechanisms to facilitate timely patient access while maintaining rigorous standards of quality, safety, and efficacy. Continued technological innovation, coupled with sustainable healthcare policies and international collaboration, will be essential to realise the full therapeutic potential of ATMPs. As evidence accumulates from clinical trials and real-world use, ATMPs are expected to play an increasingly prominent role in modern medicine and the future of personalised healthcare.
Advanced Therapy Medicinal Products (ATMPs) constitute a distinct class of innovative therapeutics that employ genes, cells, or engineered tissues to restore, replace, or modify biological functions. Unlike conventional small-molecule drugs and traditional biologics, whose effects typically depend on transient pharmacological interactions, ATMPs act through complex and often durable biological mechanisms, including gene expression, cellular integration, and tissue regeneration, thereby offering the potential for long-term or curative outcomes following a single or limited administration [1, 2].
Over the past decade, rapid technological advances have transformed ATMPs from experimental concepts into clinically approved therapies. Gene therapies have demonstrated efficacy in inherited disorders, chimeric antigen receptor T-cell therapies have achieved substantial remission rates in refractory haematological malignancies, and tissue-engineered constructs have enabled functional restoration of damaged tissues [3–5]. These achievements, however, are accompanied by significant translational challenges, including complex manufacturing processes, stringent quality requirements, interpatient variabilities in therapeutic response, long-term safety uncertainties, and exceptionally high treatment costs that raise concerns regarding sustainability and equitable access [6, 7].
The development and implementation of ATMPs require extensive interdisciplinary collaboration across molecular biology, genetic engineering, cell biology, bioprocess engineering, clinical medicine, regulatory science, health economics, and bioethics. Therapeutic performance depends not only on biological design but also on manufacturing consistency, delivery strategies, patient selection, and long-term monitoring [8]. Consequently, ATMPs challenge conventional frameworks for drug development, regulatory evaluation, and healthcare delivery.
Given their expanding clinical relevance and societal impact, a clear understanding of ATMPs classification, mechanisms of action, manufacturing considerations, safety evaluation, and regulatory oversight is essential for researchers, clinicians, regulators, policymakers, and patient communities. This review provides a structured overview of these key aspects, highlighting the distinctive features that differentiate ATMPs from conventional therapeutics and the translational factors that influence their integration into healthcare systems.
An overview of ATMPs categories and their translational contexts is presented in Figure 1.
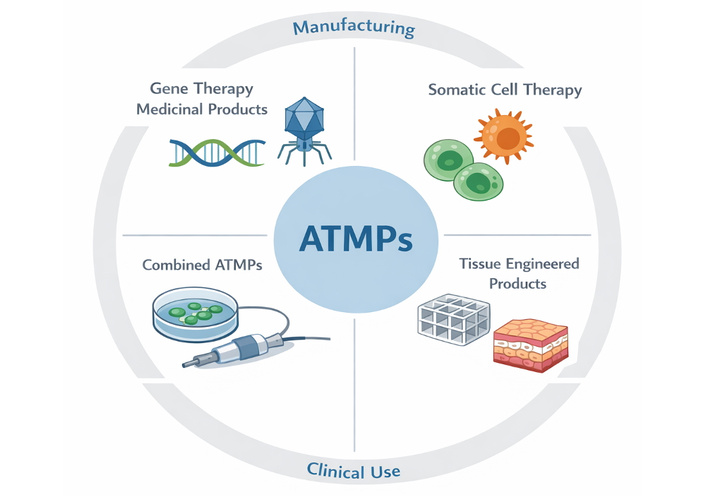
Conceptual overview of Advanced Therapy Medicinal Products and translational dimensions. Schematic representation of the four principal categories of ATMPs, including gene therapy medicinal products, somatic cell therapy medicinal products, tissue-engineered products, and combined ATMPs integrated with medical devices. The diagram also illustrates key translational domains influencing ATMP development and implementation, including manufacturing and process control, quality and potency assessment, safety evaluation, regulatory oversight, and clinical application. The interconnected structure emphasises the multidisciplinary and interdependent nature of ATMP development, where challenges in manufacturing, regulation, and clinical translation collectively influence therapeutic efficacy, safety, and patient access.
The regulatory classification of ATMPs is fundamental to determining appropriate development pathways, manufacturing standards, quality requirements, clinical evaluation strategies, and post-authorisation oversight. Unlike conventional pharmaceuticals, which are typically defined by stable chemical composition and predictable pharmacokinetics, ATMPs consist of living cells, genetic material, or engineered tissues whose biological behaviour may evolve after administration. Their inherent variability, potential for long-term persistence, and capacity to modify physiological processes necessitate regulatory approaches that extend beyond traditional medicinal product frameworks. Dedicated classification systems therefore serve not only administrative purposes but also provide the scientific foundation for risk assessment, product characterisation, and lifecycle management.
Within the European Union, ATMPs are formally defined and regulated under Regulation (EC) No. 1394/2007, implemented through the centralised procedures of the European Medicines Agency. This regulation established a specialised framework recognising the distinctive properties of gene, cell, and tissue-based therapies and introduced the Committee for Advanced Therapies to provide scientific expertise. The framework categorises ATMPs into four principal groups: gene therapy medicinal products, somatic cell therapy medicinal products, tissue-engineered products, and combined ATMPs incorporating medical devices [9, 10]. Classification is determined by the product’s mechanism of action, degree of manipulation of biological material, and intended therapeutic function rather than solely by its cellular or genetic source. This function-oriented approach acknowledges that similar starting materials may yield fundamentally different therapeutic products depending on processing and clinical intent.
Gene therapy medicinal products represent one of the most scientifically complex categories within ATMPs. These therapies introduce, remove, or modify genetic material within a patient’s cells to achieve a therapeutic effect, typically using viral vectors such as adeno-associated viruses or lentiviruses, as well as emerging non-viral delivery systems. Their therapeutic activity depends on sustained biological processes including transgene expression, genome editing, or gene silencing, distinguishing them from conventional pharmacological agents whose effects diminish as drug concentrations decline. Regulatory evaluation therefore emphasises vector design, genomic integration potential, biodistribution, persistence of gene expression, and risks such as insertional mutagenesis or off-target effects [11, 12]. Advances in genome editing technologies, particularly clustered regularly interspaced short palindromic repeats (CRISPR)-based approaches, have further expanded the scope of gene therapy, prompting regulatory agencies to refine guidance on quality control, long-term monitoring, and environmental risk assessment. The potential for durable or curative outcomes following a single administration underscores both the promise and the regulatory complexity of this category.
Somatic cell therapy medicinal products consist of cells that have undergone substantial manipulation or are intended for a different essential function in the recipient from in the donor. Substantial manipulation may involve processes such as ex vivo expansion, genetic modification, or differentiation induction that alter the biological characteristics of the cells. These therapies exert pharmacological, immunological, or metabolic effects mediated by living cells capable of interacting dynamically with host tissues. A prominent example is adoptive cell therapy using ex vivo expanded immune cells, including chimeric antigen receptor T cells, which can selectively recognise and eliminate malignant cells. Regulatory considerations include cell identity, purity, potency, viability, and stability, as well as risks related to uncontrolled proliferation, immune-mediated toxicity, or tumourigenicity [13, 14]. Because cellular therapies may persist or expand within the patient, long-term follow-up studies are often required to monitor safety and durability of response. The personalised nature of some somatic cell therapies also introduces manufacturing and logistical challenges that influence regulatory evaluation.
Tissue-engineered products are designed to regenerate, repair, or replace human tissues by combining cells with biomaterials or scaffolds that support structural integration and functional restoration. Unlike somatic cell therapies which primarily act through cellular activity, tissue-engineered constructs aim to reconstruct damaged tissue architecture and restore physiological function. Applications range from engineered skin substitutes for burn injuries to cartilage implants and bioengineered vascular grafts. Regulatory assessment focuses on the interaction between cellular and structural components, mechanical properties, biodegradation behaviour, vascularisation potential, and integration with host tissues [15, 16]. The multidisciplinary nature of tissue engineering, encompassing cell biology, materials science, and biomechanics, requires specialised evaluation to ensure that both biological safety and device-like performance characteristics are adequately addressed.
Combined ATMPs integrate an ATMP component with one or more medical devices as an integral part of the product, such as cell-seeded scaffolds, implantable matrices containing viable cells, or gene-activated biomaterials. These products exemplify the convergence of biologics and medical technology and therefore require coordinated regulatory evaluation addressing both medicinal product legislation governing the biological component and device regulations governing safety and performance. Ensuring compatibility between biological and mechanical elements is critical, as device properties may influence cell viability, distribution, or therapeutic function [9, 17]. The assessment of combined ATMPs often involves multidisciplinary expertise spanning cell biology, biomaterials engineering, clinical medicine, and regulatory science.
In the United States, products analogous to ATMPs are regulated by the Food and Drug Administration, primarily through the Center for Biologics Evaluation and Research. Regulatory oversight encompasses biological products, gene therapies, and human cells, tissues, and cellular and tissue-based products. Although terminology differs from the European framework, the underlying principles are broadly aligned, emphasising rigorous product characterisation, manufacturing controls, clinical evidence of safety and efficacy, and long-term monitoring [18, 19]. Special regulatory designations and expedited programmes have been introduced to facilitate development of regenerative therapies addressing serious conditions with unmet medical needs. Differences between jurisdictions nonetheless create challenges for global development, particularly regarding classification criteria, clinical trial requirements, and post-marketing obligations.
Beyond Europe and the United States, many countries are developing or refining regulatory pathways for advanced therapies, reflecting growing recognition of their clinical importance. International convergence initiatives seek to harmonise standards for quality control, potency assays, traceability, pharmacovigilance, and ethical oversight. Such collaboration is essential because ATMP development often involves multinational clinical trials, cross-border manufacturing, and global supply chains. Harmonisation efforts also aim to reduce duplication of regulatory requirements and facilitate timely patient access while maintaining rigorous safety standards [20, 21].
Regulatory classification has implications that extend throughout the product lifecycle. It determines applicable guidelines for non-clinical testing, clinical trial design, manufacturing validation, and risk management planning. For instance, gene therapies may require environmental risk assessments addressing potential shedding or transmission of viral vectors, whereas tissue-engineered products may necessitate evaluation of mechanical performance and degradation products. Classification also influences post-authorisation surveillance strategies, including patient registries and long-term follow-up studies designed to detect delayed adverse effects. Because ATMPs may produce durable biological changes, monitoring periods often extend for many years beyond initial treatment.
Manufacturing considerations further illustrate the importance of classification. ATMP production frequently involves complex, multi-step processes performed under stringent conditions to preserve cell viability and genetic integrity. Process variability can significantly affect product quality, making standardisation and validation critical components of regulatory assessment. Autologous therapies, produced from a patient’s own cells, present additional challenges related to batch size, logistics, and turnaround time, whereas allogeneic products raise concerns regarding immune compatibility and donor screening. Regulatory frameworks therefore emphasise traceability from source material to final product, ensuring that each batch can be linked to specific donors, manufacturing steps, and clinical outcomes.
Ethical and societal considerations are also intertwined with regulatory classification. Issues such as informed consent for donation of biological materials, equitable access to high-cost therapies, and long-term responsibilities for patient monitoring must be addressed within regulatory policies. Transparency in decision-making and public engagement are increasingly recognised as essential for maintaining trust in advanced therapies, particularly when treatments involve genetic modification or manipulation of living cells.
As the field continues to evolve, regulatory systems must remain adaptable to emerging technologies. Advances in genome editing, induced pluripotent stem cell technology, and biofabrication are likely to blur traditional boundaries between therapeutic categories, challenging existing definitions and evaluation criteria. Adaptive regulatory approaches, including conditional approvals and iterative evidence generation, are being explored to balance timely patient access with robust safety assurance. Ultimately, the effectiveness of regulatory classification frameworks will depend on their capacity to integrate scientific innovation, clinical needs, and societal expectations.
Manufacturing of ATMPs presents distinctive scientific, technical, and regulatory challenges that differ fundamentally from those associated with conventional pharmaceuticals and traditional biologics. Because ATMPs frequently involve living cells, genetic constructs, or engineered tissues, production processes must ensure not only product consistency but also preservation of biological functionality, viability, and safety. Manufacturing is therefore intrinsically linked to product performance, and even minor process variations may significantly influence clinical outcomes [10, 12].
Production typically begins with the procurement of starting materials, such as autologous or allogeneic cells, donor tissues, or genetic vectors. The quality, safety, and traceability of these materials are critical, as variability at this stage can propagate throughout the manufacturing process. Donor eligibility assessment, screening for infectious agents, and documentation of chain of custody are essential components of quality assurance systems. For autologous therapies, patient-specific manufacturing introduces additional logistical complexity, including strict timelines, controlled transport conditions, and coordination between clinical and manufacturing facilities [14].
Subsequent processing may include cell isolation, genetic modification, expansion, differentiation, and formulation under Good Manufacturing Practice (GMP) conditions. Unlike chemically synthesised drugs, ATMPs cannot be fully characterised by conventional analytical methods alone; instead, integrated phenotypic, functional, and molecular assays are required to demonstrate identity, purity, potency, and safety [16].
Potency testing represents a critical regulatory requirement. Because ATMPs act through complex biological mechanisms, potency assays must reflect the intended mechanism of action. Cell-based immunotherapies may require assays measuring cytotoxic activity or cytokine production, whereas gene therapies rely on assessments of transgene expression or vector functionality. Development and validation of such assays remain technically demanding but essential for product approval [17].
Scalability and process standardisation are major challenges, particularly for therapies originating from academic research environments. Transition to industrial manufacturing requires optimisation of culture systems, automation, closed processing technologies, and robust quality control strategies. Emerging approaches, including digital process monitoring, real-time analytics, and decentralised manufacturing platforms, aim to enhance reproducibility and efficiency while maintaining regulatory compliance [19].
Storage and distribution conditions are equally critical for maintaining product integrity. Many ATMPs require cryopreservation and controlled cold-chain logistics to preserve cell viability and biological activity. Deviations from specified conditions may compromise safety or efficacy, necessitating continuous monitoring during transport and handling [21].
Figure 2 presents a generalised manufacturing workflow for ATMPs, from starting material collection through processing, quality control, storage, and clinical administration. The workflow highlights critical control points where variability may arise and where regulatory oversight is particularly stringent, emphasising the principle that, for ATMPs, the manufacturing process is inseparable from the final therapeutic characteristics.
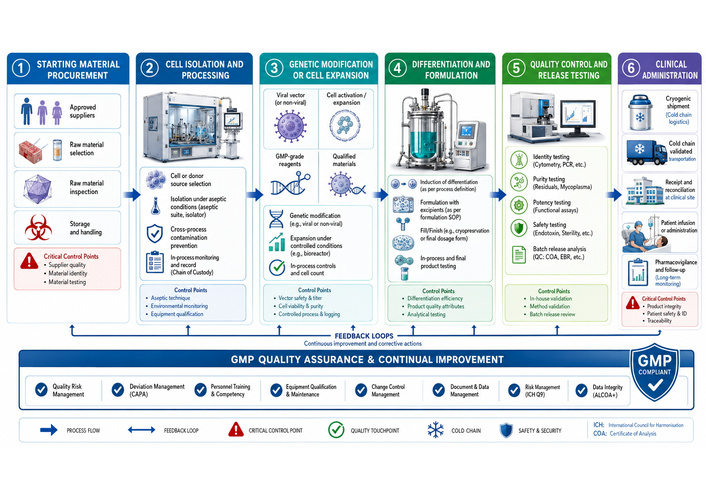
Manufacturing workflow and quality control framework for Advanced Therapy Medicinal Products. Generalised overview of the ATMP manufacturing pathway, beginning with procurement of starting biological materials and proceeding through cell or gene processing, expansion or modification under Good Manufacturing Practice (GMP) conditions, quality control testing, storage and distribution, and final clinical administration. Critical control points throughout the workflow highlight the importance of maintaining product identity, purity, potency, viability, sterility, and traceability. The figure emphasises that, for ATMPs, manufacturing processes are intrinsically linked to final therapeutic performance, safety, and regulatory compliance.
Regulatory authorities, including the European Medicines Agency and the Food and Drug Administration, require comprehensive documentation of manufacturing processes, validation procedures, and quality management systems to ensure consistent product quality and patient safety [22].
The clinical translation of ATMPs represents a major shift from experimental innovation to therapeutic reality, fundamentally redefining the objectives and possibilities of modern medicine. Unlike conventional pharmaceuticals, which typically provide symptomatic relief through transient biochemical interactions, ATMPs are designed to correct underlying disease mechanisms by restoring, replacing, or modifying biological function at molecular, cellular, or tissue levels. This approach has positioned ATMPs as therapeutic options for severe, rare, or otherwise untreatable conditions, often in patient populations with limited or no effective alternatives. As a result, clinical development programmes must balance urgent patient need with rigorous evaluation of safety, efficacy, durability, and long-term outcomes, recognising that these interventions may produce persistent or even lifelong biological effects. The transformative potential of ATMPs lies not only in their capacity to treat disease but also in their ability to alter the disease’s natural course, offering prospects for sustained remission or cure that were previously unattainable.
Early-phase clinical trials of ATMPs differ substantially from traditional drug development models. First-in-human studies must extend beyond conventional assessments of safety and dose escalation to characterise biological activity, persistence, biodistribution, and functional integration within the host organism. Gene therapies require careful evaluation of vector distribution across tissues, duration and regulation of transgene expression, and the potential for unintended genomic effects, including insertional mutagenesis or off-target editing. Cell-based therapies necessitate detailed analysis of in vivo expansion, migration to target sites, persistence over time, and risks such as uncontrolled proliferation, immune-mediated toxicity, or ectopic tissue formation. For tissue-engineered products, integration with host structures, vascularisation, and functional performance must also be assessed. Because adverse events may emerge long after administration, extended follow-up periods, often spanning years or decades, are essential to detect delayed complications and to evaluate the durability of therapeutic benefit [21]. These requirements challenge conventional clinical trial frameworks and necessitate innovative methodologies capable of capturing long-term outcomes, including adaptive trial designs, surrogate endpoints, and integration of real-world data.
Recent clinical successes demonstrate the therapeutic potential of ATMPs across diverse disease areas, illustrating the transition from proof-of-concept interventions to clinically meaningful treatments. Chimeric antigen receptor T-cell therapies have produced high remission rates in patients with refractory haematological malignancies, including those who have failed multiple prior lines of therapy. Gene replacement therapies have restored functional capacity in inherited disorders such as retinal dystrophies and neuromuscular diseases, providing tangible improvements in quality of life. Tissue-engineered constructs have enabled reconstruction of damaged tissues and organs, offering new options for patients with degenerative conditions, traumatic injuries, or congenital defects. Collectively, these achievements reflect a shift from symptomatic management towards disease modification and, in some cases, potential cure [22, 23]. Importantly, these therapies also demonstrate that biological interventions can achieve durable effects after a single administration, challenging the traditional paradigm of chronic pharmacological treatment.
In recent years, additional milestones have further validated the maturation of ATMPs as a therapeutic class. The clinical approval of CRISPR-based gene-editing therapies marks a historic transition from experimental genome engineering to routine clinical application. Trials involving ex vivo editing of haematopoietic stem cells for haemoglobinopathies have demonstrated sustained therapeutic benefit following a single intervention, including substantial reductions in disease burden, transfusion requirements, and hospitalisation rates. These developments provide compelling evidence that precise genomic modification can achieve durable correction of genetic disease and may serve as a model for future therapies targeting a wide range of monogenic disorders. Parallel advances in gene delivery technologies have enhanced the safety and efficacy of in vivo therapies. Next-generation adeno-associated viral vectors with improved tissue specificity, together with non-viral delivery systems such as lipid nanoparticles, are expanding the spectrum of treatable conditions while addressing concerns related to immunogenicity, manufacturing scalability, and off-target effects. Emerging genome-editing approaches, including base editing and prime editing, aim to increase precision while reducing genotoxic risk, further broadening the therapeutic landscape and enabling interventions for previously inaccessible mutations [24–26].
Real-world evidence is increasingly informing the evaluation of ATMP effectiveness and safety beyond controlled clinical trials. Long-term follow-up of patients receiving cellular immunotherapies has demonstrated durable remission in subsets of individuals with otherwise refractory disease, while also revealing late adverse events that necessitate ongoing surveillance. Patient registries, post-authorisation safety studies, and observational cohorts provide critical insights into treatment performance across diverse populations and clinical settings, complementing data obtained from pre-approval trials. These sources of evidence enable refinement of patient selection criteria, optimisation of dosing strategies, and development of risk mitigation measures. Integration of real-world data into regulatory decision-making is becoming increasingly important, particularly for therapies addressing rare diseases where traditional large-scale trials are impractical. The accumulation of long-term observational data will be essential for determining whether the initial benefits of ATMPs translate into sustained clinical outcomes over the lifespan of treated individuals.
Despite these advances, ATMPs present unique safety challenges that require comprehensive monitoring and proactive risk management. Immune responses to viral vectors or transplanted cells may compromise therapeutic efficacy or produce severe inflammatory reactions. Gene therapies carry the risk of insertional mutagenesis and oncogenic transformation, while immune cell therapies may induce cytokine release syndrome, neurotoxicity, or other immune-related adverse events due to excessive activation of the immune system. Tissue-engineered products may fail to integrate properly with host tissues or may provoke immune rejection. In addition, the long-term persistence of genetically modified cells raises questions regarding potential late complications, including secondary malignancies or autoimmune phenomena. Regulatory authorities therefore mandate robust risk management plans, including pharmacovigilance systems, long-term follow-up protocols, and patient registries designed to capture delayed adverse events and to monitor durability of response [27]. These measures reflect recognition that the safety profile of ATMPs cannot be fully characterised within the time frame of conventional clinical trials.
Economic and accessibility considerations constitute additional barriers to widespread implementation. The complex manufacturing processes required for ATMP production, often involving personalised or patient-specific approaches, contribute to exceptionally high treatment costs. Autologous cell therapies, for example, require individualised processing of patient-derived cells under stringent quality conditions, while gene therapies may involve sophisticated vector production and specialised delivery procedures. Although potentially curative therapies may reduce long-term healthcare expenditure by eliminating the need for chronic treatment, the substantial upfront investment poses significant challenges for reimbursement systems and healthcare budgets. Innovative payment models, including outcomes-based reimbursement schemes, annuity payments, and risk-sharing arrangements between manufacturers and payers, are being explored to align costs with therapeutic value and to improve affordability [28]. Ensuring equitable access remains a central policy challenge, particularly in low- and middle-income settings where healthcare resources and infrastructure may be limited.
Ethical considerations further complicate clinical translation. Informed consent is particularly challenging in contexts where patients face life-threatening conditions and may be willing to accept substantial uncertainty regarding risks. Paediatric applications raise additional concerns related to long-term monitoring and the potential for lifelong effects, especially when interventions are administered early in development. Genome-editing technologies introduce complex ethical questions regarding unintended germline modification and intergenerational implications, even when therapies are intended for somatic use. Issues related to fairness, prioritisation of limited resources, and global disparities in access also require careful deliberation. Transparent communication among clinicians, patients, regulators, and the public is therefore essential to maintain trust and to ensure responsible development and deployment of these therapies [29]. Ethical governance frameworks must evolve in parallel with technological innovation to address these challenges effectively.
Regulatory agencies such as the European Medicines Agency and the Food and Drug Administration have implemented adaptive pathways and accelerated approval mechanisms to facilitate timely patient access while maintaining rigorous standards of quality, safety, and efficacy. These frameworks recognise both the urgency of treating severe diseases and the limitations of traditional clinical trial designs for rare conditions. Conditional approvals, breakthrough designations, and regenerative medicine pathways allow therapies to reach patients earlier while requiring continued evidence generation through post-authorisation studies and real-world monitoring [28]. Such regulatory flexibility is essential for accommodating the unique characteristics of ATMPs, but it also necessitates robust systems for ongoing evaluation to ensure that early benefits are confirmed over time.
The rapid advancement of ATMPs has driven substantial evolution in regulatory frameworks worldwide. Conventional regulatory paradigms were originally developed for chemically defined medicinal products characterised by predictable pharmacokinetics, standardised manufacturing processes, and relatively short durations of action. In contrast, ATMPs encompass gene therapies, somatic cell therapies, tissue-engineered products, and combined products that exhibit intrinsic biological variability, complex mechanisms of action, and the potential for long-lasting or permanent effects. These distinctive characteristics challenge traditional approaches to product characterisation, clinical evaluation, manufacturing oversight, and post-authorisation monitoring. Consequently, regulatory authorities have adopted adaptive, science-based strategies designed to facilitate timely patient access while maintaining rigorous standards of quality, safety, and efficacy.
Within the European Union, the European regulatory system has established a specialised framework for ATMP evaluation that integrates multidisciplinary expertise across biological sciences, clinical medicine, and manufacturing. The centralised marketing authorisation procedure enables a single approval applicable across member states, promoting consistent regulatory standards and facilitating broader patient access within the European market. Early scientific advice procedures allow developers to engage with regulators during product development to clarify expectations regarding preclinical data, manufacturing controls, and clinical trial design. Conditional approval mechanisms permit earlier patient access in areas of high unmet medical need while requiring the completion of confirmatory studies after authorisation. Similarly, regulatory programmes in the United States provide expedited pathways for regenerative therapies that demonstrate the potential to address serious or life-threatening conditions, reflecting recognition of the limitations of conventional trial designs for rare diseases and small patient populations [24, 25]. Increasingly, regulators are also incorporating real-world evidence and long-term follow-up data into decision-making processes to better characterise the evolving benefit–risk profile of these therapies.
Despite regulatory facilitation, the economic sustainability of ATMPs remains a major concern. Development of advanced therapies requires substantial investment in specialised infrastructure, highly skilled personnel, and complex manufacturing technologies, particularly for personalised or autologous products. As a result, treatment costs often reach unprecedented levels. Although therapies with curative potential may reduce long-term healthcare expenditures by eliminating chronic treatment needs, the substantial upfront costs pose significant challenges for reimbursement systems and healthcare budgets. Innovative financing approaches are therefore being explored, including outcomes-based reimbursement linked to clinical performance, annuity payment models that distribute costs over time, and risk-sharing agreements between manufacturers and payers [26]. These mechanisms aim to balance financial sustainability with incentives for continued innovation while ensuring that patients can access transformative therapies.
Global disparities in access further complicate implementation. Most approved ATMPs are currently concentrated in high-income countries with advanced healthcare infrastructure, specialised treatment centres, and robust regulatory capacity. Patients in resource-limited settings face barriers related to cost, logistical complexity, and limited clinical expertise. Expanding access will require development of scalable manufacturing platforms, streamlined supply chains, and international regulatory harmonisation. Collaborative initiatives involving governments, international organisations, and industry stakeholders may facilitate technology transfer and capacity building to address these inequities and to promote more equitable distribution of therapeutic benefits [30–32].
Technological innovation is expected to shape the next generation of ATMPs and may help overcome many of the current limitations. Advances in genome editing, particularly CRISPR-based systems, are enabling increasingly precise modification of genetic sequences with expanding therapeutic applications demonstrated in recent clinical studies. Improvements in viral vector engineering and non-viral delivery platforms are enhancing tissue targeting, safety, and manufacturing scalability. Induced pluripotent stem cell technologies offer the possibility of generating standardised, off-the-shelf cellular products, while developments in biomaterials support the creation of more sophisticated tissue-engineered constructs. Automation, digital process control, and artificial intelligence-assisted manufacturing may improve product consistency and reduce costs, and decentralised production models could bring manufacturing closer to the point of care, thereby improving accessibility.
Ethical and societal considerations will remain central to future development. Public acceptance of genome editing and other transformative technologies depends on transparent governance and effective communication of both benefits and risks. Long-term monitoring of treated individuals raises important questions regarding data privacy, informed consent, and the responsibilities of healthcare systems to ensure ongoing surveillance. Ensuring equitable distribution of advanced therapies across populations and regions is essential to prevent widening disparities in health outcomes. Policymakers must therefore balance promotion of innovation with responsible oversight to ensure that therapeutic advances translate into meaningful public health benefits.
As ATMPs transition from experimental interventions to components of routine clinical practice, sustained collaboration among researchers, clinicians, regulators, industry stakeholders, and patient communities will be essential. Adaptive regulatory frameworks, sustainable economic models, and ethical governance structures must evolve alongside scientific progress. If these challenges can be successfully addressed, ATMPs have the potential to transform medicine by shifting the focus from chronic disease management towards restoration of biological function, long-term health, and potentially curative outcomes.
ATMPs represent a transformative frontier in modern medicine, offering therapeutic approaches that extend beyond symptomatic management towards biological repair, functional restoration, and, in some cases, potential cure. By employing genes, cells, or engineered tissues as active therapeutic agents, ATMPs redefine how diseases can be treated, particularly for conditions previously considered incurable or resistant to conventional therapies.
This review has examined the key principles underlying ATMP development, including regulatory classification, manufacturing challenges, clinical translation, safety considerations, and the evolving regulatory and economic environment. Together, these factors demonstrate that successful implementation of ATMPs depends on coordinated expertise across scientific, clinical, regulatory, and policy domains, as well as robust systems for long-term monitoring and evaluation.
Despite substantial progress, important barriers remain. Manufacturing scalability, long-term safety surveillance, affordability, and equitable access continue to constrain widespread adoption. Overcoming these challenges will require continued innovation in production technologies, adaptive regulatory strategies, sustainable reimbursement models, and international collaboration aimed at reducing disparities in access.
Future advances in gene editing, cell engineering, biomaterials, and precision medicine are expected to expand the therapeutic scope of ATMPs and improve their safety, efficacy, and scalability. As evidence accumulates from clinical trials and real-world implementation, these therapies are likely to become increasingly integrated into standard medical practice. Ensuring that this progress translates into safe, effective, and accessible treatments worldwide will be a central responsibility for the biomedical community in the coming decades.
ATMPs: Advanced Therapy Medicinal Products
CRISPR: clustered regularly interspaced short palindromic repeats
GMP: Good Manufacturing Practice
The authors would like to express their gratitude to the laboratory staff and research colleagues at the Food and Drug Investigation Laboratory, The Indonesian Food and Drug Authority, for their technical support and assistance during the preparation of this manuscript. The figures (Figure 1 and Figure 2) in this manuscript were generated with the assistance of ChatGPT for image visualization. The authors take full responsibility for the accuracy, originality, and integrity of all content presented in these figures.
AS: Conceptualization. DS and Normasari: Supervision. AH: Visualization, Writing—original draft & editing. All authors have read and approved the final manuscript.
The authors declare they have no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1803
Download: 37
Times Cited: 0
