
Open Access
Original Article
Heart rate variability as an objective biomarker for work-related musculoskeletal disorder risk: a cross-sectional study of Hong Kong (China) professionals
Adrian Low, Benny Lam
Published: July 09, 2026 Explor Musculoskeletal Dis. 2026;4:1007128
This article belongs to the special issue Prevalence and Risk Factors of Work-related Musculoskeletal Disorders

Open Access
Review
Innovations in paediatric orthopaedics: a narrative review of precision, personalisation, and biological restoration
Davor Bojić ... Filip Murn
Published: July 09, 2026 Explor Musculoskeletal Dis. 2026;4:1007129
This article belongs to the special issue Innovation in Orthopedics

Open Access
Original Article
Distal locking of intramedullary nails with reduced radiation exposure and operating time using standard equipment
Sebastien D’ulisse ... Sagi Martinov
Published: June 30, 2026 Explor Musculoskeletal Dis. 2026;4:1007127
This article belongs to the special issue Innovation in Orthopedics
Open Access
Original Article
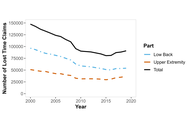
Work-related upper extremity and back injury claims across Canada from 2000–2019
Youssef Habib ... Peter J. Keir
Published: June 22, 2026 Explor Musculoskeletal Dis. 2026;4:1007125
This article belongs to the special issue Prevalence and Risk Factors of Work-related Musculoskeletal Disorders
Open Access
Mini Review
AI-driven spine-focused musculoskeletal management
Helen Gharaei ... Sonal Mahalwar
Published: June 22, 2026 Explor Musculoskeletal Dis. 2026;4:1007126
This article belongs to the special issue Data-Driven and AI-Based Approaches for Musculoskeletal Disease Monitoring and Rehabilitation
Open Access
Meta-Analysis
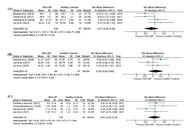
The impact of chronic low back pain on walking patterns: a systematic review and meta-analysis
Jianhong Gao ... Chen Soon Chee
Published: June 04, 2026 Explor Musculoskeletal Dis. 2026;4:1007124
Open Access
Case Report
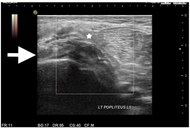
An unusual mechanism of isolated injury to the popliteus muscle related to dance: a case report
Jason Kok Kiong Chia
Published: May 08, 2026 Explor Musculoskeletal Dis. 2026;4:1007123
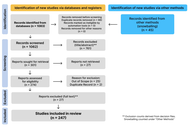
Open Access
Review
Standardizing infrared thermography for occupational applications: an integrative narrative review of protocol quality, thermal metrics, and multimodal integration in work-related musculoskeletal disorders
João Alberto de Souza Ribeiro, Luciana Aparecida Giacomini
Published: April 01, 2026 Explor Musculoskeletal Dis. 2026;4:1007122
This article belongs to the special issue Prevalence and Risk Factors of Work-related Musculoskeletal Disorders
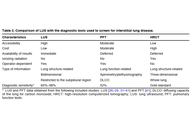
Open Access
Review
Advances in the validation of lung ultrasound for the detection of interstitial lung disease associated with rheumatoid arthritis
Esther F. Vicente-Rabaneda ... Ingrid Möller
Published: April 01, 2026 Explor Musculoskeletal Dis. 2026;4:1007121
This article belongs to the special issue Ultrasound as Outcome Measure in Rheumatic Diseases Trials
Open Access
Editorial
My take on innovation
Ashok N. Johari
Published: March 29, 2026 Explor Musculoskeletal Dis. 2026;4:1007120
This article belongs to the special issue Innovation in Orthopedics

Open Access
Review
Paediatric orthopaedics is changing—the significant innovations in recent years!
Ashok N. Johari, Ritesh A. Pandey
Published: March 29, 2026 Explor Musculoskeletal Dis. 2026;4:1007119
This article belongs to the special issue Innovation in Orthopedics

Open Access
Original Article
Association of neutrophil-to-lymphocyte ratio with peripheral neuropathy in rheumatoid arthritis
Binay Kumar Singh ... Shweta Tanwar
Published: March 26, 2026 Explor Musculoskeletal Dis. 2026;4:1007118
Open Access
Review
Work-related musculoskeletal disorders: prevalence and contributing risk factors—a narrative review
Mário Lopes, Marisa Lages
Published: March 13, 2026 Explor Musculoskeletal Dis. 2026;4:1007117
This article belongs to the special issue Prevalence and Risk Factors of Work-related Musculoskeletal Disorders
Open Access
Meta-Analysis
Work-related musculoskeletal disorder prevalence among African nurses: systematic review and meta-analysis
Philippe Gorce, Julien Jacquier-Bret
Published: February 12, 2026 Explor Musculoskeletal Dis. 2026;4:1007116
This article belongs to the special issue Prevalence and Risk Factors of Work-related Musculoskeletal Disorders

Open Access
Editorial
Biosimilars: state of the art in the treatment of rheumatic diseases
Valderilio Feijó Azevedo
Published: February 03, 2026 Explor Musculoskeletal Dis. 2026;4:1007115
This article belongs to the special issue Biosimilars: State of the Art in the Treatment of Rheumatic Diseases

Open Access
Editorial
Magnetic Resonance Neurography: redefining the diagnostic frontier in musculoskeletal disease
Theodoros Soldatos
Published: February 02, 2026 Explor Musculoskeletal Dis. 2026;4:1007114
This article belongs to the special issue Magnetic Resonance Neurography: Advances, Techniques, and Clinical Applications

Open Access
Case Report
Xanthoma simulating gouty tophus (case report of atypical cholesterol crystal deposition)
Maxim Sergeevich Eliseev ... Maria Nikolaevna Chikina
Published: December 24, 2025 Explor Musculoskeletal Dis. 2025;3:1007113
This article belongs to the special issue Evaluation and Outcomes in the Management of Gout

Open Access
Mini Review
Lycopene supplementation in rheumatic diseases: a comprehensive review
Jozélio Freire de Carvalho, Ana Tereza Amoedo Martinez
Published: December 09, 2025 Explor Musculoskeletal Dis. 2025;3:1007112
This article belongs to the special issue Complementary and Integrative Medicine in Rheumatology: Evidence, Therapies, and Clinical Impact
Open Access
Case Report
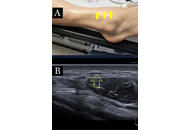
Ultrasound-guided dextrose hydrodissection for multiple peripheral entrapment neuropathies in scleroderma: a case presentation
Helen Gharaei ... Ziba Bagherian
Published: December 08, 2025 Explor Musculoskeletal Dis. 2025;3:1007111

Open Access
Mini Review
Current concepts on the intervention for adhesive capsulitis
Abeer Alomari, Philip Peng
Published: December 05, 2025 Explor Musculoskeletal Dis. 2025;3:1007110
