 Review
Review

Affiliation:
1Division of Rheumatology and Clinical Immunology, Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, PA 15232, USA
Email: Bhp39@pitt.edu
ORCID: https://orcid.org/0000-0002-7022-7177
Affiliation:
2Division of Rheumatology, Department of Medicine, NYU Grossman School of Medicine, New York, NY 10016, USA
3Rheumatology Section, VA New York Health Care System, Margaret Cochrane Corbin Campus, New York, NY 10010, USA
ORCID: https://orcid.org/0000-0003-4152-0232
Affiliation:
4Arthrosi Therapeutics, Inc., San Diego, CA 92121, USA
ORCID: https://orcid.org/0000-0002-2341-7314
Affiliation:
2Division of Rheumatology, Department of Medicine, NYU Grossman School of Medicine, New York, NY 10016, USA
3Rheumatology Section, VA New York Health Care System, Margaret Cochrane Corbin Campus, New York, NY 10010, USA
ORCID: https://orcid.org/0000-0003-3168-1542
Explor Musculoskeletal Dis. 2025;3:1007104 DOI: https://doi.org/10.37349/emd.2025.1007104
Received: May 06, 2025 Accepted: September 15, 2025 Published: September 29, 2025
Academic Editor: Blanka Stiburkova, Charles University and General University Hospital, Czech Republic
The article belongs to the special issue Pharmacological and Non-Pharmacological Management of Gout
In the past decade, the metabolic syndrome has been recast as a chronic inflammatory disease whose mechanisms involve macrophage and neutrophil activation, initiation of the nod-like receptor protein 3 (NLRP3) inflammasome, and IL-1β secretion. Colchicine, an inhibitor of NLRP3, has been linked to the prevention or amelioration of diseases associated with the metabolic syndrome, including diabetes and cardiovascular disease. Its underlying therapeutic mechanisms extend beyond direct suppression of NLRP3, and include sirtuin and AMP-activated protein kinase (AMPK) pathway regulation, and downregulation of cellular stress signals, which promote atherosclerotic plaque rupture, insulin resistance, and obesity. Colchicine’s proven efficacy in preventing cardiovascular disease is a promising new development recognized by its inclusion in the 2023 American College of Cardiology treatment guidelines. As colchicine’s effects are better understood, along with a clearer understanding of metabolic syndrome’s pathophysiology, promising new applications and uses for this old drug may be on the horizon and are worthy of further investigation. In this review, we discuss colchicine’s pharmacology and explore its established and emerging anti-inflammatory mechanisms, and the role these could play in disrupting the chronic inflammation in metabolic syndrome and associated diseases.
Introduced to the United States by Benjamin Franklin [1] but not approved by the US Food and Drug Administration (FDA) until 2009, colchicine has been used medicinally for millennia, treating and preventing gout and calcium pyrophosphate disease (CPPD) flares in rheumatology, and in cardiology, treating pericarditis. Recently, its versatility as an anti-inflammatory agent has been recognized, including its FDA approval in 2023 for reducing cardiovascular (CV) events in high-risk patients.
Colchicine’s range of uses, from familial Mediterranean fever (FMF) to Behçet’s disease, relates to its ability to modify leukocyte and endothelial functions, including but not limited to its ability to inhibit the nod-like receptor protein 3 (NLRP3) inflammasome. The NLRP3 inflammasome is increasingly associated with diseases of the metabolic syndrome. Colchicine’s effects both on and independent of the inflammasome may have clinical implications for treating patients with metabolic syndrome, which is increasingly appreciated as a systemic inflammatory condition. Here, we review colchicine’s basic biology and highlight its potential in treating and preventing diseases associated with the metabolic syndrome. Colchicine, a drug as old as medicine itself, may guide us towards a deeper understanding of the pathophysiology and treatment of metabolic syndrome-associated diseases.
Colchicine is a lipid-soluble tricyclic alkaloid derived from plants of the genus Colchicum, notably the autumn crocus. A plant resembling Colchicum autumnale was described in the oldest existing medical text, the 1,500 BCE Ebers Papyrus manuscript, where it was used to treat pain and swelling [2, 3]. Colchicine-like treatment of podagra was first reported around the 6th century. It was extracted from Colchicum autumnale in 1820 by French chemists Pelletier and Caventou and later, in purified form, in 1833 by the German pharmacist Geiger, who coined the name colchicine, and today remains in use as a purified natural product [3].
Single doses of colchicine range from 0.5 to 1.2 mg. After rapid absorption in the jejunum and ileum, it undergoes first-pass metabolism in the liver and is largely excreted via bile [4]. Colchicine reaches peak plasma concentrations approximately 1 hour after ingestion. A delay in peak concentration with consumption of higher doses suggests a saturable influx transporter in the gut [5]. Colchicine’s bioavailability is broad (24–88%), and 40% of the drug binds to albumin when in circulation. Bioavailability does not differ between the young and elderly, but colchicine’s clearance is reduced in older populations [6]. About 5% is metabolized by cytochrome P450 (CyP) 3A4, 10–20% is excreted unchanged in the kidneys [halved in individuals with a glomerular filtration rate (GFR) between 10 and 30], while the majority is eliminated as the parent drug or metabolites in the gastrointestinal tract [5, 7]. Colchicine preferentially binds three proteins: CyP 3A4, P-glycoprotein (P-gp), and tubulin. P-gp is a transmembrane efflux pump that removes colchicine from cells and the gut, facilitating colchicine’s export out of the body. Other transporters, such as multidrug resistance-associated protein 2 (MRP2) and organic anion transporting polypeptide 2B1 (OATP2B1), have more recently been proposed as important efflux pumps, but their role is less well established [7].
Colchicine has a narrow therapeutic index, and for millennia has been used not only as a medication but as a poison. The commonly accepted safe upper limit serum concentration of colchicine is 3.0 µg/L. Safe levels of colchicine are typically maintained by 0.5 or 0.6 mg taken orally once or twice daily in those with normal or mild to moderate renal impairment. However, serum concentrations correlate poorly with colchicine’s anti-inflammatory effects since it preferentially accumulates inside neutrophils, which lack the P-gp efflux pump [4, 8, 9].
Gastrointestinal side effects are common, though usually mild, affecting 5–20% of individuals taking colchicine at normal doses. Myalgias are less common but well recognized. Lower doses or longer duration of treatment may attenuate colchicine’s side effects. Acute overdoses can cause multiorgan failure and disseminated intravascular coagulation, while chronic overdosing can lead to myotoxicity, neurotoxicity, and cytopenias. In modern times, most colchicine-related deaths stem from intentional self-overdoses [5]. Unintentional overdoses may occur with concomitant use of CyP 3A4 and/or P-gp inhibiting medications (Table 1), with higher daily doses of colchicine (e.g., up to 2.4 mg, sometimes used in FMF treatment), with dosing unadjusted for renal disease, or with several of these factors simultaneously [10]. Overdosing was more common in the setting of IV boluses, a practice banned by the FDA in 2008. In 2010, the AGREE trial demonstrated that a low-dose, single-time regimen (1.2 mg, then 0.6 mg 1 hour later) was non-inferior to the previous traditional regimen (4.8 mg given over 6 hours) to treat gout attacks, demonstrating that the anti-inflammatory effects of colchicine can be achieved with relatively low-dose therapy [11].
Common inhibitors of colchicine metabolism.
| Strong CyP 3A4 inhibitors | Moderate CyP 3A4 inhibitors | P-gp inhibitors |
|---|---|---|
| Antimicrobials | ||
| Clarithromycin | Ciprofloxacin | Ritonavir |
| Ketoconazole | Fluconazole | Clarithromycin |
| Voriconazole | Erythromycin | Azithromycin |
| Itraconazole | Itraconazole | Erythromycin |
| Ritonavir | - | Ketoconazole |
| Cobicistat | - | Quinidine |
| Cardiac medications | ||
| Diltiazem | Verapamil | Amiodarone |
| - | Amiodarone | Carvedilol |
| - | - | Verapamil |
| - | - | Diltiazem |
| - | - | Atorvastatin |
| Miscellaneous | ||
| Grapefruit | Fluoxetine | Tacrolimus |
| - | Imatinib | Grapefruit |
| - | Cimetidine | Cannabidiol |
| - | Paroxetine | Cyclosporine |
| - | Avacopan | - |
| - | Cyclosporine | - |
CyP: cytochrome p450; P-gp: P-glycoprotein; -: no data.
Overdose or poisoning by colchicine is managed strictly through supportive care, due to colchicine’s large volume of biodistribution, rendering it resistant to removal by plasma exchange or dialysis. A colchicine-specific Fab fragment has been used effectively in a case of colchicine overdose but never advanced to commercial availability [12]. Gastric lavage and N-acetylcysteine have been studied for colchicine overdose, with unclear benefit, and to our knowledge, cholestyramine has not been studied despite its action as a bile sequestrant.
Colchicine’s anti-inflammatory effects are largely a consequence of its ability to concentrate in neutrophils. However, it also has anti-inflammatory effects on the endothelium and macrophages. Many of these effects are mediated by its capacity to bind in a poorly reversible manner to soluble α- and β-tubulin heterodimers, destabilizing the microtubule lattice and inhibiting further microtubule elongation [13, 14]. Higher concentrations promote toxic microtubule depolymerization and disruption of the mitotic spindle, blocking mitotic cells in metaphase [15]. Colchicine’s inhibition of microtubule polymerization prevents vesicle transport, cytokine secretion (thereby modulating signal transduction), gene expression, and phagocytosis [4]. For example, colchicine suppresses protein tyrosine phosphorylation in neutrophils with subsequent inhibition of both intracellular mobilization and extracellular release of granular enzymes, which contain matrix metalloproteinases, elastase, and α-defensins [16, 17].
NLRP3 is expressed intracellularly in monocytes and macrophages, and to a lesser extent in neutrophils [18]. NLRP3 is a pattern-recognition receptor (PRR) and acts in the innate immune system to recognize and transduce external danger signals communicated through pathogen- and damage-associated molecular pattern (PAMP and DAMP, respectively) receptors, and internal signals such as reactive oxygen species (ROS) and potassium shifts (Figure 1). When activated, NLRP3 organizes an inflammasome, a multimeric protein platform that drives the proteolytic maturation of cysteine aspartate protease-1 (caspase-1), leading to activation and release of IL-1β and IL-18. Among multiple NLRPs, NLRP3 is the most studied and is broadly activated. DAMPs acting upstream to stimulate NLRP3 include monosodium urate (MSU) crystals, cholesterol crystals, free fatty acids, and oxidized low-density lipoprotein (LDL). The NLRP3 inflammasome consists of NLRP3 itself, the adaptor protein apoptosis-associated Speck-like protein containing caspase recruitment domain (ASC), and caspase-1. Microtubules mediate the activation of the NLRP3 inflammasome by facilitating the spatial approximation of mitochondria, the source of ASC, to the endoplasmic reticulum, the source of NLRP3. Colchicine, by inhibiting microtubules from pulling mitochondria to NLRP3, prevents NLRP3 from recruiting ASC and activating caspase-1 [19]. Canonical NLRP3 activation also requires a preceding priming step to upregulate NLRP3 and pro-IL-1β expression (often designated “Signal 1” to distinguish it from subsequent direct inflammasome activation, designated “Signal 2”) (Figure 1) [20]. For example, gouty inflammation is significantly enhanced by PRR priming molecules that are food or infection-derived [including free fatty acids and lipopolysaccharide (LPS)]. These priming molecules engage with toll-like receptors to facilitate the generation of NLRP3 substrates, which may explain why certain food ingestion or infections can trigger gout attacks [21].
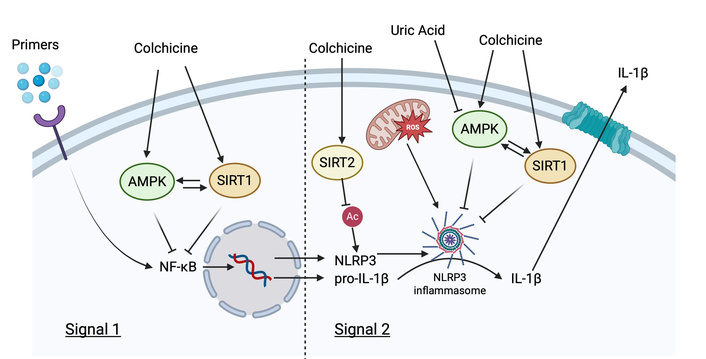
AMPK and SIRT1 actions on Signal 1 and 2 of NLRP3 inflammasome activation. Pointed arrows = induction/activation; flathead arrows = inhibition; attenuating arrow tails = further upstream interactions not specifically defined. AMPK: AMP-activated protein kinase; IL-1β: interleukin-1β; NF-κB: nuclear factor-κB; NLRP3: nod-like receptor protein 3; SIRT1: sirtuin 1; SIRT2: sirtuin 2. Created in BioRender. Plotz, B. (2025) https://BioRender.com/2dy4z0u.
In addition to microtubule inhibition, other purported mechanisms exist for colchicine to inhibit NLRP3 activation. It is currently believed that NLRP3 inflammasome activation is highly dependent on K+ concentrations and will only form when [K+]intracellular is < 70 mM. P2X purinergic receptor 7 (P2X7) receptors allow rapid and massive outflows of K+ out of the cytoplasm when activated. An in vitro model has demonstrated that colchicine can inhibit P2X7, preventing intracellular K+ depletion, which prevents NLRP3 activation [22, 23]. Others have demonstrated that colchicine may directly reduce levels of pro-caspase and caspase-1 proteins, thereby preventing NLRP3 inflammasome assembly [24]. Other in vitro studies have demonstrated that varying concentrations of colchicine directly decreased mRNA concentrations for the gene encoding pyrin (MEFV), a known potent activator of the NLRP3 inflammasome (see Established therapeutic uses section below) [25]. While colchicine concentrations used to inhibit NLRP3 activation in the initial work by Martinon and colleagues were higher than those achieved with usual low-dose therapeutic treatment, the true intracellular concentrations of colchicine in polymorphonuclear leukocytes (PMNs) are unknown but thought to accumulate given the absence of an efflux pump (see Pharmacology, dosing, and toxicity section above) [26]. To summarize, there are at least four proposed mechanisms for the inhibition of NLRP3 inflammasome formation by colchicine: (1) inhibition of cytoplasmic co-localization due to tubulin inhibition; (2) inhibition of P2X7, resulting in decreased K+ cellular efflux; (3) direct caspase-1 blockage; and (4) inhibition of MEFV, thereby decreasing pyrin expression.
Colchicine has multiple effects on neutrophil migration to sites of inflammation. It inhibits the ability of neutrophils to express proinflammatory molecules such as leukotriene B4 and IL-8, modulates the distribution of E-selectin adhesion molecules on endothelial cells and L-selectin expression on neutrophils to reduce neutrophil adhesion to vessel walls [27], and prevents neutrophil deformability, reducing neutrophil extravasation out of circulation. The net effect of these measures is decreased neutrophil accumulation at sites of inflammation [4].
Colchicine also throttles down neutrophil inflammatory actions. In addition to inhibiting lysosomal granzyme release, it reduces superoxide anion production and increases intracellular cAMP, which suppresses neutrophil function [28]. Colchicine may additionally inhibit actin filament polymerization [29–31], a process crucial to the formation of neutrophil extracellular traps (NETs), in which the formation of F-actin promotes the translocation of neutrophil elastase to the nucleus and subsequent NET formation [32]. Anti-NET properties have also been suggested in a mouse model in which colchicine reduced NADPH-oxidase 2 (NOX2) production and Ca2+ influx, which otherwise generate ROS required for NETosis [33]. However, a fully elucidated mechanism remains an area of active research.
Colchicine has many uses in rheumatic and non-rheumatic diseases. In gout, MSU crystals drive inflammasome assembly. Consequently, colchicine is well-established as effectively treating and preventing gout flares. Based on extensive clinical studies, the American College of Rheumatology (ACR) and the European Alliance of Associations for Rheumatology (EULAR) recommend colchicine for treating gout flares and for preventing flares while lowering serum urate levels [34, 35].
FMF is a hereditary autoinflammatory disease characterized by recurrent fevers, arthritis, and serositis caused by neutrophil-mediated inflammation. FMF results from genetic mutations in the pyrin gene domain, which encodes the pyrin protein. Pyrin forms its own inflammasome, which, like the NLRP3 inflammasome, activates caspase-1, leading to IL-1β secretion. Pyrin inflammasome activation depends on microtubule assembly and is inhibited by colchicine [36]. Colchicine’s effects in FMF are thought to mirror those in gout: blocking inflammasome activation, neutrophil activation, and migration. In multiple double-blind trials, colchicine has been shown to effectively induce FMF remission and/or decrease flare frequency [37]. Doses from 1 to 2.4 mg daily are used as first-line therapy to prevent flares. Importantly, colchicine use also largely prevents amyloid A amyloidosis, even when incompletely suppressing FMF flares [38].
Several diseases not presently linked to dogmatic NLRP3 inflammasome activation also benefit from colchicine treatment, potentially due to colchicine’s anti-neutrophil migratory properties. These include neutrophilic dermatoses, including Sweet’s syndrome, small vessel vasculitis, Behçet’s disease, and pericarditis, reviewed elsewhere [17, 37, 39].
Metabolic syndrome is defined as a cluster of clinical risk factors that significantly increase the probability of developing many common morbid conditions, including diabetes, CV disease, and stroke. These risk factors include central (visceral) obesity, hypertension, elevated serum triglycerides, low serum high-density lipoprotein (HDL), and insulin resistance [40]. The pathogenic link between metabolic syndrome and its associated diseases is multifactorial and complex. One possible connection may be the strong correlation between hypertension and elevated serum urate seen in obesity and in gout. The significant effect of urate-lowering therapy on improving hypertension has been demonstrated in numerous epidemiological, genetic, and meta-analyses [41, 42]. In fact, the association between gout, metabolic syndrome, and hyperuricemia is well documented. Uric acid is the final oxidation product of purine degradation. It is a known biological danger marker that increases as a result of local inflammation and tissue damage and helps prime cells for stress response [43]. Despite hyperuricemia being independently associated with risk of coronary heart disease, stroke, kidney disease, hyperglycemia, hyperlipidemia, obesity, and hypertension, it is not part of any broadly accepted metabolic syndrome criteria [44]. It is very difficult to untangle the role of uric acid as a causal agent or byproduct in the complex web of biological interactions that occur in metabolic syndrome [42]. However, a possible link between hyperuricemia and the development of metabolic syndrome associated diseases may be inflammation.
In the context of metabolic syndrome, a possible link between its associated diseases may be chronic low-grade inflammation caused by excess adipose tissue in obesity. This low-grade inflammation, in turn, helps drive and predispose to the metabolic syndrome-associated diseases. Studies have demonstrated that obese mice fed a high-fat diet secrete more inflammatory cytokines and chemokines, including IL-1β, IL-6, IL-18, and TNF, compared to lean mice [45, 46]. Similarly, cross-sectional studies have shown that obese humans have higher circulating markers of inflammation than lean ones. Innate immune cells, including neutrophils and macrophages, have been implicated in the regulation of adipose tissue homeostasis and as drivers of low-grade inflammation in obesity [47].
Macrophages, central to robust NLRP3 activation, have emerged as important regulators of adipose tissue homeostasis. In obese individuals, increased circulating fatty acids and ceramides bind to PRRs on adipose tissue macrophages (ATMs), shifting them to an M1 (pro-inflammatory) phenotype and helping to prime NLRP3 inflammasomes [45, 48]. The increase in M1 macrophages in obese adipose tissue facilitates further recruitment of peripheral monocytes, which themselves differentiate into an M1 phenotype. Peripherally, obese individuals have higher white blood cell counts compared to lean individuals [45].
ATMs acquire mitochondria from neighboring adipocytes in a heparan-sulfate-dependent manner as a mechanism of crosstalk to facilitate fat tissue homeostasis [49]. Mitochondria become dysfunctional in obesity, with impairments including a reduced oxidative capacity and elevated ROS levels [50]. Decreased mitochondrial transfer from adipocytes to macrophages is associated with obesity in mice. Instead of being transferred to macrophages, ATM-derived mitochondria may enter the circulation, potentially affecting distant organ metabolism [49]. Although studies are currently lacking, it is possible these “sickly” mitochondria may help prime NLRP3 through increased ROS release or by release of their contents to act as DAMPs, which has been shown to promote NLRP3 inflammasome activation in mice [51], but may also modulate stress responses in distant tissues, promoting homeostasis [52]. The full significance that mitochondrial transfer and shedding may have on gout or other metabolic syndrome diseases is unknown. However, next-generation sequencing has demonstrated potentially pathogenic gout-associated alleles in mitochondrial DNA [53].
The serine/threonine AMP-activated protein kinase (AMPK) is a master energy biosensor and regulator activated by stressors that increase the AMP: ATP ratio (e.g., starvation and exercise). Its role is thought to restore energy balance by activating catabolic processes, which generate ATP while downregulating anabolic processes that consume ATP [54]. It has also been shown to be a negative regulator of NLRP3 activation in multiple disease states [55]. In its phosphorylated active state, it inhibits inflammation partially through inhibition of nuclear factor-κB (NF-κB). AMPK activity is diminished in metabolic syndrome, type 2 diabetes, and in hyperuricemic states [42]. Researchers have demonstrated that colchicine promotes activation of AMPK and an M2 phenotype in bone-marrow-derived macrophages exposed to MSU crystals in vitro [56]. If colchicine can also activate AMPK in hepatocytes, it is likely to have salutary effects on lipid metabolism and gluconeogenesis, although it does not appear to reduce, and may actually increase, atherogenic lipoprotein particle concentrations in adults [42, 57]. Although colchicine may modulate the quality of fat, in a single human study, it has not been shown to alter the amounts of tissue fat or body weight, although it was shown to decrease the pro-inflammatory adipokine, resistin, which is positively associated with metabolic syndrome [58].
Sirtuins are a family of ubiquitously expressed NAD+-dependent deacetylases involved in many diverse regulatory biological roles. They play a role in apoptosis, oxidative stress, metabolism, and aging, amongst other important processes [59]. Sirtuin 1 (SIRT1) is recognized for its role in promoting cell longevity in response to calorie restriction. AMPK and SIRT1 appear to co-regulate each other while sharing common biological roles, including the downregulation of NF-κB (Figure 1) [54]. SIRT1 activation leads to improved insulin sensitivity, increased mitochondrial capacity in adipose tissue, and lower levels of plasma glucose. A direct mechanism of SIRT1 inhibition of NLRP3 is currently being investigated; SIRT1 knockout mice have increased NLRP3 inflammasome activation in the lungs and liver [60, 61], and SIRT1 agonists prevent NLRP3 activation in the heart [61]. In gout, SIRT1 also appears to inhibit NLRP3: activation of SIRT1 in mice injected with MSU crystals and an LPS primer demonstrates significantly reduced acute inflammation, IL-1β, and M1 polarization [62], while others have shown treatment with a SIRT1 agonist reduces inflammation in a murine gout model [63].
Sirtuin 2 (SIRT2) is also a metabolic sensor, which shuttles between the cytoplasm and nucleus. SIRT2’s role in metabolic syndrome is still being elucidated [64]. However, recent work has demonstrated that NLRP3 is post-translationally modified by acetylation in macrophages and is targeted by SIRT2 for deacetylation, reducing NLRP3 activation. In in vitro studies, mutations of the NLRP3 gene to prevent acetylation (mimicking the deacetylating action of SIRT2) led to significantly reduced IL-1β production (Figure 1). SIRT2 knockout mice fed a high-fat diet had increased levels of IL-18, obesity, and insulin resistance compared to controls, implicating SIRT2 as a negative regulator of NLRP3 inflammasome activation and the metabolic syndrome [65].
Neutrophils are recruited into adipose tissue by resident macrophages, as well as by adipocytes via byproducts of lipolysis. One study demonstrated that upon exposure to the saturated fatty acid palmitate (an extracellular metabolic danger signal), macrophages release nucleotides and nucleotide derivatives (i.e., AMP, ADP, adenosine, xanthine) that act as neutrophil chemoattractants through the pore-forming channel protein pannexin-1 [66]. Lipolysis of adipocytes and leukotriene B4 production induce neutrophil accumulation in adipose tissue and subsequent IL-1β secretion mediated through the NLRP3 inflammasome [67]. The inflammation-adipose environment facilitates further priming of macrophage NLRP3 inflammasomes and their activation. Indeed, increased expression of NLRP3 and IL-1β has been observed in the visceral tissues of obese individuals and is confirmed by genetic studies [68].
Together, these data demonstrate the pro-inflammatory effects of obesity in activating and recruiting M1 macrophages and neutrophils, which in turn facilitate NLRP3 activation in a complex process that involves signaling from AMPK, SIRT1 and 2, and mitochondrial modulation. If inflammation is an important driver of metabolic syndrome-related disease or even precursor states, then anti-inflammatory medications such as colchicine could theoretically reverse or prevent disease.
Systemic inflammation is closely tied to the development of insulin resistance and subsequent diabetes mellitus. M1 macrophage-enriched adipose tissue leads to decreased IL-10 and transforming growth factor-β (TGF-β), which otherwise act on fat and muscle to enhance insulin sensitivity [45]. Systemic inflammation impairs β-islet cell function, decreasing insulin secretion and eventually leading to β-islet cell apoptosis. Persistent depletion can transition an at-risk individual into overt diabetes. Substantial evidence links AMPK dysregulation to metabolic syndrome and insulin resistance, while several diabetes medications increase AMPK and SIRT1 activation [69]. In this context, the role of colchicine on the AMPK/SIRT1 pathway demands investigation.
In murine models, mice fed a high-fat diet to increase systemic inflammation were treated with a neutralizing IL-1β antibody. These mice demonstrated better glycemic control and β-islet cell function despite no effect on weight gain or dietary intake [70, 71]. Prophylactic treatment helped prevent fasting hyperglycemia and insulin resistance. These data suggest a role for IL-1β in regulating diabetes. IL-1β blockade in human studies has yielded mostly positive results, with some but not all studies showing significant decreases in hemoglobin A1c and β-islet cell function [72]. One meta-analysis of almost 3,000 patients showed that IL-1β inhibition significantly lowered HbA1c levels, with results approaching those of traditional anti-diabetic drugs (decreases of 0.8–1.2%) in select populations, including those with type II diabetes, advanced CV disease, and rheumatoid arthritis [72].
Colchicine’s pluripotent inhibition of inflammatory pathways, when applied persistently over time, could theoretically generate a chronic anti-inflammatory response to abrogate the development of diabetes. In a retrospective study of over 1,000 pairs of propensity-matched adults with gout taking or not taking colchicine, the incidence of diabetes was numerically but non-significantly reduced in the presence of colchicine. A time-varying model also demonstrated a non-significant trend towards a longer duration of colchicine use and reduction in diabetes risk [73]. In the LoDoCo2 trial [74], a large-scale clinical trial designed to measure colchicine’s effects on CV prevention, a secondary analysis again found a non-significant trend towards reduction of diabetes incidence (1.5% diabetes incidence in those taking colchicine compared to 2.2% in placebo, p = 0.10) [75]. More dramatically, a retrospective study of a large gout population in East Asia using and not using colchicine demonstrated a significantly lower risk of incident diabetes [18.8% in colchicine users compared to 25% in non-users, adjusted hazard ratio (HR), 0.74; 95% confidence interval (CI), 0.36–0.87] [76].
To date, a single prospective clinical trial has studied the effects of colchicine on obesity and metabolic syndrome. Demidowich and colleagues randomized 40 adults with metabolic syndrome to receive colchicine 0.6 mg twice daily or placebo for three months. Colchicine significantly reduced C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), white blood cell, and neutrophil counts. Although no difference was shown in insulin sensitivity, secondary endpoints for insulin resistance were improved overall, suggesting colchicine improved metabolic syndrome-associated inflammatory variables [57]. A secondary analysis of this cohort further demonstrated that colchicine improved insulin regulation of lipolysis. Interestingly, there was no difference seen in neutrophil or macrophage distribution in the subcutaneous fat in the treated vs. placebo groups [77]. A separate analysis by the same group demonstrated different populations of peripheral leukocytes in those taking colchicine compared to controls, finding reductions in multiple monocyte lineages that produce pro-inflammatory cytokines known to be upregulated in obesity-induced inflammation, decreased levels of myeloperoxidase, which is secreted by activated neutrophils, and decreased natural killer (NK) cells, which may play a role in activating neutrophils in obese humans [78]. Given the brevity of the study and the progressive colchicine benefit in long-term studies of CV risk (reviewed in the following section) and in diabetes, longer duration studies will be needed to assess the potential long-term benefits of colchicine on metabolic syndrome.
Altogether, colchicine may be effective at limiting the development of diabetes in certain at-risk populations. Animal models and human studies using colchicine or IL-1β inhibition suggest targeting colchicine-specific pathways may improve insulin resistance. However, these benefits may be mild, and further research is needed to identify specific patient subgroups that may derive the most benefit.
Coronary artery disease (CAD) is a natural consequence of atherosclerosis of the coronary arteries and is highly associated with metabolic syndrome. Colchicine’s efficacy in CAD has been robustly examined from multiple pathological domains, including endothelial dysfunction, smooth muscle dysfunction, lipid deposition, and macrophage dysregulation. LDL particles embed in arterial walls, become oxidized and otherwise modified, crystallize, and activate the NLRP3 inflammasome [79]. Leukocyte chemotaxis and adhesion contribute to vascular inflammation. Through atherosclerosis-induced inflammatory signals, neutrophils are activated and further contribute to plaque destabilization through the release of granular enzymes, metalloproteinases, elastase, and NET formation, which further adhere to chromatin, which accumulates in rupture-prone plaques [17].
Atherosclerosis can be characterized at least in part as a crystal-driven disease that mimics the pathogenesis of gout; a “gout” of the arteries (Figure 2) [79]. Interestingly, a small number of studies suggest that in addition to cholesterol crystals, gout patients may deposit MSU crystals within the vasculature [80]. Researchers, perhaps inspired by early gout studies, which found that intraperitoneally injected MSU crystals induced NLRP3 inflammasome activation, studied the effects of intraperitoneally injected cholesterol crystals. The inflammatory response to cholesterol crystals was impaired in mice deficient in components of the NLRP3 inflammasome, reducing neutrophil recruitment. Atherosclerosis-prone LDL knockout mice whose bone marrow was reconstituted with NLRP3, IL-1β, or ASC-deficient bone marrow had markedly reduced atheroma size after being fed a high-fat diet [81]. Animal and human atherosclerotic plaques also demonstrate abundant IL-1β and IL-18 expression [82]. These cytokines are known to enhance the instability of atherosclerotic plaques [17].
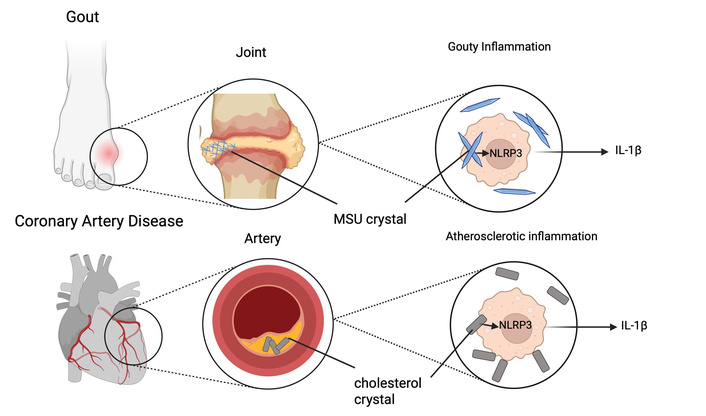
Atherosclerosis and gout both activate the NLRP3 inflammasome via crystals. Straight lines = identifying labels; pointed arrows = induction/activation; attenuating arrow tails = further upstream interactions not specifically defined. IL-1β: interleukin-1β; MSU: monosodium urate; NLRP3: nod-like receptor protein 3. Created in BioRender. Plotz, B. (2025) https://BioRender.com/m9698hk.
The AMPK pathway, SIRT1, and SIRT2 have also been directly implicated in CV disease models. Researchers have examined the effects of colchicine pretreatment in a coronary microembolization (CME) mouse model. CME is a common complication of acute coronary syndrome (ACS) and percutaneous coronary intervention (PCI). In these experiments, colchicine pretreatment increased activated AMPK and SIRT1 expression, and inhibited NLRP3 expression, resulting in decreased microinfarction area and improved left ventricular function [83]. Separately, researchers investigating the role of colchicine in a doxorubicin-induced dilated cardiomyopathy mouse model found that colchicine treatment upregulated the expression of SIRT2, leading to decreased acetylated NLRP3, decreased NLRP3 inflammasome activation, reduced cardiac fibrosis, and reduced neutrophil infiltration into the failing myocardium [84].
Although documentation of a common pathway is currently lacking, exposure of macrophages to both MSU and cholesterol crystals has been shown to decrease activation of AMPK. When human umbilical vein endothelial cells were treated with cholesterol crystals and colchicine, colchicine decreased the uptake of the crystals, suppressed the expression of NLRP3, and amplified AMPK and SIRT1. These effects—which were reversed through silencing AMPK and SIRT1—suggest that colchicine may regulate NLRP3 through AMPK/SIRT1 signaling [85]. In vitro and murine studies have also demonstrated that hyperuricemia may promote NLRP3-driven inflammation and IL-1β secretion caused by cholesterol crystals in a dose-dependent manner, while hyperuricemia also suppresses AMPK phosphorylation (activation) [86]. Additionally, IL-1β secretion from human and murine-derived pBMCs becomes attenuated in the presence of an AMPK inducer [56, 86, 87]. Collectively, these studies implicate AMPK and SIRT1 as potentially important negative regulators of NLRP3 signaling, which themselves are beneficially affected by colchicine (Figure 1).
Human trials have demonstrated that inhibition of the NLRP3 inflammasome and IL-1β reduces the risk of CV disease. The CANTOS trial confirmed the ability of canakinumab, an anti-IL-1β antibody, to reduce major adverse CV events while cholesterol levels remained unaffected [88]. Colchicine’s cardioprotective effects were first demonstrated retrospectively in gout and FMF populations, where chronic colchicine use reduced the incidence of CAD [17] and reduced myocardial infarction (MI) incidence in gout patients compared to counterparts who did not take it [89, 90].
These observations led to large-scale randomized and blinded prospective trials examining the effect of colchicine in patients at risk for coronary events. The LoDoCo2 [74] (stable CAD population) and COLCOT [91] (post-MI population) trials both demonstrated an approximately 1/3 reduction in a major adverse CV composite endpoint over approximately two years of daily 0.5 mg colchicine use. A subsequent trial, COPS [92], using a higher dose of colchicine for the first month (0.5 mg twice daily), did not find a significant reduction in their composite endpoint at 12 months, but a post-hoc analysis using the same endpoint as in LoDoCo2, which excluded non-CV death, did. Moreover, follow-up of COPS at 24 months demonstrated a significantly reduced rate of the composite endpoint in the colchicine group despite study treatment ending at 12 months, suggesting a persistent colchicine effect [93]. An aim of a separate multinational placebo-controlled trial, CLEAR SYNERGY [94], was to assess the effect of colchicine on the reduction of recurrent CV events in patients presenting with acute ST-elevation MI managed with PCI. Median follow-up was 3 years. The study found no colchicine benefit. However, these conclusions have been called into question because enrollment during the COVID-19 pandemic suggested under-reporting of cardiac events, whereas the pre-COVID-19 phase of the study suggested a probable colchicine benefit [95]. These trials have been reviewed extensively elsewhere [16] and are summarized in Table 2.
Clinical trials of colchicine in cardiovascular disease.
| Study | Intervention | Duration | Population | Primary outcome | Result(s) | Reference |
|---|---|---|---|---|---|---|
| LoDoCo2 | 0.5 mg/day | Median follow-up 28.6 months | Patients with stable CAD ≥ 6 months (n = 5,522) | Composite of cardiovascular death, myocardial infarction, ischemic stroke, or ischemia-driven coronary revascularization | Colchicine 6.8%, placebo 9.6%, HR 0.69 (0.57–0.83) p < 0.001 | [74] |
| COLCOT | 0.5 mg/day | Median follow-up 22.6 months | Patients within 30 days after myocardial infarction with preserved ejection fraction (n = 4,745) | Composite cardiovascular death, resuscitated cardiac arrest, myocardial infarction, stroke, or coronary revascularization | Colchicine 5.5%, placebo 7.1%, HR 0.77 (0.61–0.96) p = 0.02 | [91] |
| COPS | 1st month 0.5 mg twice a day, 0.5 mg/day for 11 months | 12 months | Patients with acute coronary syndrome and CAD on angiography (n = 795) | Composite of all-cause mortality, acute coronary syndrome, ischemia-driven unplanned revascularization, and non-cardioembolic ischemic stroke | Colchicine 24 events, placebo 38 events (p = 0.09*, log-rank). Higher total death rate in colchicine (8:1; p = 0.017), and non-cardiovascular death (5:0; p = 0.024) | [92] |
| CLEAR SYNERGY | 0.5 mg/day, 0.5 mg twice daily for the first 90 days in those ≥ 70 kg with and without 25 mg spironolactone | Median follow-up 36 months | Patients following percutaneous intervention with myocardial infarction (n = 7,062) | Composite of death from cardiovascular causes, recurrent myocardial infarction, stroke, or unplanned ischemia-driven coronary revascularization | Colchicine 9.1%, placebo 9.3%, HR 0.99* (0.85–1.16) p ≤ 0.93 | [94] |
*: denotes non-significant result; CAD: coronary artery disease; HR: hazard ratio.
The potential for colchicine to reduce CV morbidity specifically in those with diabetes compared to those without has been examined separately in LoDoCo2 and the COLCOT studies with mixed results: no additional benefit was seen in a secondary analysis of LoDoCo2 [75], but a greater post-MI benefit of colchicine in the diabetes subgroup of the COLCOT trial (HR 0.65; 95% CI 0.44–0.96; p = 0.03) has led the investigators to propose a COLCOT-T2D trial [96].
In addition to a reduction of CV events when taking colchicine, studies suggest a benefit on atherosclerotic plaque remodeling. In one nonrandomized observational trial, 80 patients with recent ACS were given colchicine 0.5 mg daily for 1 year, while the change in low attenuation plaque volume was measured by coronary computed tomography angiography (CTA) after 1 year. The authors found a significant reduction of plaque volume in the colchicine group (–40.9% vs. –17.0%; p = 0.008) that correlated linearly with reductions in hsCRP levels. This helped to inform the COLOCT trial, a prospective, single-center, randomized, double-blind clinical trial of 128 patients with ACS whose coronary plaques were assessed by optical coherence tomography. Patients who were treated with colchicine vs. placebo over a year improved in all 6 predefined measures of coronary plaque stabilization [97]. Currently, the EKSTROM trial is underway to explore the effect of 1 year of 0.5 mg daily colchicine on stable coronary plaques [98].
The beneficial effects of colchicine on vascular function—a feature seen in both CV disease and metabolic syndrome—have been suggested since at least 1985. Lagrue and colleagues demonstrated that 3–4 months of 1 mg of daily colchicine significantly improved conjunctival biomicroscopy scores [99]. Colchicine treatment duration in stable FMF patients was associated with lower carotid femoral pulse wave velocities measured by ultrasound [100]. One prospective study of gout patients initiating urate-lowering therapy and colchicine found that brachial artery flow-mediated dilation was improved in those taking colchicine alone after 4 weeks, and subsequently in those who successfully reduced their urate levels without certain CV risk factors (but not in those with risk factors) [101]. These also correlated with decreases in peripheral markers of inflammation, including hsCRP and IL-1β.
Overall, colchicine has been shown to improve endothelial function, stabilize coronary plaques, and reduce CAD. Often these effects correlate with reductions in systemic inflammation and IL-1β. Results from in vivo mouse models and in vitro studies using MSU and cholesterol crystals to model gout and atherosclerosis, respectively, implicate AMPK and SIRT1 signaling as negative regulators of NLRP3, whose expression is enhanced by colchicine. Additionally, SIRT2 may inhibit NLRP3 through deacetylation. However, future studies are needed to explore this pathway in humans.
Despite colchicine’s narrow set of drug targets, the central role of those targets—neutrophil activation and migration, and NLRP3 activation—gives it a broad scope of potential therapeutic uses in human disease. As more attention is given to the role of NLRP3 activation in diseases associated with metabolic syndrome, we are learning more about the benefits of inhibiting it. By inhibiting chronic (and acute) inflammation, colchicine may reduce the incidence of diabetes and CV events. Its long-term use has been shown to be cardioprotective; it reduces morbid CV outcomes and stabilizes coronary plaques while promoting healthier endothelial function.
However, challenges in understanding the true benefit of colchicine remain. The role of NLRP3 in many metabolic syndrome-related diseases remains to be fully established and elucidated. Colchicine increases the negative regulators of NLRP3 in AMPK and SIRT1 and 2 signaling (Figure 1), as demonstrated in gout and CV disease models, but a gap remains connecting these pathways to the improved insulin resistance measures and mild reduction in diabetes incidence seen in humans. Potential mechanisms of action of colchicine on these signaling pathways have not been elucidated. Further study of these biomechanical pathways may identify new drug targets or therapeutic pathways for colchicine or other drugs that target NLRP3 directly or indirectly.
As we better understand how the NLRP3 inflammasome facilitates a chronic inflammatory state, which in turn drives and incites metabolic syndrome-associated diseases and poor patient outcomes, prospective trials using colchicine and other drugs targeting NLRP3 regulation should be conducted. Colchicine’s chronic use in gout and CV disease may serve as the paradigm highlighting the importance of NLRP3 in the transformation of metabolic syndrome into disease, revealing new drug targets and therapeutic strategies along the way.
ACS: acute coronary syndrome
AMPK: adenosine monophosphate-activated protein kinase
ASC: apoptosis-associated Speck-like protein containing caspase recruitment domain
ATMs: adipose tissue macrophages
CAD: coronary artery disease
caspase-1: cysteine aspartate protease-1
CI: confidence interval
CME: coronary microembolization
CRP: C-reactive protein
CV: cardiovascular
CyP: cytochrome P450
DAMP: damage-associated molecular pattern
FDA: Food and Drug Administration
FMF: familial Mediterranean fever
HR: hazard ratio
LDL: low-density lipoprotein
LPS: lipopolysaccharide
MI: myocardial infarction
MSU: monosodium urate
NETs: neutrophil extracellular traps
NF-κB: nuclear factor-κB
NLRP3: nod-like receptor protein 3
P-gp: P-glycoprotein
P2X7: P2X purinergic receptor 7
PCI: percutaneous coronary intervention
PRR: pattern-recognition receptor
ROS: reactive oxygen species
SIRT1: sirtuin 1
SIRT2: sirtuin 2
BP: Conceptualization, Writing—original draft, Writing—review & editing, Visualization. MT: Writing—review & editing. RTK: Writing—review & editing. MHP: Conceptualization, Writing—review & editing, Supervision. All authors read and approved the submitted version.
Robert T. Keenan, who serves as Editorial Board Member of Exploration of Musculoskeletal Diseases, had no involvement in the decision-making or review process of this manuscript. The authors declare that they have no other conflicts of interest regarding this manuscript. For completeness, they note the following financial disclosures: BP has nothing to declare. MT has consulting fees from Amgen Inc. and Scilex Pharmaceuticals. RTK is an employee of Arthrosi Therapeutics, Inc. MHP is supported by grants from the National Institutes of Health (1UL1 TR001445) and the VA (I01 CX002358), and has served as a consultant for Amgen, Federation Bio., Fortress Bioscience, Convergence Bio., and Scilex.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Hamish Farquhar ... Lisa K. Stamp
Naomi Schlesinger, Dan Kaufmann
Mark D. Russell, James B. Galloway
Robert T. Keenan ... Michael H. Pillinger
Robin Christensen ... Lisa K. Stamp
Edward Roddy ... Christian D. Mallen
Philip L. Riches ... Amrey Krause
Emilie Schurenberg ... Kenneth G. Saag
Enrique Calvo-Aranda ... Marta Novella-Navarro
Orsolya I. Gaal ... Tania O. Crișan
