 Review
Review

Affiliation:
1Department of Medical Genetics, “Iuliu Haţieganu” University of Medicine and Pharmacy, 400437 Cluj-Napoca, Romania
ORCID: https://orcid.org/0000-0002-4726-915X
Affiliation:
1Department of Medical Genetics, “Iuliu Haţieganu” University of Medicine and Pharmacy, 400437 Cluj-Napoca, Romania
2Department of Internal Medicine, Radboudumc, 6525GA Nijmegen, the Netherlands
ORCID: https://orcid.org/0000-0001-6166-9830
Affiliation:
1Department of Medical Genetics, “Iuliu Haţieganu” University of Medicine and Pharmacy, 400437 Cluj-Napoca, Romania
2Department of Internal Medicine, Radboudumc, 6525GA Nijmegen, the Netherlands
Email: Tania.Crisan@umfcluj.ro
ORCID: https://orcid.org/0000-0002-3282-5627
Explor Musculoskeletal Dis. 2025;3:1007103 DOI: https://doi.org/10.37349/emd.2025.1007103
Received: May 08, 2025 Accepted: August 12, 2025 Published: August 28, 2025
Academic Editor: Naomi Schlesinger, University of Utah, USA
The article belongs to the special issue Pharmacological and Non-Pharmacological Management of Gout
Gout is a chronic inflammatory arthritis driven by monosodium urate crystal deposition and the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome activation, leading to interleukin-1β (IL-1β)-mediated inflammation. Recent studies reveal that hyperuricemia induces a state of immunological memory in innate immune cells through persistent epigenetic and metabolic reprogramming of monocytes and macrophages. These alterations enhance the responsiveness of innate immune cells, leading to exaggerated inflammatory reactions upon subsequent stimulation. This review synthesizes recent studies that elucidate how metabolic shifts (e.g., increased glycolysis and fumarate accumulation) and epigenetic changes (e.g., altered histone methylation and DNA methylation) reinforce this pathogenic memory. Importantly, these mechanistic insights provide the rationale for emerging therapeutic strategies: IL-1β inhibitors aim to interrupt the central inflammatory axis; metabolic modulators (e.g., metformin, statins) seek to reverse the trained metabolic state; and epigenetic therapies [e.g., histone deacetylase (HDAC) or DNA methyltransferase (DNMT) inhibitors] hold potential to reset dysregulated immune programming. Collectively, this review argues that multi-layered intervention, such as cytokine blockade for acute control, coupled with metabolic or epigenetic remodeling for long-term reprogramming, could yield sustained disease suppression and reduced flare frequency in gout.
Gout represents a widespread chronic inflammatory disorder affecting millions worldwide [1, 2]. At its core, it is primarily caused by needle-like monosodium urate (MSU) crystals, which are formed in the joints when serum urate (SUA) concentrations rise too high (hyperuricemia) [2]. It is characterized by sudden, severe attacks of joint pain, swelling, redness, and warmth, most commonly affecting the first metatarsophalangeal (MTP) joint, but also other joints such as the ankles, knees, wrists, and elbows [3]. It is characterized by self-limiting flares, which are attributed to the eventual clearance of crystals by macrophages and their secretion of anti-inflammatory mediators. However, subclinical inflammation may persist between flares, leading to chronic synovitis and progressive joint damage [4].
The pathogenesis of gout is multifactorial, with hyperuricemia representing a central but not exclusive prerequisite. Uric acid is the end product of purine metabolism in humans, and its systemic concentration is tightly regulated through a balance between production and excretion [5]. Renal excretion accounts for approximately two-thirds of uric acid elimination, primarily through complex processes of glomerular filtration, reabsorption, and tubular secretion mediated by specific urate transporters such as URAT1 (SLC22A12), GLUT9 (SLC2A9), OAT1/3 (SLC22A6/8), and ABCG2 [5]. Dysregulation or genetic variation in these transporters can impair renal urate handling, contributing to elevated SUA levels. In parallel, the gastrointestinal tract plays a complementary role in urate excretion, with intestinal transporters, particularly ABCG2, facilitating extra-renal clearance [6]. Emerging evidence also implicates gut microbiota in urate metabolism [7, 8], suggesting a broader interplay between host genetics, renal and intestinal excretion pathways, and microbial function in the pathophysiology of hyperuricemia and gout. Clinically, gout is characterized by acute inflammatory flares triggered by innate immune responses to MSU crystals, which activate the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome and drive the release of interleukin-1β (IL-1β), a central mediator of gouty inflammation, where neutrophil infiltration further amplifies the inflammatory cascade [9]. Moreover, chronic tophaceous gout, marked by the accumulation of tophi formed by the accumulation of MSU crystals, represents the end stage of uncontrolled disease. However, for a more comprehensive discussion of the immunologic, genetic, and environmental factors contributing to gout pathogenesis, the reader is referred to recent in-depth reviews on the topic [3, 9].
Traditionally considered an episodic crystal-induced arthritis, gout is now increasingly recognized as a disease with underlying dysregulation of innate immune memory mechanisms. Growing evidence supports the central hypothesis of this review: that trained immunity, defined as persistent epigenetic and metabolic reprogramming of innate immune cells, plays a critical role in gout pathogenesis and progression, contributing to heightened inflammatory responsiveness even in the presence of urate-lowering therapy (ULT). Understanding these mechanisms opens new therapeutic opportunities by directly targeting the maladaptive immune memory that sustains chronic inflammation [10].
Trained immunity is a phenomenon that involves a set of myeloid cells, like monocytes, macrophages, or natural killer cells, which establish a type of innate immune “memory” by altering their response following exposure to certain stimuli [11–13]. Up to this date, observational studies have demonstrated that innate immune cells can undergo epigenetic, transcriptional, and metabolic reprogramming resulting in a heightened and sustained response to future triggers/stimuli [14–16]. Metabolism and epigenetics serve as fundamental pillars of trained immunity, engaging in a dynamic and reciprocal interplay [17]. Altered metabolic pathways not only provide the energy and biosynthetic precursors for immune activation but also generate key metabolites, such as acetyl-CoA and fumarate, which directly modify the epigenetic landscape by influencing histone acetylation and methylation, thereby regulating pro-inflammatory gene expression [18]. Consequently, recent research increasingly implicates these interconnected pathways in gout and related rheumatic diseases [10].
In gout, hyperuricemia can induce transcriptional and epigenetic reprogramming of monocytes, resulting in maladaptive immune responses and chronic inflammation [19, 20]. Once “trained”, these cells exhibit an enhanced pro-inflammatory response upon secondary exposure, even if the stimulus differs from the original one. Thus, in the context of gout, external stimuli, such as soluble urate, prime the innate immune system for heightened responsiveness to MSU crystals by inducing epigenomic reprogramming [21, 22], resulting in sustained upregulation of NLRP3 inflammasome components, glycolytic enzymes, and pro-inflammatory cytokines such as IL-1β [23]. In addition to this, a new line of research points to the implications of long non-coding RNAs (lncRNAs) as pivotal regulators in the modulation of trained immunity, highlighting their involvement in the epigenetic reprogramming that underpins this process [24].
Clonal hematopoiesis of indeterminate potential (CHIP) is a phenomenon characterized by the expansion of hematopoietic stem and progenitor cell (HSPC) clones harboring somatic mutations in genes involved in epigenetic regulation [e.g., ten-eleven translocation-2 (TET2), DNA methyltransferase 3A (DNMT3A)], DNA damage repair, and inflammatory signaling components such as NLRP3 [25]. While initially linked to hematologic malignancies, CHIP is now recognized as a key driver of systemic inflammation, contributing to cardiovascular disease [26], autoimmunity [27, 28], and other inflammatory conditions such as gout [29, 30].
In this review, we synthesize recent mechanistic insights into the integrated roles of metabolic dysregulation, epigenetic reprogramming, and innate immune memory in driving gout pathogenesis. We also explore emerging therapeutic strategies aimed at targeting these maladaptive immune memory mechanisms to achieve more durable disease control and reduce flare recurrence. We conclude the review by discussing CHIP as a representative example of hematopoietic reprogramming that reflects similar principles of persistent immune activation and may offer broader insight into chronic inflammatory diseases such as gout.
IL-1β is a pro-inflammatory cytokine that plays a central role in gout pathogenesis and trained immunity [31]. As a product of NLRP3 inflammasome activation, IL-1β is released in response to MSU crystals, driving acute inflammation [32, 33]. However, beyond this acute phase, IL-1β also contributes to long-term immune reprogramming, a process characteristic of trained immunity [34, 35]. Therefore, upon activation by various stimuli, such as MSU crystals, the NLRP3 inflammasome assembles and activates caspase-1. Activated caspase-1 then processes pro-IL-1β into its mature form, IL-1β, which is subsequently secreted to mediate inflammatory responses [36]. Moreover, evidence from the literature suggests that IL-1β can independently induce a trained immunity phenotype in human monocytes, similar to oxidized low-density lipoprotein (oxLDL), β-glucan, or Bacillus Calmette-Guerin (BCG) [37, 38]. This process is driven by epigenetic reprogramming, marked by reduced H3K9me3 and increased H3K4me3 at the promoter regions of IL-1β, IL-6, and TNF [39]. In line with this, Arts et al. [39] previously reported that, under in vitro conditions, exposing human monocytes to IL-1β at concentrations of 1 ng/mL and 10 ng/mL for 24 h induces trained immunity. Consequently, a recent study provides evidence that IL-1β exposure induces trained immunity in hematopoietic progenitor cell (HPC)-derived monocytes, leading to increased inflammatory cytokine production, metabolic activity, and atherogenic gene expression after restimulation [40]. Hence, it can be assumed that IL-1β, which is the main cytokine described in gout, may induce trained immunity by sensitizing the innate immune cells in a way that makes them hyper-responsive to MSU crystals and additional gout triggers. While conventional gout therapies focus on symptom control [non-steroidal anti-inflammatory drugs (NSAIDs), colchicine] or SUA reduction (allopurinol, febuxostat), they do not directly reset trained immunity. However, because IL-1β is a key mediator of both acute gout inflammation and trained immunity, blocking this cytokine with IL-1 inhibitors such as anakinra or canakinumab may theoretically help modulate trained immunity and prevent excessive inflammatory priming. The mode of action of these therapies is to interrupt the IL-1β-driven inflammatory cycle [41, 42], which might prevent epigenetic and metabolic reprogramming, and offer long-term immunomodulation. Therefore, by targeting IL-1β, these agents do not merely suppress symptoms; they may also modulate the underlying immune memory that contributes to recurrent inflammation. This represents a shift from symptomatic treatment to modifying the disease’s immunopathology.
Central of this inflammatory event lies the NLRP3 inflammasome, a multiprotein complex that mediates the activation of caspase-1 and the subsequent maturation and release of pro-inflammatory cytokines in response to MSU crystals [36]. Persistent or excessive activation leads to the production of high concentrations of IL-1β, which recruits and activates additional immune cells, resulting in the characteristic painful flares and joint damage associated with the disease [32]. Therefore, in order to mitigate this, the use of NLRP3-specific inhibitors such as MCC950 or OLT1177 (dapansutrile) holds great potential. MCC950 is a small-molecule inhibitor that selectively targets NLRP3 inflammasome activation, preventing its assembly and subsequent inflammatory cascade [43, 44]. Preclinical studies have demonstrated the potential of MCC950 in blocking the inflammatory process observed in gout. In murine models of MSU crystal-induced arthritis, MCC950 significantly reduced IL-1β levels, neutrophil infiltration, and joint swelling [44, 45]. In the same line, OLT1177 is an orally active NLRP3 inhibitor that blocks caspase-1 activation and IL-1β secretion [46, 47]. In LPS-stimulated human blood-derived macrophages, OLT1177 has been shown to decrease IL-1β levels by 60% and IL-18 by 70% at concentrations significantly lower than those safely achieved in human plasma [47]. By inhibiting the NLRP3 activation, OLT1177 can potentially reduce cytokine production, thereby alleviating inflammation and modulating the trained immune response associated with recurrent gout flares. Hence, these molecules offer a promising therapeutic approach for managing gout by addressing both acute inflammation and the underlying trained immunity that contributes to disease recurrence with a unique potential to prevent trained immunity-related inflammation in gout.
Metabolites serve as substrates, intermediates, or end products of metabolic reactions that govern cellular function. Consequently, they offer critical insights into the interplay between gene expression and environmental influences, making them highly informative and sophisticated biomarkers for disease [48]. In gout, MSU crystals have been implicated in long-lasting changes in monocytes and macrophages, including alterations in cellular metabolism and histone modifications [49–52]. These changes result in heightened inflammatory signaling, reinforcing the cycle of recurrent flares. Additionally, it is known that a hallmark of trained immunity is the shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, commonly referred to as the Warburg effect [16]. Therefore, this metabolic shift is supported by upregulated expression of key enzymes that drive increased glycolysis, such as [hexokinases (HKs; HK1 and HK2), lactate dehydrogenase A (LDHA), or pyruvate kinase M2 (PKM2)] and glutaminolysis [glutaminase 1 (GLS1) or glutamate dehydrogenase (GDH)]. Further, this shift is facilitated by factors such as mechanistic target of rapamycin (mTOR) and hypoxia-inducible factor 1-alpha (HIF-1α) [17, 53, 54]. In gout, this glycolytic phenotype correlates with enhanced production of IL-1β and other pro-inflammatory cytokines.
To further elaborate on the idea of modulating the metabolic pathways characteristic of trained immunity, experimental models have shown that metformin can reduce inflammatory responses in monocytes by dampening glycolysis and altering the epigenetic landscape that drives trained immunity [38]. Moreover, some epidemiological studies suggest that patients taking metformin for type 2 diabetes may have a lower incidence of gout flares, potentially reflecting metformin’s anti-inflammatory effects [55]. In line with this, a large cohort study demonstrated that metformin users had a significantly lower incidence of gout compared to non-users, with a 32% relative risk reduction. However, metformin did not significantly alter serum uric acid or C-reactive protein levels, suggesting its protective effect may be independent of urate-lowering or anti-inflammatory actions [56].
Also, as mTOR represents another key player and critical regulator of cellular metabolism that supports the shift towards glycolysis in trained immune cells, for which rapamycin emerged as a very effective inhibitor in other inflammatory conditions [57]. In diet-induced obese mice, rapamycin treatment led to beneficial metabolic effects, including enhanced energy expenditure and insulin sensitivity, associated with increased regulatory T-cells and myeloid-derived suppressor cells in adipose tissue [58]. Nevertheless, studies have demonstrated that rapamycin can downregulate dendritic cell activation markers and decrease the secretion of inflammatory cytokines such as IL-6, TNF, and IL-1β in rheumatoid arthritis (RA) models [59]. Moreover, clinically, rapamycin has been utilized in combination therapies, such as SEL-212 (a combination of pegadricase and rapamycin), to mitigate immunogenicity and enhance the efficacy of uricase treatments in refractory gout patients [60]. Hence, based on this rationale, targeting metabolic reprogramming by using metformin or rapamycin to shift immune cells back to their normal OXPHOS as well as reducing the excessive inflammatory responses of the immune cells may represent an accessible approach in the treatment of gouty inflammation and by targeting the mTOR pathway offering a promising strategy in order to modulate the observed trained immunity phenotype in gout. This metabolic approach could reduce flare frequency, prevent chronic inflammation, and complement ULTs.
In addition, the mevalonate pathway, responsible for cholesterol biosynthesis, is another critical metabolic axis in trained immunity; it regulates monocyte differentiation, inflammasome activation, and cytokine production [37]. Statins, which inhibit this pathway, have been shown to have immunomodulatory effects beyond their cholesterol-lowering properties, making them potential candidates for treating autoimmune diseases [61]. Statins can suppress the activation of innate immune cells, disrupt the inflammasome signaling, and alter the epigenetic memory, phenomena observed in the pathogenesis of gout. While direct evidence linking statin use to reduced inflammation specifically in hyperuricemic individuals is limited, several studies have explored the relationship between statin therapy, SUA concentrations, and gout risk [62, 63]. Therefore, statins have been shown to significantly reduce SUA concentrations, with atorvastatin demonstrating the most substantial effect [63]. Moreover, there is evidence suggesting that statins reduce systemic inflammation and may alter immune responses implicated in gouty arthritis, including decreased production of pro-inflammatory cytokines (IL-1β, IL-6, TNF) [64]. In line with this, statins can shift macrophages towards an anti-inflammatory phenotype, resulting in decreased secretion of pro-inflammatory cytokines [64]. Additionally, statin use has been associated with a reduced risk of mortality in gout patients, suggesting cardiovascular benefits in this population [65].
In summary, while metformin and statins may offer indirect benefits in gout management through risk reduction and uric acid modulation, rapamycin’s role appears more directly linked to therapeutic strategies, especially in combination treatments for refractory cases. Further research is warranted to fully elucidate their roles and optimize treatment protocols for gout patients.
The above presented mechanisms were shown to contribute to epigenetic rewiring, which is the molecular mechanism of trained immunity. Ultimately, trained immunity is mediated by epigenetic modifications that induce chromatin remodeling, thereby exposing promoter and enhancer regions governing immune-related gene expression to transcription factors [66]. The persistence of these epigenetic marks maintains cells in a “trained” state, characterized by increased accessibility of pro-inflammatory gene loci, which in turn facilitates a more rapid and robust transcriptional response upon subsequent stimulation [66]. Metabolic shifts, including increased aerobic glycolysis, glutamine metabolism, and cholesterol synthesis, are key drivers of the epigenetic changes underlying trained immunity [67]. Therefore, epigenetic mechanisms, particularly chromatin modifications, are critical in sustaining cellular memories and supporting adapted phenotypes of monocytes and macrophages in trained immunity [18, 68].
In gout, urate can induce such transcriptional and epigenetic reprogramming of monocytes and macrophages, which contributes to the persistent inflammation observed [19]. Moreover, there is evidence that differential DNA methylation patterns in gout patients reveal altered signaling pathways related to immunity and osteoclastogenesis [69]. Methylation patterns are written and maintained by DNMTs and can be removed or altered by TET enzymes or passive demethylation. Notably, epigenetic changes may be reversible, making them attractive targets for therapeutic intervention.
Recent research underscores the significance of epigenetic modifications, particularly DNA methylation, in maintaining inflammation and promoting trained immunity in monocytes/macrophages [70]. DNA methylation, mediated by DNMTs, controls gene expression, typically leading to transcriptional repression. In gout, dysregulated DNA methylation may alter the expression of key inflammatory mediators, including cytokines like IL-1β, TNF. Moreover, cell lineage-specific methylation alterations were observed in gout, with several loci associated with IL-1β regulation in monocytes [71]. Tseng et al. [71] identified seven CpG loci linked to IL-1β production and gouty inflammation, independent of hyperuricemia, including novel genes such as geranylgeranyltransferase type I subunit beta (PGGT1B), insulin-induced gene 1 (INSIG1), and angiopoietin-like protein 2 (ANGPTL2). Also, another study revealed differential DNA methylation patterns in signaling, immunity, and osteoclastogenesis pathways in gout patients [59].
Based on this evidence, the use of DNMT inhibitors such as 5-azacytidine or decitabine, having the potential of leading to either global or specific hypomethylation [72], might be taken into account for new treatment strategies in controlling gouty inflammation. These inhibitors, already used in cancer treatment, could potentially reactivate silenced anti-inflammatory genes or correct the methylation imbalance that sustains the pro-inflammatory responses observed in gout as well. Furthermore, by enhancing TET enzyme activity, it may be possible to restore a balanced immune response by facilitating active DNA methylation. Thus, TET2 activation could counteract trained immunity and excessive monocyte priming, for which small molecules such as α-ketoglutarate (α-KG) or 2-hydroxyglutarate (2-HG) are potential candidates to enhance TET activity or DNA demethylation [53, 73]. Several lines of evidence have shown that α-KG and 2-HG not only have a role in promoting an anti-inflammatory phenotype by regulating gene expression in immune cells, but also inhibit pro-inflammatory macrophage (M1) polarization and promote an anti-inflammatory (M2) macrophage phenotype [74] in other inflammatory diseases such as RA [75]. A review by Liu et al. [76] highlighted the regulatory role of α-KG in macrophage polarization. The authors emphasized that α-KG’s influence on macrophage function could have therapeutic implications for various inflammation-related diseases [76].
Given that these diseases share common inflammatory and metabolic pathways with gout, it is plausible that therapeutic strategies effective in these conditions may also be beneficial in the treatment of gout. Further, by targeting DNA methylation, this could provide new therapeutic approaches for gout treatment, focusing on the regulation of inflammatory pathways rather than solely addressing hyperuricemia.
Histone modifications such as acetylation, methylation, and phosphorylation are key epigenetic regulators that alter gene expression without changing the DNA sequence [18]. These modifications influence chromatin accessibility, impacting inflammatory pathways relevant to gout, and play a crucial role in regulating the inflammatory responses. The exposure to MSU crystals and chronic hyperuricemia has been shown to induce aberrant histone modifications in monocytes and macrophages. These changes can result in a “trained” pro-inflammatory state, characterized by sustained upregulation of cytokines like IL-1β, TNF [22, 49]. Histone deacetylase (HDAC) inhibitors have demonstrated efficacy in various inflammatory conditions, including RA, psoriasis, inflammatory bowel disease, and multiple sclerosis [77–79]. A previous study has observed that romidepsin (an HDAC1/2 inhibitor) turned out to be very effective in reducing C16.0 + MSU-induced IL-1β production compared to other specific class I HDAC inhibitors [80]. It is intriguing to consider that these inhibitors could potentially suppress the epigenetic imprinting that maintains inflammation in gout. Furthermore, preclinical studies in other inflammatory models have demonstrated that modulating histone methylation can significantly alter cytokine production and immune cell phenotype [81, 82]. A good example for this is a murine model of collagen-induced arthritis, where the enhancer of zeste homolog 2 (EZH2) inhibitor GSK126 was used to modulate the inflammatory phenotype of macrophages. Inhibition of EZH2, which catalyzes the repressive mark H3K27me3, led to a significant reduction in joint inflammation and lower concentrations of pro-inflammatory cytokines [83]. In addition, research in experimental models of colitis demonstrated that modulation of the histone demethylase Jumonji domain-containing protein-3 (JMJD3) can regulate macrophage activation. In one key study, JMJD3 was shown to link inflammation with epigenetic control by demethylating H3K27me3, thereby facilitating the expression of pro-inflammatory genes. Inhibition of JMJD3 resulted in reduced cytokine production and improved disease outcomes in murine colitis [82].
These examples form a scientific foundation for considering histone modification—targeting approaches as potential therapeutic interventions in gout. Also, knowing that abnormal methylation patterns may sustain the expression of pro-inflammatory genes, histone methylation modifiers might represent a promising alternative approach in treating gouty inflammation.
LncRNAs are key regulators of gene expression and chromatin remodeling in trained immunity [24]. Specific lncRNAs modulate monocyte/macrophage function in response to inflammatory stimuli, influencing cytokine secretion and metabolic reprogramming [84]. Moreover, recent studies have highlighted the involvement of lncRNAs in the epigenetic reprogramming that underpins trained immunity. A notable example is the identification of “immune-gene priming lncRNAs (IPLs)”, which facilitate the accumulation of the histone modification H3K4me3 at promoters of pro-inflammatory genes, thereby enhancing their transcriptional readiness [85].
Therefore, by targeting specific lncRNAs that regulate the epigenetic landscape of innate immune cells, it may be possible to attenuate the heightened inflammatory responses characteristic of gout flares. For instance, inhibiting lncRNAs that promote pro-inflammatory gene priming could reduce the severity and frequency of acute gout episodes. However, research in this area is still in its early stages. Comprehensive profiling of lncRNA expression in gout patients has been initiated to shed light on their potential roles in disease pathogenesis. A study utilizing microarray analysis identified distinct lncRNA expression patterns in individuals with primary gout, suggesting their involvement in the disease process [86]. Moreover, lncRNAs such as HOX transcript antisense RNA (HOTAIR) and small nucleolar RNA host gene 8 (SNHG8) have been found to regulate inflammatory processes in gout. HOTAIR knockdown alleviates gouty arthritis by upregulating miR-20b and downregulating NLRP3, thereby suppressing inflammatory cytokine secretion [87]. Similarly, SNHG8 accelerates acute gouty arthritis development by sponging miR-542-3p and upregulating adaptor-related protein complex 3 subunit delta 1 (AP3D1), leading to increased paw swelling and pro-inflammatory factor production in mice [88]. Their selective upregulation during flares and their functional role in sustaining IL-1β production make them attractive therapeutic targets for modulating trained immunity. Therefore, these lncRNAs could be developed into novel therapeutic targets, leading to improved treatment strategies by addressing the underlying epigenetic mechanisms that contribute to gout’s chronic and recurrent nature. However, several critical challenges must be addressed before clinical application. The primary barrier to therapeutic lncRNA modulation is the effective and cell-specific delivery of antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), or CRISPR-based epigenetic editors. Current strategies being explored include the use of lipid nanoparticles (LNPs)—clinically validated for mRNA vaccines and siRNA therapies—which allow for targeted delivery to the liver and immune cells [89, 90]. In addition, engineered exosomes are gaining attention as natural, immunologically inert vehicles capable of delivering RNA-based therapeutics to monocytes and macrophages [90]. Other promising approaches involve N-acetylgalactosamine (GalNAc)-conjugation or aptamer-based targeting, which may enable more precise delivery to inflamed tissues or specific immune cell subsets [90]. Still, as lncRNAs often have complex and pleiotropic functions, including interactions with chromatin regulators, transcription factors, and miRNAs, the risk of off-target effects needs to be taken into consideration as well. Furthermore, patient-specific variability in lncRNA expression driven by genetic polymorphisms, epigenetic modifications could influence treatment responses.
Accordingly, while conventional treatments primarily address acute inflammation and urate concentrations, emerging epigenetic-based strategies could provide long-term immunomodulation, reducing flare recurrence and chronic gout progression.
CHIP is a condition in which a subset of hematopoietic stem cells (HSCs) in the bone marrow acquire somatic mutations, leading to the expansion of mutant blood cell clones [25, 91]. The presence of CHIP-associated somatic mutations in genes such as TET2, DNMT3A, ASXL1, and JAK2 leads to persistent immune activation through mechanisms resembling trained immunity, in which innate immune cells exhibit long-term epigenetic and metabolic reprogramming that enhances their inflammatory response [92, 93]. Notably, evidence suggests that CHIP carriers have higher concentrations of inflammatory markers like C-reactive protein [94], therefore CHIP can be seen as an amplifier of inflammation through abnormal immune activation and excess IL-1β production [30], meaning that even without infections or other triggers (such as urate or MSU crystals), subjects with CHIP can have a baseline higher level of inflammation, this leading to a stronger inflammatory reaction in the joints. Moreover, mechanistically, loss-of-function mutations in TET2 impair active DNA demethylation, leaving permissive chromatin marks (H3K4me3, H3K27ac) on the IL-1β, NLRP3, and TNF promoters, where the resulting monocytes and macrophages enter a “primed” state that constitutively overexpresses these cytokine genes. Parallel gain-of-function variants in NLRP3 (e.g., p.Q705K) lower the activation threshold of the NLRP3 inflammasome, expediting apoptosis-associated speck-like protein containing a CARD (ASC) speck formation, caspase-1 cleavage, and release of mature IL-1β and IL-18 [95]. In TET2-knockout mouse models, this dual hit—heightened transcriptional priming plus hyper-responsive inflammasome activation—translates into markedly increased MSU-induced joint inflammation and a higher flare frequency [30]. Hence, in individuals with TET2- or NLRP3-mutant CHIP, otherwise asymptomatic hyperuricemia and crystal deposition are converted into disproportionately intense IL-1β-driven responses, providing a clear mechanistic rationale for considering CHIP a catalyst for gout pathogenesis and flare severity [29]. This dual molecular hit promotes excessive IL-1β production and heightened MSU-induced inflammation, even in the setting of asymptomatic hyperuricemia.
In this context, RG108, a non-nucleoside DNMT1 inhibitor, emerges as a potential therapeutic agent [96]. Evidence from the literature indicates that RG108 can mitigate cellular damage induced by oxidative stress by restoring abnormal methylation patterns in aging-related genes, thereby enhancing treatment effectiveness in age- and inflammation-associated diseases [97]. Additionally, by inhibiting DNMT activity and thereby reversing the epigenetic modifications that underpin trained immunity, RG108 could theoretically dampen the hyperinflammatory state observed in CHIP carriers. Therefore, its ability to modulate DNA methylation offers a promising approach to reprogram maladaptive innate immune memory, potentially reducing IL-1β production and inflammatory flares associated with gout. Because CHIP-mutant immune cells release more pro-inflammatory cytokines, CHIP is now recognized as a contributor to chronic systemic inflammation [98]. In line with this, CHIP mutations may induce a form of innate immune memory, where these cells are in constant readiness, leading to increased IL-1β production and prolonged inflammation. Consequently, it is reasonable to propose that CHIP mutations may contribute to the persistence of trained monocytes/macrophages in gout, sustaining inflammation even in the absence of acute crystal deposition. Moreover, given that CHIP mutations sustain a pro-inflammatory state through epigenetic modifications, metabolic reprogramming, and increased myeloid cell activation [92], targeting these pathways may offer novel therapeutic opportunities for gout (Table 1).
Potential therapeutic targets within trained immunity pathways in gout
| No. | Category | Modulator | Mechanism of action |
|---|---|---|---|
| 1 | Cytokine targeting therapies | IL-1 inhibitors (e.g., anakinra, canakinumab) | Neutralize IL-1β, preventing trained monocyte/macrophage hyperactivity [41, 42] |
| 2 | Inflammasome inhibitors | NLRP3 inhibitors (e.g., MCC950, OLT1177) | Prevent NLRP3 inflammasome assembly, reducing IL-1β secretion [43, 44, 46, 47] |
| 3 | Metabolic modulators | AMPK activators (e.g., metformin, AICAR) | Activate AMPK, shifting metabolism away from inflammatory glycolysis [38] |
| 4 | mTOR inhibitors (e.g., rapamycin, everolimus) | Inhibit mTOR signaling, suppressing glycolysis-dependent immune activation [58, 59] | |
| 5 | Statins (e.g., atorvastatin, simvastatin) | Reduce mevalonate pathway flux, disrupting monocyte/macrophage training [61, 64] | |
| 6 | Epigenetic modulators | DNMT inhibitors (e.g., 5-azacytidine; α-ketoglutarate) | Inhibit DNA methylation, reversing epigenetic priming in monocytes [71, 72] |
| 7 | HDAC inhibitors (e.g., vorinostat, panobinostat) | Suppress histone deacetylation, reducing inflammatory gene expression [81–83] | |
| 8 | LncRNA modulators | HOTAIR (HOX transcript antisense RNA) inhibitors | Regulates chromatin remodeling, influencing inflammatory gene expression in trained monocytes [87] |
| 9 | SNHG8 (small nucleolar RNA host gene 8) modulators | Modulates metabolic pathways involved in monocyte activation and cytokine production [88] | |
| 10 | Non-nucleoside DNMT1 inhibitor | RG108 | Restores normal DNA methylation by inhibiting DNMTs, reducing oxidative stress-induced damage [97] |
IL-1: interleukin-1; mTOR: mechanistic target of rapamycin; lncRNA: long non-coding RNA; DNMT: DNA methyltransferase; NLRP3: NACHT, LRR, and PYD domains-containing protein 3; HDAC: histone deacetylase
Furthermore, given the presence of histone modifications in the context of CHIP and trained immunity, HDAC inhibitors are emerging as promising therapeutic agents for inflammatory diseases due to their epigenetic regulatory effects on the immune system [99]. Nevertheless, given their heightened inflammasome activation, patients with CHIP-associated inflammation may preferentially respond to IL-1 inhibitors (e.g., anakinra, canakinumab). Therefore, the intersection of clonal hematopoiesis, trained immunity, and gout represents a novel framework for understanding persistent inflammation and recurrent flares in hyperuricemia-driven disease (Figure 1). Moreover, given the shared pathways between CHIP and inflammatory myeloid activation, we foresee that future therapies targeting epigenetic regulation, inflammasome activation, and metabolic reprogramming hold promise for modulating CHIP-driven immune priming in gout. Likewise, as CHIP detection becomes more accessible, identifying CHIP-positive gout patients may lead to personalized treatment strategies, leveraging epigenetic and metabolic interventions to disrupt the persistent inflammatory phenotype that sustains gout pathology. Further clinical studies are required to validate these approaches and assess their efficacy in modifying disease progression.
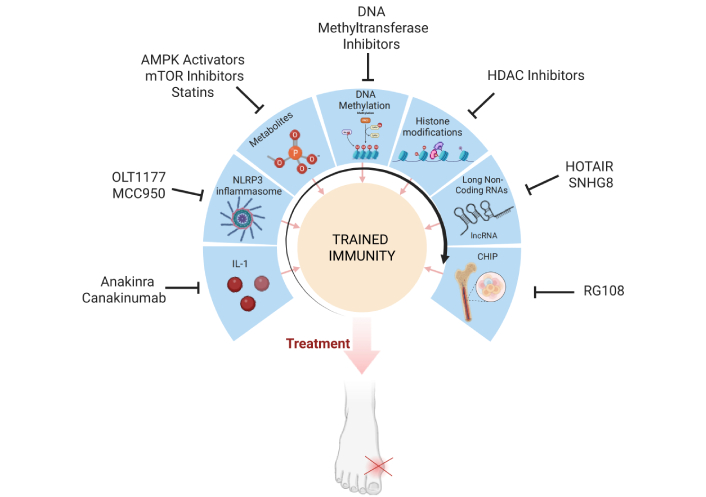
Emerging therapeutic strategies targeting trained immunity pathways in gout. Multiple levels of trained immunity regulation are therapeutically targetable to modulate gout inflammation, including IL-1 blockade (anakinra, canakinumab), inflammasome inhibitors (OLT1177, MCC950), metabolic modulators (AMPK activators, mTOR inhibitors, statins), and epigenetic interventions (DNMT inhibitors, HDAC inhibitors, lncRNA regulators such as HOTAIR and SNHG8). Targeting clonal hematopoiesis of indeterminate potential (CHIP) represents an additional upstream strategy to suppress sustained myeloid-driven inflammation. IL-1: interleukin-1; mTOR: mechanistic target of rapamycin; lncRNA: long non-coding RNA; HOTAIR: HOX transcript antisense RNA; SNHG8: small nucleolar RNA host gene 8; DNMT: DNA methyltransferase; HDAC: histone deacetylase; NLRP3: NACHT, LRR, and PYD domains-containing protein 3. Created in BioRender. Cabau, G. (2025) https://BioRender.com/lw9ba8g
This review highlights the evolving understanding of gout as a disease fundamentally linked to trained immunity. Research increasingly demonstrates that epigenetic and metabolic reprogramming in innate immune cells, such as monocytes and macrophages, results in heightened inflammatory responses upon subsequent stimulation. Mounting evidence shows that hyperuricemia not only initiates crystal-driven inflammation but also imprints a lasting pro-inflammatory memory on monocytes and macrophages. This maladaptive immune priming leads to exaggerated production of cytokines such as IL-1β, driven in part by sustained activation of the NLRP3 inflammasome and other inflammatory pathways. Despite the growing interest in trained immunity and immune modulation, it is important to emphasize that hyperuricemia remains the essential upstream driver of gout. The innate immune cascade cannot be activated in the absence of elevated uric acid levels, as no MSU crystals form in normouricemic conditions. Therefore, ULT remains the cornerstone of gout management. In parallel with therapies targeting immune pathways, ongoing research into novel agents that modulate uric acid synthesis, renal excretion, and metabolism, including xanthine oxidase inhibitors, uricosurics, recombinant uricase, and newer dual-mechanism agents, offers additional opportunities for comprehensive disease control.
In conclusion, the trained immunity concept reshapes our understanding of gout, not just as a disorder of uric acid metabolism but as a persistent, epigenetically driven inflammatory syndrome. This can explain why patients experience progressive worsening of flares, even with ULT, and why targeting immune memory mechanisms may be a key to long-term disease control. Moreover, given the chronic, relapsing nature of gout and the role of epigenetically reprogrammed innate immune cells, it is likely that trained immunity, once established, can persist for an extended period and repeated flares or continuous hyperuricemia potentially prolong this trained phenotype. Therefore, interrupting or reversing the trained immune state represents a promising adjunctive strategy to achieve better long-term disease control. Although no current intervention has been shown to fully “reset” trained immunity, promising strategies such as metabolic modulators (e.g., metformin, rapamycin) and epigenetic therapies offer the potential to partially reverse or dampen the trained inflammatory state. These approaches may serve as valuable adjuncts to conventional therapy, particularly in patients with recurrent flares or signs of persistent systemic inflammation. Nonetheless, significant challenges remain, especially in identifying which patients will benefit most, optimizing drug delivery, and ensuring long-term safety.
Future longitudinal studies that follow patients with gout and assess monocyte or macrophage gene signatures over months or years are needed to pin down the precise duration of this trained state and how it might be reversed.
2-HG: 2-hydroxyglutarate
CHIP: clonal hematopoiesis of indeterminate potential
DNMT3A: DNA methyltransferase 3A
EZH2: enhancer of zeste homolog 2
HDAC: histone deacetylase
HKs: hexokinases
HOTAIR: HOX transcript antisense RNA
IL-1β: interleukin-1β
JMJD3: Jumonji domain-containing protein-3
lncRNAs: long non-coding RNAs
MSU: monosodium urate
mTOR: mechanistic target of rapamycin
NLRP3: NACHT, LRR, and PYD domains-containing protein 3
OXPHOS: oxidative phosphorylation
RA: rheumatoid arthritis
siRNAs: small interfering RNAs
SNHG8: small nucleolar RNA host gene 8
SUA: serum urate
TET2: ten-eleven translocation-2
ULT: urate-lowering therapy
α-KG: α-ketoglutarate
OIG: Conceptualization, Writing—original draft. LABJ: Conceptualization, Writing—review & editing, Funding acquisition. TOC: Conceptualization, Writing—review & editing, Funding acquisition.
The authors declare no competing interests.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by a Romania’s National Recovery and Resilience Plan grant of the Romanian Ministry of Investments and European Projects [PNRR-III-C9-2022-I8, CF 85/15.11.2022]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Benjamin Plotz ... Michael H. Pillinger
Hamish Farquhar ... Lisa K. Stamp
Naomi Schlesinger, Dan Kaufmann
Mark D. Russell, James B. Galloway
Robert T. Keenan ... Michael H. Pillinger
Robin Christensen ... Lisa K. Stamp
Edward Roddy ... Christian D. Mallen
Philip L. Riches ... Amrey Krause
Emilie Schurenberg ... Kenneth G. Saag
Enrique Calvo-Aranda ... Marta Novella-Navarro
