 Review
Review

Affiliation:
1Department of Biotechnology, National Institute of Technology Durgapur, Durgapur 713209, India
ORCID: https://orcid.org/0000-0003-4114-9461
Affiliation:
2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India
ORCID: https://orcid.org/0000-0001-7155-2257
Affiliation:
2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India
ORCID: https://orcid.org/0000-0001-6767-8383
Affiliation:
2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India
Affiliation:
2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India
Email: rubydhar@gmail.com
ORCID: https://orcid.org/0000-0003-3600-6554
Affiliation:
2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India
Email: subhradip.k@aiims.edu
ORCID: https://orcid.org/0000-0002-4757-8729
Explor Immunol. 2024;4:1–33 DOI: https://doi.org/10.37349/ei.2024.00126
Received: August 30, 2023 Accepted: November 09, 2023 Published: January 31, 2024
Academic Editor: Vladimir N. Uversky, University of South Florida, USA
The article belongs to the special issue Old and New Paradigms in Viral Vaccinology
The coronavirus disease 2019 (COVID-19) pandemic cost 7–8 million deaths worldwide, creating an unprecedented health and economic crisis. Affecting 700 million people globally, the magnitude of this pandemic is far from anything that humanity has encountered in recent times. A detailed investigation revealed that more than the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus, the hyperactive immune system mediated injury as the real cause of mortality. Cytokine storm following viral infection leads to the surge of proinflammatory cytokines resulting in acute respiratory distress syndrome (ARDS) and lung injury. Anti-inflammatory intervention with anti-interleukin-6 (anti-IL-6) receptor monoclonal antibodies (mAbs; e.g., sarilumab and tocilizumab) and anti-IL-6 mAbs (i.e., siltuximab) and/or steroid-based approach leads to substantial protection and prevent death thereby implying the role of inflammation in COVID-19. In this review, the authors have summarized the dysregulated immune system in COVID-19 infection, investigating in detail the virus-host immune cross talks and presenting the possibilities of therapeutic intervention.
Coronavirus disease 2019 (COVID-19) is an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and has been affecting the world since 2019. As of January 2022, the outbreak of SARS-CoV-2 and its varied variants has spread to all the continents, with more than 600 million registered cases and over 6 million mortalities worldwide. Despite the observed low mortality rate of COVID-19 when compared with other severe acute respiratory syndrome (SARS) epidemics, its broadcasting has led to devastating effects on the healthcare system and the harmonization of national economics worldwide.
It’s been just 2.2 years since the first case of COVID-19, but the response by both the medical personnel and the scientific community has been humongous, with a varied number of scientific research reports, meta-analyses, prospective cohorts, etc. However, in order to curb the pandemic, there have been numerous decisions taken in a hurry based on some anecdotal or poor evidence [usage of ibuprofen, hydroxychloroquine, angiotensin (Ang)-converting enzyme (ACE) inhibitors, and lopinavir-ritonavir], inciting misperception among medical personnel and the general public [1–3]. These challenges are potentially aggravated, subjecting to the gap in the knowledge in the understanding of COVID-19 pathophysiology and the complexity of treatment validation. However, the world is still learning the dynamics of SARS-CoV-2 infection as a persuasive goal to understand the emergence of the various new variants that come with multiple COVID-19 phenotypes. Though all of the new variants (as of now) manifest with acute respiratory disorders, several non-respiratory subjects and post-COVID complications have also been reported. With the emerging points to consider, this clinical picture directs to the fact that SARS-CoV-2 dysregulates the host-immune response towards the infection, thereby fueling misleading research and biased hypotheses.
So as to implement optimal management strategies in order to improve the medical outcomes regarding COVID-19, similarities in pathophysiology, symptoms, autopsy reports, and in-depth analysis of COVID-19-positive patients are deemed necessary. This paper aims to summarize the pathophysiology of COVID-19 and the related and unrelated disease mechanisms that underline several respiratory and non-respiratory manifestations. We also try to solve the question of whether the virus contributes towards the host’s death or if it is the complex array of cellular and humoral immune-inflammatory mechanisms that underlie the misfortune. Moreover, several systematic investigations and autopsy studies of SARS-CoV-2-associated deaths are still considered necessary to provide evidence for the epidemiological clusters and the organotropism of the virus and its variants.
As a phylogenetic member of the large coronavirus family, SARS-CoV-2 shares many similarities with SARS-CoV and Middle East respiratory syndrome (MERS), which were responsible for the two previous epidemics in the early 21st century. Alike SARS-CoV, the primary route of virus entry and transmission is expelled respiratory droplets that are absorbed by the mucous membranes. Both viruses exhibit similar coding domains (S1 and S2) of the spike protein (S protein), which facilitates its entry into the human host. The receptor-binding domain (RBD) in S1 interacts with the human host cell surface’s ACE2, thus mediating SARS-CoV internalization [4]. Several biophysical and structural evidence have stated the extensive affinity of ACE2 with SARS-CoV-2, with respect to that of its other members, thus hypothesizing the contagious nature of the virus [5]. Viral entry also has been seen to be associated with transmembrane protease serine 2 (TMPRSS2) protease activity, and it’s the synergy between ACE2 and TMPRSS2 that leads to virus entry into the host cell membrane. Expression of both TMPRSS2 and ACE2 has been seen to occur largely in alveolar epithelial cells, with the former being extensively dispersed compared to ACE2 receptors, signifying that ACE2 receptors might play a rate-determining factor in the entry of the virus [6]. However, ACE2 expression is found to be higher in adults when compared with children, thus infection rate is seen higher in adults [7, 8]. Other proteases like cathepsin B/L also play a vital role in contributing to viral entry in the absence of TMPRSS2 [9]. Extensive bioinformatics analyses have also deduced that furin, a type I membrane-bound protease, has also been seen associated with several viral entries including coronavirus [10]. Furin’s action on the S protein of SARS-CoV-2 influences its cellular entry by revealing the fusion domains and increasing the chances of pathogenesis, thus idealizing as an additional pathway for SARS-CoV-2 to enter [10–12]. Furin has also been reported in several circulating cells like T cells, which work in a forward-feedback loop, further facilitating in contributing towards the replication of the virus and inducing cytokine storm in several patients [12].
Although SARS-CoV-2 is well known for targeting the lung epithelial cells, it has been seen in several extra pulmonary manifestations, including myocardial dysfunctions, thrombotic complications, hepatocellular injuries, neurologic illnesses, ocular and dermatological complications, etc. [13]. Considering the importance of the cellular surface proteins, like ACE2, TMPRSS2, cathepsins, and furin, in aiding SARS-CoV-2 entry into the human host, several transcriptomic gene analyses have been reported. In a Genotype-Tissue Expression project data, ACE2 has been found to be expressed highly in the ileum and testis (> 10 transcripts per million) and low in other organs like the heart, kidney, thyroid, and adipose tissues (> 5 transcripts per million) [14]. Single-cell RNA-sequence data analysis by Zou et al. [15] revealed that ACE2 expression was also highly expressed in cell types like alveolar type 2 epithelial cells, respiratory epithelial cells, myocardial cells, esophageal epithelial cells, etc., thereby forming physiological barriers against viral entry. However, ACE2, albeit serving as an entry point for SARS-CoV-2, increases the susceptibility of the human host to COVID-19 infection (as explained in Figure 1). Findings by Xu et al. [16] also supplemented that ACE2 expressions in oral mucosal epithelial cells are highly notable, and SARS-CoV-2 infection is also susceptible in the oral cavity.
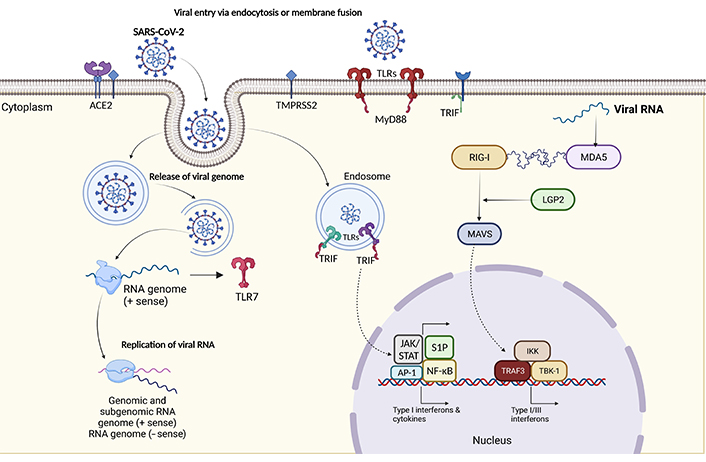
Schematic representation of pathogenesis of SARS-CoV-2 infection. The pathogenesis of COVID-19 begins with viral entry, as SARS-CoV-2 infects respiratory cells. Once inside, the virus is detected by host cells through pattern recognition receptors (PRRs) like Toll-like receptors (TLRs). TLRs, particularly TLR3, recognize viral RNA and trigger the expression of Toll/interleukin-1 (IL-1) receptor (TIR) domain-containing adaptor-inducing interferon-β (IFN-β; TRIFs), which play a critical role in inducing IFN responses. Viral RNA may also be detected in endosomes, activating TLRs to initiate antiviral signalling. In addition, cytoplasmic sensors like melanoma differentiation-associated protein 5 (MDA5) and laboratory of genetics and physiology 2 (LGP2) recognize viral RNA, further stimulating IFN production. These IFNs activate the immune system to combat the virus, making these processes pivotal in COVID-19 pathogenesis. AP-1: activator protein 1; IKK: IκB kinase; JAK: Janus kinase; MAVS: mitochondrial antiviral signalling; MyD88: myeloid differentiation primary response 88; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; RIG-1: retinoic acid inducible gene-1; S1P: sphingosine-1-phosphate; STAT: signal transducers and activators of transcription; TBK-1: TANK-binding kinase-1; TRAF3: tumor necrosis factor (TNF)-receptor associated factor-3
Neurological symptoms associated with COVID-19 patients also speculated the tendency of SARS-CoV-2 to invade the brain barriers and trigger neurological damage [17, 18]. In a prospective study conducted in New York, neurologic disorders conferred a high risk of in-hospital mortality [18]. Further genome sequencing has verified the existence of the virus in the cerebrospinal fluid (CSF). Possible explanations could be that the SARS-CoV-2 virus could have breached the olfactory mucosa lining the cribriform plate and must have transversed along the perineuronal space to access the CSF in the subarachnoid space [19]. Drainage of the virus-loaded CSF also might have triggered the immune response, thereby triggering COVID-19-affiliated encephalitis [20, 21].
Gastroscopy examinations have shown that though viruses usually cannot survive the acidic environment of the stomach, there have been several shreds of evidence stating that the gastrointestinal (GI) tract might be a potential transmission route of the SARS-CoV-2 virus [22]. Several case studies have also proved the point by showing that COVID-19 patients present GI symptoms like diarrhoea and nausea, prior to the development of fever [23]. As mentioned above, the expression of ACE2 and TMPRSS2 are high in absorptive enterocytes and the epithelial cells of the ileum, the GI symptoms perceived in COVID-19 patients might be due to the invasion of the virus across the gut-epithelial barrier. Elevated levels of faecal calprotectin and IL-6 along with the clinical symptoms of diarrhoea in COVID-19 patients also suggest an acute inflammatory response in the GI tract [24, 25].
Though lung tissues were one of the first and most critical organs affected by COVID-19, they expressed modest levels of ACE2. However, the presence of alveolar type 2 epithelial cells and their affiliated higher expression of ACE2 provided the platform for SARS-CoV-2 to replicate and upregulate cytokines like transforming growth factor-beta 1 (TGF-β1) and connective tissue growth factor (CTGF), extracellular matrix (ECM) proteins and infection, further resulting in pulmonary fibrosis [26, 27].
The most common clinical manifestations constitute cough (45–80%), fever (75–98%), fatigue, muscle pain, anorexia, and loss of smell and taste [28]. However, apart from these, other symptoms like sore throat, rhinorrhea, headache, and GI symptoms, have also been seen to precede the respiratory symptoms. Some patients who show severe symptoms like dyspnea (around day 5), often require hospitalization by days 7–8 and manifest hypoxemia and bilateral pneumonia. Out of the hospitalized patients, some deteriorate abruptly after the inception of dyspnea and succumb to respiratory failure [29, 30]. COVID pneumonia, with clinical symptoms like fever, cough, tachypnoea, higher tidal volume, and respiratory distress, in adults has also been described as a potentiating reason for patients registering in hospitals with hypoxia [23, 31]. Though clinical management for hypoxia remains a contentious issue, several hypotheses have been proposed to explicate hypoxia, reduced diffusion capacity, oxygen receptor chemosensitivity, and loss of hypoxic vasoconstrictive mechanism [32–35]. In fact, an unusual phenomenon that was observed in the earlier months of COVID-19, which played a significant role in major mortalities, was silent hypoxemia, also known as happy hypoxia, characterized by low partial pressure of arterial oxygen, yet low or mild respiratory discomfort [36, 37]. Moreover, altered mechanisms of the lungs, associated with alveolar collapse, pulmonary inflammation, fibrosis, and atelectasis further impair lung functions, causing lung tissue hypoxia.
Acute respiratory distress syndrome (ARDS), an impairment of oxygenation, is one of the most severe complications of COVID-19. Though respiratory support is crucial and high-flow oxygen is often provided via invasive or non-invasive ventilation, ARDS is concomitant with sustained hospitalization and high mortality [28]. Associated with high D-dimer concentrations and low compliance, intermittent prone positions help in improving gaseous exchange, reducing the ventilation/lung-perfusion mismatch, however, this technique hasn’t been proved clinically.
One of the initial mechanisms for ARDS is the cytokine storm, which uncontrollably stimulates an inflammatory response resulting from the enormous amounts of pro-inflammatory cytokines (IL-1β, IL-6, IL-12, IL-18, IL-33, IFN-α, IFN-γ, TNF-α, TGF-β, etc.) and chemokines [C-C motif chemokine ligand 2 (CCL2), CCL3, CCL5, C-X-C motif chemokine ligand 8 (CXCL8), CXCL9, CXCL10, etc.] by the immune-effector cells in the viral infection [29, 38–40]. Characterized by the deterioration of the function of the lungs and respiratory failure, lung fibrosis is correlated with ARDS, but with a poor prognosis. Driven by pro-fibrotic factors like TGF-β, secretion of the same promotes repair and perseverance of infection-mediated damage in COVID-19 injured lungs. However, severe damage causes excessive TGF-β signalling causing epithelial-to-mesenchymal transition (EMT) and endothelial-to-mesenchymal transitions, thereby causing pulmonary fibrosis [41–45]. Pulmonary fibrosis, being a common complication of SARS and MERS, several post-mortems have revealed a high section of COVID-19 cases with fibrosis [46, 47]. The progression of ground-glass opacities in computed tomography (CT) scans due to thick reticular pattern, irregular interface, and interstitial thickening is suggested to be the early sign of COVID-19 pulmonary fibrosis [48].
A stage of coagulopathy and endothelial damage also arises in severe COVID-19 cases, with frequent reports of microvascular thrombosis or disseminated intravascular coagulation. Several pieces of evidence also state the association of pulmonary failure with deranged coagulation. Hypercoagulability symptoms like high circulating D-dimer concentrations, elevated prothrombin time, increased fibrinogen, and thrombocytopenia are some of the common ones found in patients with severe COVID-19 [29, 49]. Though the pathobiology of hypercoagulability in COVID-19 cases remains unclear, numerous autopsy reports have identified diffused alveolar damage, severe endothelial damage, and coagulopathic symptoms in the pulmonary microvasculature in patients with severe COVID-19, hence establishing the relation [50, 51]. Evidence of alveolar damage accompanied by thrombotic microangiopathy also supports the fact that endothelial injury stimulates pulmonary circulation thrombosis [52, 53].
Endotheliitis, as referred by Varga et al. [54], can also be associated with refractory COVID-19-related ARDS with loss of hypoxic vasoconstriction, intra-alveolar fibrin, alveolar damage, edema, haemorrhage, and hyperfused intrapulmonary blood flow [55]. However, being not just restricted to the lungs, other vascular disorders including stasis, disturbed endothelial barrier, endothelium localized inflammations, disturbed permeability control, and prothrombotic endothelial cell state also act as proactive factors localised to brain, heart, gut, liver, and kidney, thus revealing several other extrapulmonary manifestations [13, 51, 54, 56, 57].
Though the pathophysiology of SARS-CoV-2 isn’t fully comprehensive, relative literature about SARS-CoV and MERS and recent literature have shown that SARS-CoV-2 has quite a lot of defence mechanisms, which makes it challenging to eradicate. Alike SARS-CoV, SARS-CoV-2 contains 4 structural proteins, envelope protein (E protein), membrane protein (M protein), nucleocapsid protein (N protein), S protein, and 14 open reading frames (ORFs) [58]. Following the entry of SARS-CoV-2 into the cell by binding to the ACE2 on the epithelial cell surfaces, the viral genome is transferred into the cytoplasm and is translated into two polyproteins and structural proteins, after which they replicate. Post replication, the viral particles germinate into the endoplasmic reticulum-Golgi apparatus complex and fuse with the plasma membrane to release the virus [59].
Subsequent antigen production stimulates the human body’s immune response (both cellular and humoral). Inflammatory responses are also triggered by pathogen-associated molecular patterns (PAMPs) in correlation with the cytosolic and endosomal PRRs to limit infection and promote viral clearance [60]. Armed with an array of PRRs that recognize PAMPs or damage-associated molecular patterns (DAMPs), to stimulate inflammatory pathways and immune responses, the immune system, in accordance with the PRR-affiliated inflammasomes, TLRs, nucleotide-binding oligomerization domain-like receptors (NLRs), and RIG-1-like receptors (RLRs), has been seen to stimulate signalling pathways against COVID-19 [60]. The infected lung and the alveolar epithelial cells trigger the innate immune response by secreting IL-8, which acts as a chemoattractant for T-lymphocytes and neutrophils. TLRs, in coordination with MyD88 and TRIF, possess a heterogeneous expression of the immune cell population [61]. Several analyses have suggested that TLR2 recognizes the E protein of the virus and induces innate immune cell activation [62–64]. Other TLRs like TLR1, TLR4, and TLR6 have been seen to show affinity towards the S protein of SARS-CoV-2, thereby contributing towards the release of pro-inflammatory cytokines [65]. Moreover, X chromosomal TLR7 genetic anomalies have been observed in several young patients suffering from COVID-19, suggesting the pro-active role of TLR7 in the infection [66, 67]. The single-stranded RNA in the virus aggravates the immune response post-recognition by TLR7, which is expressed on dendritic cells and macrophages. TLR7, further stimulates several transcriptional factors, pro-inflammatory cytokines like IL-1, IL-6, monocyte chemoattractant protein-1 (MCP-1), TNF-α, and IFN-1; and putative signalling pathways like NF-κB, JAK/STAT, S1P, AP-1, IFN response factor 3 (IRF3), and IRF7 [68]; thus establishing the correlation of type I IFN with cytokine secretion in CD4+ T cells polarization [69]. Activation of these pathways further endures other pathways like extracellular signal-related kinase (ERK)/rat sarcoma (RAS), PI3K/Akt/endothelial nitric oxide synthase (eNOS), and phospholipase C (PLC)/Ca2+ downstream pathways which regulate the migration and trafficking of numerous immune cells, including natural killer (NK) cells, B and T lymphocytes, and dendritic cells [70]. JAK1 tyrosine kinase 2 (TYK2) kinases phosphorylate STAT1/2 and form a complex with IRF9 to commence transcription of IFN-stimulated genes (ISGs) [71]. Following this, neutrophils get rapidly recruited to the infection sites and respond by their neutrophil extracellular traps (NETs) and defensin secretion to kill the viruses [72]. Early expressions of IFN-α, IFN-γ, CXCL10, CCL12, and ISG-encoded proteins were also noted in patients who recovered from SARS [71].
RNAs derived from the genomic intermediates of the virus can be detected by RLRs, namely LGP2, RIG-1, and MDA5. RIG-1 and MDA5 are key regulators and are associated with pathways like IFN and MAVS signalosome, which further activate TRAF3, IKK, and TBK-1 to mediate transcription of type I and III IFNs. Subsequent production and stimulation of IFNs activate downstream signalling via IFN receptors in an autocrine and paracrine manner to secrete vivid ISGs of different antiviral functions [73–76]. However, silencing of the genes encoding MDA5 or LGP2 in human lung epithelial (Calu-3) cells causes reduced type I IFN expression during COVID-19 infection [77]. On the contrary, small interfering RNA (siRNA) silencing for gene, encoding for RIG-1 did not show any difference in Calu-3 cells, though this study remains quite contentious [78, 79]. Type I IFNs and other cytokines are also stimulated by NLRs in SARS-CoV-2 infections, with nucleotide-binding domain leucine-rich-containing family, pyrin-domain-containing-3 (NLRP3) being one of the most characterized inflammasome sensors. Triggered in response to DAMPs and PAMPs, caspase-1 gets activated, thereby stimulating the release of IL-1β, IL-18, and cleavage of gasdermin D, thus leading to membrane ruptures [80, 81]. Increased levels of IL-1β and IL-18 have also been correlated with disease severity and morality in patients suffering from SARS-CoV-2. PAMPs derived from ORF-3a (viroporin), ORF-8b, viral RNA, and the E protein of SARS-CoV-2 have been demonstrated to stimulate NLRP3 inflammasome [82]. The IFN pathways are also hindered by innate-signalling proteins like non-structural protein 13 (NSP13), NSP15, and ORF-9b. NSP13 interacts with transducin-like enhancer of split-1 (TLE1), TLE3, and TLE5, and on the other hand, ORF-9c interferes with NLRX1, Nedd4 family interacting protein 2 (NDFIP2), and F2R-like trypsin receptor 1 (F2RL1), which are also involved in the NF-κB pathways [83]. Further, NLRC1 has also been noted to contribute to COVID-19 response and cytokine release, and silencing the gene encoding for NLRC1 has been seen to reduce IFN-1β expression [77].
Active viral replication, however, opens the window for immune intervention, but prolonged replication later results in hyperproduction of type I IFN with an influx of macrophages and neutrophils. This heavy dependence upon type I IFN responses further culminates in controlling the replication and induces an effective adaptive immune response. In general, both the B and T cells play a vital role in viral clearance, wherein most patients try to develop a humoral response within the first 10–14 days of the infection. Poor humoral responses (developed by B cells) have been associated with ineffective COVID-19 virus clearance in some patients, thereby suggesting its importance in SARS-CoV-2 neutralization [84]. Since the polarization of B cell follows a linear process, through pro-B cell, pre-B cell, immature B cell, and finally, matured B cell, studies have shown the partial reliance of variability, diversity and joining (VDJ) gene segments during the making of these virus-specific antibodies. Concerning SARS-CoV-2, antibody development towards immunoglobulin G (IgG) heavy variable 3-23 (IGHV3-23) and IGHV3-7 was identified, thereby helping us with antibody and vaccine development [85]. Nevertheless, certain types of mature B cells, known as double-negative (DN) B cells, which are known to lack expressions of memory markers (CD27) and IgD, have also been shown to promote cytokine release and autoantibody (AAb) progression, thus enhancing COVID-19 pathogenesis [86, 87]. Studies have reported that CD8+ T cell responses demonstrated in a better and more frequent manner, with respect to CD4+ cells, thereby correlating with disease severity [88]. Additionally, the virus-specific T cells from severe patients were associated with a higher frequency of polyfunctional CD4+ T cells (IL-2, IFN-γ, and TNF-α) and CD8+ T cells (IFN-γ and TNF-α) in comparison with moderate patients, thus postulating that strong T cell responses correlate with higher neutralizing antibodies (NAbs) [88]. Furthermore, T helper 17 (Th17) cells, neutrophils, and other granulocytes mediate the production of ILs (IL-1, IL-6, IL-8, and IL-17), MCP-1, granulocyte colony stimulating factor (G-CSF), granulocyte macrophage colony stimulating factor (GM-CSF), TNF-α, and prostaglandin E2 (PGE2), which further stimulate the production of monocytes, neutrophils, and other immune cells, thus trying to eradicate SARS-CoV-2 [89–91].
Concerning viral clearance, antibodies also play a multifactorial role by binding to viral proteins expressed on the infected cell surface and recruiting NK cells to kill the infected cell by antibody-dependent cell cytotoxicity mechanism [92]. Antibodies bind to the RBD of the S protein of SARS-CoV-2 and effectively block virus-ACE2 interaction, thereby neutralizing the virus [93–95]. However, the NAb responses have poor durability, thereby putting patients on a high susceptibility to re-infection after a few years of infection [96, 97]. Typical production of IgM and IgG constitute the antibody profile against the virus; however, it has been reported that IgM disappears by the end of week 12, but IgG persists longer [98]. Numerous studies have also suggested that SARS-CoV-2-based humoral immunity also doesn’t last long [99–101]. A steep decline of anti-SARS-CoV-2 antibodies has also been observed in asymptomatic COVID-19 patients, postulating their minimal protection against the viral infection [99–101].
As mentioned above, the magnitude and durability of COVID-19 infection correlate with the immune response. Several pieces of evidence have stated the defence mechanism of SARS-CoV-2, wherein it inhibits IFN-1 by regulating IFN-β synthesis, which further results in managing or aggravating viral replication or induction of an adaptive immune response [68, 102]. In several circumstances, it has also been observed that the virus dampens the attack by virtue of immune dysregulation caused by the same infection, thus derailing into a cytokine storm and T-cell exhaustion, further weakening the overall body’s immune response. This also partly clarifies the longer incubation period (2–11 days) when compared with influenza which usually has an incubation period of 2–4 days [103]. Indeed, the virus has also been seen to reduce antigen presentation in both major histocompatibility complex class I (MHC-I) and II [104–106].
Several autopsies and hospital reports have reported that patients with pneumonia abruptly succumb to respiratory failure and require mechanical ventilation [107]. Lethal sepsis arising from bacterial community-acquired pneumonia and sudden deterioration after 7–8 days of the first symptom derived the hypothesis of immune dysfunction [108]. Further analysis also stated that there was a depletion in CD4+ and CD8+ frequency cells, γδ T cells, and features of lymphopenia with increased D-dimers and hepatic dysfunction were noted [109]. There have been several hypotheses behind the cause of lymphopenia, with one being the inhibition of T cell circulation and apoptosis of memory CD8+CD44high T cells by cytokines like class I IFN and TNF-α, thereby promoting retention in lymphoid organs [109–111]. Necroptosis has also been associated with T cell death in which, NSP12 of the SARS-CoV-2 interactome interacts with receptor-interacting serine/threonine-protein kinase 1 (RIPK1) [112]. Though studies have suggested retention of T cells as a primary component in the deterioration of immune response, other investigations also have demonstrated that surviving T cells get functionally exhausted and express high-level T-cell Ig mucin-3 (TIM3) and programmed cell death protein 1 (PD-1) in SARS-CoV-2 patients [113]. Decreased productions of IFN-γ and IL-21 also support the exhaustion of CD8+ and CD4+ T cells [114, 115]. Other immune cells like cytotoxic lymphocytes and NK cells possess an upregulation of NK group 2 member A (NKG2A), an inhibitor of CD8+ T cells, and a downregulation of CD107a+, granzyme B+, IFN-γ+, and IL-2, consistent with the functional exhaustion of the immune cells [114, 116].
Release of pro-inflammatory cytokines, including IL-2, IL-6, IL-7, G-CSF, CXCL10, MCP-1, CCL2, macrophage inflammatory protein 1α (MIP-1α), and TNF-α in severe COVID-19 patients, dictates the severity of the infection. Circulating cytokines like IL-6 and IL-1β were also found in large quantities in the patient’s immune profile, thereby mediating Th17 cell differentiation and promoting further IL-6 and IL-17 production, thus worsening the clinical status of the patient [29, 117]. IL-17, produced by Th17 cells, also plays a major role in recruiting monocytes and neutrophils, and activating other downstream cytokines and cascades like IL-1, IL-6, IL-8, IL-21, TNF-α, and MCP-1, thereby exacerbating the inflammation [16, 118]. IL-22 also has been seen to enhance the expression of mucins, fibrinogens, and anti-apoptotic proteins, thereby increasing the chances of life-threatening edema and ARDS [119]. These cytokines further drive inflammation in the lungs and promote virus persistence by expressing B cell lymphoma 2 (Bcl-2) and Bcl-xL [120, 121]. All of this also results in an elevated D-dimer, C-reactive protein (CRP), procalcitonin, coagulopathy, and hyperferritinemia profile, which further turns into a typical macrophage activation syndrome [122–124]. Consistent with the cytokine profile of the other coronaviruses, this cytokine storm leads to severe clinical phenotypes, namely ARDS, tissue hypoxia, and death [125]. Ironically, the very immune system that usually acts to fight the infection potentially harms the host in doing so.
With the dysregulated secretion of inflammatory cytokines like TNF and IFN-γ, the combination of the same further characterizes a life-threatening condition, which is mediated by inflammatory cell death [pyroptosis, apoptosis, and necroptosis (PANoptosis; Figure 2)] [126]. Dependent on PANoptosomes, caspases, inflammasome components, and the synergism of TNF and IFN-γ, PANoptosis also depends upon several signalling pathways and transcriptional factors like STAT1 and IRF1, which further activates caspase-8, Z DNA binding protein 1 (ZBP1), TGF-β activated kinase-1 (TAK1) and RIPK1-affiliated cell death [127–135]. As a whole, PANoptosis works on a positive feedback loop wherein cytokine secretion causes PANoptosis which further results in more cytokine secretion, thus culminating into a cytokine storm, thereby promoting inflammation and disease progression [82].
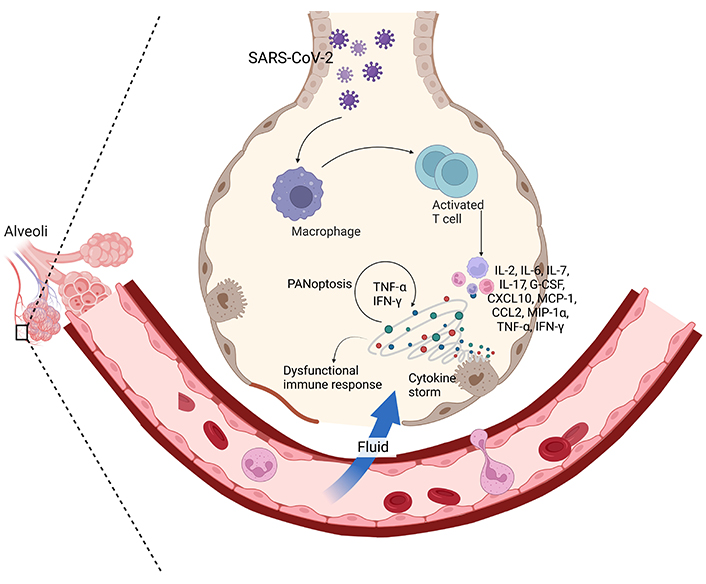
Clinical manifestations of COVID-19. The clinical manifestations of COVID-19 encompass a spectrum of symptoms. Initially, patients may experience fever, cough, and shortness of breath, as SARS-CoV-2 infects respiratory cells, primarily targeting alveoli. In severe cases, a cytokine storm, characterized by an excessive release of pro-inflammatory cytokines, including IFNs, can lead to severe inflammation and organ damage, a process akin to PANoptosis. This can result in ARDS and multi-organ dysfunction. The interplay of cytokines and the viral load contributes to the disease’s severity, making an understanding of these immunological responses vital for effective management and treatment of COVID-19
The cytokine release syndrome plays a vital and decisive role in COVID-19 patients. Occurring via 2 pathways, cytokine production either recognizes the virus by PRRs like TLR3, TLR7, TLR8, and TLR9 or through induction of DAMPs released from SARS-CoV-2 damaged cells [136, 137]. Upon lung or alveolar injury, the epithelial or endothelial or parenchymal cells release inflammatory mediators that mutually elicit multiple inflammatory cytokines and chemokines. Other than the inflammatory cytokines mentioned above, the type I and type III IFN responses, along with the IL-1-IL-6 axis further constitute pertinent biological signalling pathways [85, 138–142]. However, there are other controversies regarding the signatures of IL-6, IL-8, and TNF across varied acute conditions. Apart from the supporting evidence cited above, few studies have reported varying messenger RNA (mRNA) expressions of IL-1β and IL-6 in the autopsy reports of 4 individuals who succumbed to COVID-19 [143]. The same was in fact reproduced in a SARS-CoV-2-infected lung organoid using human induced pluripotent stem cells (iPSCs) [144].
Cytokine responses have also been observed in other organs, given that ACE2 is robustly found in other organs and epithelial cells. Cytokine storms in COVID-19 infections have been seen to trigger violet assaults to the body, causing massive excitation of the immune system, triggering an uncontrolled systemic inflammation (Figure 2), leading to ARDS and multi-organ failure and finally causing death [145]. Encompassing a broad range of severity, several neurological disorders have been associated with cytokine storm, e.g., immune effector cell-associated neurotoxicity syndrome, traumatic brain injury, encephalopathy, hemophagocytic lymphohistiocytosis, cytokine storm-associated encephalopathy (as proposed by Pensato et al. [146]), etc. [147]. Concerning the renal system, conditions from mild proteinuria to advanced acute kidney injury have been noted due to cytokine storms and COVID-19 [148]. Altered liver chemistry with progressive cases like cirrhosis has also been reported with respect to cytokine storms [149, 150]. Several meta-analyses have also deduced that heart failure is deemed to be one of the common complications occurring in COVID-19 cases, with cytokine storm being the primary inducer [151]. Other conditions like myocarditis, cardiac fibrosis, calcium dyshomeostasis, and cardiomyocyte pyroptosis amongst a few are also some of the manifestations observed as a result of cytokine storm [151].
Lifestyle comorbidities like hypertension, obesity, and diabetes have also been associated with COVID-19 complications [152]. ACE2, being expressed in metabolic tissues and organs like the thyroid, pancreas, adrenal, pituitary, ovaries, etc. infection in any of the organs, leads to disruption in the endocrinal system. Evidence confirms the dysregulation of glycemic homeostasis as an outcome in COVID-19 patients, suffering from diabetes [153]. A retrospective study stated that patients with controlled glycemia showed less mortality, in comparison to patients with high glycemic variability [153]. Associated with hyperinflammation and enhanced oxidative stress, diabetes in addition to infection further exacerbates endothelial or organ damage via TLR4 and receptor for advanced glycation end-products (RAGE) [154, 155]. In addition, COVID-19 infection is also known to dysregulate the potassium axis, leading to hypokalemia, further affecting glucose homeostasis in diabetic patients [156]. TMPRSS2 has also been correlated with androgenic hormones, which further describes the disparity in COVID-19 infection on the basis of gender [9]. Though more or less, SARS-CoV-2 infects both men and women equally, women’s mortality was found to be twice as high as that of men [157]. The renin-Ang-aldosterone system (RAAS) has also been reported to be highly active in COVID-19 patients [158]. Ang II promotes hyperinflammation by stimulating IL-6 in vascular and endothelial muscle cells via the type 1 Ang II (AT1) receptor [159].
Recent research has identified vitamin D as a key regulator of the RAS, although the precise molecular mechanism behind its down-regulation of the RAS is not fully understood [160, 161]. Vitamin D acts as a negative regulator of renin biosynthesis, mediated by the vitamin D receptor (VDR). It has been reported that vitamin D inhibits the formation of cyclic adenosine monophosphate (cAMP) response element-binding protein 1 (CREB1) and nuclear receptor corepressor 1 (NCOR1), which enhance renin protein expression [162]. Therefore, vitamin D may suppress RAS activity by down-regulating the renin transcript and inhibiting the conversion of angiotensinogen to Ang I and the ACE/Ang II cascade [163]. Vitamin D also triggers the development of regulatory T cells (Tregs) and inhibits the development of Th17 cells, subsequently helping the host cells to maintain the balance between COVID-19-mediated damage and immune response [160].
Mild peripheral neutrophilia has been documented in patients with COVID-19, wherein highly stimulated CD11β+, CD38+, human leukocyte antigen (HLA)-DR+ (an MHC-II cell surface molecule), and myeloid-derived suppressor cells (MDSCs) [164, 165]. Increased MDSCs have also been correlated with inflammation, hematopoiesis, and increased activation of peripheral immature neutrophil granulocytes, namely CD10– and CD16low [166, 167]. Neutrophilic infiltrations were also associated with the presence of SARS-CoV-2, where chemokine production and an activated Th17 response were reported, which likely contributed to the pathogenesis of the virus [168]. Circulating monocytes and other leukocyte subsets were also seen to undergo fluctuations and trajectories. Dysregulation of monocytes also led to a decrease in CD16+ and a shift towards CD14+ monocytes, though this transfer resolves in later stages of the infection [85, 105]. An enhanced CD169 expression with elevated productions of IL-1 and IL-6 also was shown in both COVID-19 patients’ lungs and mouse models of SARS-CoV-2 infection [55]. Though monocytes haven’t been reported to substantially alter in COVID-19, the feedback loop between T cells and macrophages drives the limited alveolitis towards the development of persistent alveolar inflammation and further lung damage [169].
Numerous studies have reported the prevalence of AAbs in COVID-19 patients, specifically those which neutralize type I IFNs, especially IFN-α2 and IFN-ω [170–173]. These AAbs were identified in approximately 10% of patients and were associated with critical COVID-19 pneumonia. Notably, these were absent in asymptomatic or mildly affected patients and were rare found in only 0.33% of healthy individuals before the pandemic and in a small fraction of individuals tested before contracting SARS‐CoV‐2 [170]. A significant discovery was that these AAbs were observed to neutralize IFN‐α and/or IFN‐ω at lower, more physiological concentrations (100 pg/mL), with a prevalence of 6.5% and 13.6% in patients with severe and critical COVID-19, and 18% in deceased patients [172]. These were more common in critical patients aged over 65 and were more prevalent in men. Furthermore, testing a broader age range of individuals from the general population revealed a marked increase in AAbs against IFN-α and/or IFN-ω after the age of 70, which may contribute to the increased risk of critical COVID-19 in the elderly [172]. IFN AAbs were also identified in the upper respiratory tract (nasopharyngeal swabs) and lower respiratory tract (bronchoalveolar lavage fluid) of COVID-19 patients [174]. These AAbs in nasopharyngeal swabs were associated with reduced IFN-stimulated responses in nasal epithelial cells in severe COVID-19 cases, potentially facilitating higher or prolonged viral replication in the respiratory tract and exacerbating excessive respiratory inflammation leading to severe pneumonia. These were also shown to hinder the antiviral activity of IFN-α against SARS‐CoV‐2 infection in vitro and in vivo, offering a possible explanation for the weakened antiviral defense observed in some severe patients during the acute phase [171]. A similar presence of systemic lupus erythematosus (SLE)-related AAbs were also found in COVID-19 cases [175]. Complement consumption is a well-established characteristic of SLE, with immune complexes primarily affecting the classical pathway [176]. Likewise, in severe instances of COVID-19, there is clear evidence of an exaggerated complement activation, resulting in thrombotic microangiopathy [177]. In addition, NETs release results in exposure to self-antigens and boost of AAb production, intensifying the autoinflammatory response [178]. COVID-19 patients exhibit elevated levels of anti-NET antibodies and other AAbs, with anti-NET IgG and IgM significantly higher in hospitalized patients compared with healthy individuals, correlating with poor clinical outcomes [179]. Additionally, those with higher anti-NET antibodies are more likely to have other AAbs, including antinuclear antibodies (ANAs) and anti-neutrophil cytoplasmic antibodies (ANCAs), suggesting that COVID-19 NETs may trigger autoimmune responses [180].
Moreover, COVID-19 has been associated with the development of AAbs targeting various tissues, including the skin, heart, and muscles, leading to conditions like myositis [181]. Hematological abnormalities, such as thrombocytopenia, lymphopenia, neutrophilia, and coagulopathy, have also been observed in COVID-19 patients [182]. The virus can also infect the central nervous system (CNS), leading to neuroinflammation and neurodegeneration through AAb production, molecular mimicry, and epitope spreading. Thyroid function can be impaired by COVID-19, with the virus affecting the CNS and leading to T-lymphocyte activation. Disorders such as Hashimoto’s and Graves’ thyroiditis have also been associated with COVID-19 potentially due to the high levels of ACE2 expression in the thyroid [183]. Autoimmune neurological disorders like multiple sclerosis and Guillain-Barré syndrome have been reported in COVID-19 patients [184]. Moreover, the virus can trigger autoimmune diseases in the GI system, including celiac disease, inflammatory bowel diseases, autoimmune hepatitis, and primary sclerosing cholangitis, possibly through long-term antigenic stimulation and T-cell activation [182]. In fact, an increased COVID-19 prevalence and mortality rate was recorded in patients with systemic sclerosis and inflammatory rheumatic disease, however, the efficacy of vaccines in terms of improved outcomes was less evident in the same [185–187].
During the initial stages of the pandemic, with no vaccine or acquired immunity from infection, the primary factor influencing viral evolution was the pursuit of efficient transmission. The emergence and recognition of variants of concern (VOCs), each with greater transmissibility than ancestral SARS-CoV-2 and varying abilities to cause more severe disease and to evade host immunity, has led to primarily identifying 5 variants, namely Alpha, Beta, Gamma, Delta, and Omicron (as given in Figure 3).
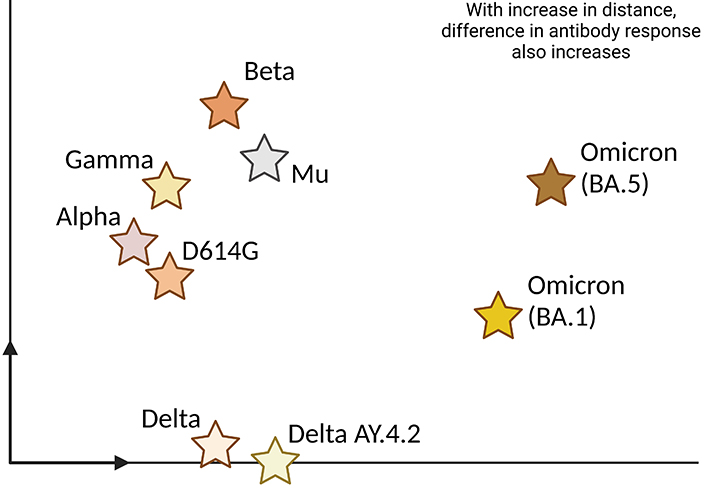
Antigenic map of SARS-CoV-2 variants. SARS-CoV-2 variants can be visualized on the antigenic map, which represents their differences in terms of antigenicity. This map shows how various viral strains respond to the immune system’s antibodies. Each star on the map represents a specific viral variant, and the distance between points reflects the degree of antigenic variation. Variants that are closer together on the map share more similarities in their antigens, while those farther apart exhibit greater differences
The Alpha variant or B.1.1.7 VOC, identified in the UK in 2020, exhibited an increase in disease severity, when compared with that of D614G, as evidenced by undesirable outcomes such as the need for critical care or death [188]. However, there was no clear indication of significant immune evasion, meaning it did not display resistance to antibodies from natural infection or vaccine-induced immunity [189]. Notable genetic changes in the Alpha variant include the 69/70 deletion, which has been observed independently and repeatedly. This deletion has been proposed to assist in evading N-terminal domain (NTD)-specific antibodies, which play a crucial role in immunity to SARS-CoV-2, and it has also been suggested as a compensatory mechanism for RBD mutations that contribute to immune escape [189]. Additionally, the N501Y substitution in the RBD of the Alpha variant has emerged repeatedly and has been shown to enhance viral fitness and transmissibility by increasing its affinity for the ACE2 host receptor [190]. Notably, genetic variation in the RBD of the S protein is of great significance, as it is an immunodominant region, essential for ACE2 receptor binding, and most currently used vaccines stimulate the production of S protein as the immunogen, which includes the RBD.
The Beta variant, also known as B.1.351, first detected in South Africa in May 2020, swiftly triggered a significant surge in infections in South Africa, which raised global concerns. Preliminary analyses of S protein mutations in Beta suggested substantial immune evasion, evident from reduced NAb levels in individuals who had previously been infected with other variants [191]. Furthermore, the Beta variant was found to cause more severe disease and exhibit higher transmissibility than the original SARS-CoV-2 strain [192]. Surprisingly, it did not spread globally to the extent initially feared, with the reasons for this phenomenon remaining unclear. The immune resistance of Beta is attributed to three key substitutions in the RBD: K417N, E484K, and N501Y [193]. The E484K and K417N substitutions have been shown to decrease antibody neutralization, while N501Y enhances binding to the ACE2 receptor, promoting increased transmissibility, as mentioned earlier.
The Gamma variant, also known as P.1, was first identified in Brazil in November 2020 and was designated as a VOC in January 2021. Similar to the Beta variant, Gamma led to a significant local surge in cases but did not cause widespread global transmission. Analyses indicated that Gamma was more transmissible and had a higher likelihood of resulting in severe outcomes, including death, compared with other variants circulating at the time [194]. Notably, Gamma shares substitutions in the same trio of amino acids in the RBD as Beta, although the specific substitutions in Gamma are K417T (instead of K417N), E484K, and N501Y, which contribute to immune evasion and increased infectivity. These characteristics were further substantiated by a high rate of SARS-CoV-2 reinfections in the Manaus region of Brazil [195]. The presence of the triple mutation in the RBD (K417N/T, E484K, and N501Y) in at least two distinct VOCs suggested convergent evolution [194, 195], leading some to propose limited flexibility in the SARS-CoV-2 spike glycoprotein. However, this hypothesis has been disproven by the emergence and global spread of highly divergent VOCs.
Delta, also known as B.1.617.2, was the first variant following Alpha to exhibit rapid global transmission, displacing Alpha as the predominant variant in most regions. Delta’s increased transmissibility was immediately apparent, given its rapid global spread, and numerous studies have conclusively established, that at the time of its emergence, Delta was the most transmissible VOC to date [196, 197]. An early study linked this greater transmissibility to higher virus levels in the respiratory tract, based on cycle threshold (Ct) values obtained through quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays [197]. Laboratory investigations of Delta viral isolates demonstrated accelerated growth kinetics, elevated virion release, and increased cleaved S protein levels compared to Alpha [198], offering an explanation for the higher virus levels. Analysis of the RBD and other S protein substitutions in Delta, combined with a higher rate of breakthrough infections in vaccinated individuals, led to the hypothesis that Delta was evading host humoral immune responses [198]. This hypothesis gained further support from laboratory experiments using blood from previously infected and vaccinated individuals to neutralize authentic or pseudo-typed virus [198, 199]. The two substitutions in the Delta spike RBD, L452R and T478K, did not enhance ACE2 binding; however, the L452R substitution was shown to confer approximately a fivefold reduction in susceptibility to polyclonal antibodies in plasma from vaccinated individuals [200]. Additionally, several substitutions in the NTD that are located within antigenic supersites (G142D, E156G, and deletion at positions 157/158) reduced antibody binding of NTD-specific monoclonal antibodies (mAbs) by at least tenfold [200].
With the recent breakthrough of the new variant of SARS-CoV-2, Omicron, formerly designated as B.1.1.529, a VOC is known to have 32 mutations located within the S protein [201]. Consisting of all the key mutations of the previous variants, including K417N, E48A, N501Y, and others, this variant can also change the sensitivity of the virus to NAbs [202, 203]. Zhang et al. [204] further showed that the newly made mutations did cause significant changes, concerning neutralization sensitivity, against numerous samples of human sera, pre-infected with the previous variants of COVID-19. The mutations also enhance the S protein’s affinity to ACE2 receptors, further contributing to its risk of transmissibility (3 mutations at cleavage site) and evasion in the immune system [205]. However, a hypothesis by Dejnirattisai et al. [206], states that changes found in RBD-62, which also happen to serve as an anchorage for ACE2, are more prone to mutational changes including those to reduce ACE2 affinity, so as to evade antibody neutralization. Recent research has stated the mutation of NSP6 in the Omicron variant [207]. NSP6 has been known to interfere with type I IFN pathways, by occluding the stimulation of TBK, thus presenting a huge number of autophagosomes. In addition, NSP13, NSP15, and ORF-9b also hinder IFN pathways [207]. Though not much research has been done in this field, we can speculate that mutations in NSP6 can lead to an appreciable amount of modifications, establishing a critical host-virus relationship, signifying immune escape, and SARS-CoV-2 pathogenicity.
Several investigations have been made in order to analyse the immune response towards this new variant. When compared with its older lineages, Omicron’s neutralization has been found to be significantly low (explained via an antigenic map in Figure 3), however, the higher rate of transmission and infectivity of Omicron possesses a major concern [208, 209]. Lung pathology reports of Omicron-infected hamsters suggested that NAbs produced after Omicron infection demonstrated substantial reduction or negligible neutralization against its other variants [207]. However, in spite of the broad neutralization escape against Omicron, 70–80% of the T cell responses were found to be cross-reactive [210]. Peptide pool simulation results detected no differences in typical cytokine production by virtue of both the wildtype and Omicron variants of COVID-19, suggesting that T cell epitopes remain minimally affected irrespective of the variant. In fact, booster vaccinations also led to the maintenance of variant-specific CD4+ and CD8+ T cell responses [211]. Apart from T cell production, a decreased IL-2, and IL-2 plus other cytokine release was also noted [212].
Taking the world by storm, this new Omicron variant has a very high transmission and infection rate, compared to the rest of the COVID-19 variants. It has also been seen to evade natural and vaccine-induced immunity [213]; however, booster vaccinations (mechanism explained in Figure 4) have been seen to improve protection against Omicron. In comparison with the prior Delta variant, a thumping 91% decrease in mortality was noticed in a study, when associated with Omicron infections [214]. Nevertheless, in spite of its mild severity, the sheer increase in the number of Omicron cases has put the world on a high alert with the global economy at stake. In fact, the further newer variants of Omicron, BA.1 and BA.2 have also replaced the previous Delta variants on their ability of immune escape and more transmissibility. A real-world study in Denmark showed that BA.2 is more infectious than BA.1, as it possesses more immune escape properties [215, 216]. The numerous mutations in the N-terminal and RBD region of the Omicron variants attribute to the immune escape properties [217]. However, the latest lineages of Omicron, BA.4 and BA.5’s neutralization capacities have reduced by 7 folds and 3 folds in unvaccinated and vaccinated individuals respectively [218]. NAbs titres against these sub-variants have been observed to be lower than that of their initial ancestors, BA.1 and BA.2, suggesting their increased neutralization escape [219, 220]. In context of immune evasion, BA.4 and BA.5 have been demonstrated to escape from BA.1- and BA.2-induced immunity, pertaining to the reason for re-infections in several cases [219, 221].
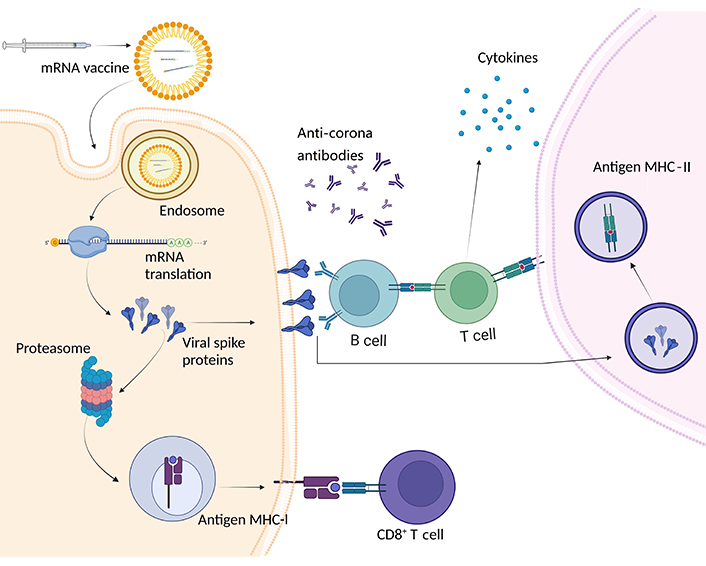
Scheme of mechanism of action of mRNA vaccines. mRNA vaccines for COVID-19 contain synthetic mRNA encoding the viral S protein. Once administered, host cells take up the mRNA and translate it, producing S proteins. These S proteins are then displayed on the cell surface via MHC-I, acting as antigens recognized by CD8+ T cells. Additionally, CD4+ T cells are activated, promoting a robust immune response. The proteasome plays a role in breaking down S protein fragments. This process triggers the release of cytokines and the production of antibodies, establishing immunity against COVID-19 without the presence of live virus
However, several analyses have shown that people subjected to Omicron variants had significantly lower odds in terms of hospitalization or mortality when compared with the other variants [214, 215, 222–224]. A high rate of asymptomatic cases was also observed, suggesting the milder symptoms of the variant, thereby confirming the lower pathogenicity of the same. In vitro studies have demonstrated the Omicron stimulated multinuclear syncytia in several cell lines when compared with other variants [225–227]. Activation of signalling pathways like NF-κB seemed to be less efficient in terms of the new variant, implying the slighter inflammatory response and extremely low mortality [228–230]. Though some of the countries have initiated booster doses as a resort to control the spread of this variant, more research is required on its influence upon cytokines, vaccine-based immune responses, post-COVID-19 complications, and mortality rates associated with Omicron. In fact, as per data of August 2, 2023, about 769 million cases have been reported globally signifying the revival of new variants of SARS-CoV-2 in the world [231].
Amidst the backdrop of successful vaccinations and improved social management [232], the virus persists albeit in attenuated form. Recent global statistics have revealed over 1 million new COVID-19 cases in 1 month, with an increasing trend in prevalence especially for newer variants. Public health attention is now focused on EG.5, a burgeoning subvariant of the Omicron lineage, gaining prominence in countries like the US and the UK. Though surpassing other Omicron descendants, EG.5 does not currently exhibit heightened transmissibility and is currently designated by the World Health Organization (WHO) as a “variant of interest”, and not yet a matter of concern [233]. EG.5’s adaptability is noteworthy, characterized as “competitive” in navigating vaccine-induced antibodies. Despite its prevalence, EG.5’s impact remains consistent with other Omicron descendants. Its association with a mutation at spike position 465, seen in various other variants, suggests an evolutionary advantage [234, 235]. This dynamic scenario underscores the evolving nature of the virus and the continuous need for vigilance and adaptation. Thus, further research is on demand as to whether this stays as the last variant of its kind or if it is going to script yet another deadly chapter in the history of the pandemic.
In a progressive evolutionary pattern, each new viral variant originates from the previous one, reflecting genetic drift in response to the host’s immune pressure. However, in the case of SARS-CoV-2, there have been instances of divergent strategies to enhance transmission and evade humoral immune responses, particularly in the Omicron and post-Omicron lineage. The most optimistic scenario for the future of SARS-CoV-2 evolution envisions a return to a more progressive pattern, now that substantial human immunity has been achieved. This scenario is less likely to yield a variant with significantly increased virulence. Nevertheless, as exemplified by the emergence of Omicron, the possibility of a highly divergent variant arising from prolonged infection in an immunocompromised host or an animal reservoir cannot be ruled out. Such a variant could potentially combine increased virulence with immune evasion.
Variants of SARS-CoV-2 continue to pose significant challenges in the realms of treatment, prevention, and COVID-19 diagnosis. An unanswered question is whether previous VOCs might resurface over time, either from the human population or an animal reservoir, or whether they have become extinct. Additionally, the development of methods to predict new mutations and mutation combinations that could appear in variants may aid in readiness for their emergence. For instance, a recent report outlines a new approach to identifying impactful mutations based on genomic and epidemiological surveillance data [236]. This method has successfully predicted mutations present in VOCs in retrospect [236]. Functional assays have also been employed to explore the mutational landscape of the RBD, assessing the potential for immune response and therapy evasion. While these methods offer valuable insights, predicting future variants remains challenging due to the intricate host-virus interactions and the adaptable nature of the virus. Further research aimed at enhancing the prediction and detection of SARS-CoV-2 variants, coupled with rapid implementation of measures to control their spread, is crucial for the future of global public health.
The current understanding of SARS-CoV-2 pathogenesis in human can be divided into two phases. The initial phase is marked by high-level viral replication and diminished immune responsiveness, while the second phase sees a reversal of this balance. Both of these phases can be addressed using immunotherapeutic strategies. In the first phase, these strategies aim to bolster immunity, while in the second phase, the focus shifts to reducing immunomodulatory responses. Two crucial lessons drawn from historical experiences have been swiftly incorporated into treatment design: Firstly, the window for effective antiviral immune intervention may be brief and early; and secondly, it is recognized that cytokines can have both harmful and beneficial effects, necessitating the potential inhibition of cytokine responses to mitigate immunopathology.
Drawing from historical knowledge of the antiviral properties of IFNs against hepatitis C virus and influenza, and supported by retrospective studies highlighting the pivotal role of type I IFNs in safeguarding against SARS-CoV-2, the therapeutic application of type I IFN in the early stages of SARS-CoV-2 infection was rapidly considered at the outset of the pandemic [170]. It is, however, important to note that IFN levels are closely associated with the severity of COVID-19. The timing of IFN response appears to be a critical determinant, a factor observed in previous influenza research and more recently in preclinical studies related to SARS, MERS, and COVID-19 [237]. A retrospective multicohort study in the early stages of the pandemic suggested that early IFN-α treatment (within 5 days of hospitalization) increased the likelihood of patient survival [238]. In contrast, delayed initiation of IFN therapy led to reduced benefits, reflecting findings reminiscent of those in patients with MERS [238]. Prospective studies involving hospitalized COVID-19 patients further corroborated the importance of timing and disease severity in the effects of IFN therapy. Interestingly, the WHO Solidarity trial revealed that subcutaneous IFN-β1a led to a slight increase in mortality risk for patients requiring supplemental oxygen therapy compared to controls, indicating that these patients might have progressed too far into the disease course to benefit from IFN therapy [239]. Conversely, a small randomized controlled trial among a similar population of hospitalized COVID-19 patients demonstrated that aerosolized IFN-β1a facilitated faster recovery [240]. Additional trials in different patient groups and IFN subtypes are currently ongoing, with the goal of providing further insights into this complex issue.
Corticosteroid treatment has been proven to offer substantial benefits to severely ill COVID-19 patients [241]. Notably, corticosteroids effectively reduced the risk of death in patients receiving invasive mechanical ventilation and those receiving oxygen support without mechanical ventilation. This stands in contrast to influenza, where corticosteroids were ineffective and even detrimental in severe cases. The underlying reasons for this discrepancy remain uncertain but might be related to the significantly lower incidence of secondary bacterial infections in COVID-19. This reduced incidence is linked to SARS-CoV-2’s induction of elevated levels of the cytokine IL-6, which possesses strong pro-inflammatory and potent antibacterial effects.
One characteristic of severe COVID-19 is the presence of a heightened cytokine response that contributes to immunopathology [49]. It is noteworthy that the absolute levels of cytokines in COVID-19, while impactful, are only a fraction of those found in other life-threatening syndromes unrelated to COVID-19. Managing the virus-induced hyperinflammatory response has posed a challenge, despite ongoing investigations in severe influenza. Multiple anti-cytokine approaches are under current examination. Notably, IL-6 became an early target due to its successful application in treating immune-mediated inflammatory disorders such as rheumatoid arthritis and its effectiveness in addressing hyperinflammatory complications associated with chimeric antigen receptor (CAR) T cell therapy [242]. Two classes of anti-IL-6 agents have been under scrutiny: those that target the IL-6 receptor (e.g., tocilizumab and sarilumab) and those direct against IL-6 itself (e.g., siltuximab) [243]. Tocilizumab has undergone extensive evaluation in COVID-19 and has shown improved clinical outcomes, including reduced progression to invasive mechanical ventilation and reduced mortality. Nevertheless, discrepancies in trial results might be attributed to the concurrent use of corticosteroids, which were more frequently administered and earlier in some trials. Essentially, it appears that corticosteroids and IL-6 blockade act on complementary inflammatory pathways and demonstrate more significant benefits when used in combination.
Investigations into treatments that block signalling by GM-CSF, IL-1, and other cytokines are also ongoing for COVID-19 [244]. Early findings suggest improved patient outcomes with therapies such as the IL-1 receptor antagonist anakinra and GM-CSF antibodies [83]. However, conclusive evidence necessitates larger-scale trials. The pleiotropic functions of these cytokines underscore the importance of comprehending their roles in severe COVID-19 and identifying patient groups that might derive the most benefit from cytokine-directed therapies. Another approach involves the use of JAK inhibitors, which function downstream of various cytokine receptors. Originally designed for patients with chronic inflammatory conditions such as rheumatoid arthritis and inflammatory bowel diseases, these orally bioavailable small molecules offer a practical alternative to large-molecule therapeutics that require injection, like anti-TNF agents. Combination therapies, including baricitinib (a JAK inhibitor) and remdesivir (a direct antiviral), have demonstrated shorter recovery times and reduced mortality compared with remdesivir alone [245]. Similarly, trials involving the JAK inhibitor tofacitinib in COVID-19 patients have indicated improved survival, albeit in a relatively small sample size [246]. The exact contribution of JAK inhibitors to this effect remains unclear, necessitating further investigation. These oral therapies are more suitable for widespread use due to practical considerations, offering a potential alternative to intravenous mAbs. Furthermore, two small-scale studies investigating the serotonin reuptake inhibitor fluvoxamine found it to prevent clinical deterioration in early COVID-19 patients [247, 248]. This effect might be attributed to the drug’s ability to dampen pro-inflammatory cytokine production, although the precise relationship between these factors remains to be fully elucidated.
Severe COVID-19 often exhibits excessive coagulation and thrombosis in conjunction with hyperinflammatory responses [13]. Endothelial dysfunction, coagulopathy, and overactivation of the complement system, in addition to inflammation, contribute to the development of thrombotic complications that likely play a role in the onset of ARDS. While the previously mentioned anti-inflammatory interventions can aid in mitigating this pathological process, additional therapeutic strategies, including antithrombotic medications like heparin, garadacimab, nafamostat mesylate, tissue-type plasminogen activator, and inhibitors targeting the complement cascade, such as C5 inhibitors, eculizumab and ravulizumab, and the C3 cleavage inhibitor, AMY-101, may act synergistically with anti-inflammatory treatments [243].
Another pressing therapeutic challenge, with some similarities to post-tuberculosis (TB) lung disease, is “long COVID”. This condition is characterized by persistent symptoms such as fatigue, anhedonia, muscle weakness, cognitive deficits, anxiety, depression, myalgia, and arthralgia [249]. As the underlying mechanisms of long COVID remain unclear, immunotherapeutic strategies have yet to be developed. However, since persistent inflammation and a prothrombotic state are frequently observed in these patients, it is likely that they will require anti-inflammatory and antithrombotic therapies similar to those described above for acute COVID-19. Notably, clinical trials are underway to investigate the beneficial effects of dissolving NETs through the use of deoxyribonuclease (DNAse), given the presence of neutrophil activation and NET release in patients with COVID-19 [72]. Several clinical trials (e.g., ClinicalTrials.gov: NCT04402944, NCT04355364, NCT04432987, NCT04359654, NCT04445285, and NCT04402970) are aimed at validating initial observations regarding the advantages of NET dissolution.
In summary, more substantial progress has been made in mitigating the immunopathological aspects of severe COVID-19 by employing cytokine inhibitors and signalling pathway inhibitors than in bolstering immunity. The latter approach requires early implementation during infection, when the pathogen load is low, to achieve success.
Being obligatory cellular parasites and infecting their host cells since the dawn of life, the coevolution of viruses and hosts is an intricate arms race that has shaped the biological landscape. This dynamic process fuels a continual adaptation: Viruses mutate to escape immune responses, while hosts refine immune strategies to counteract new viral tactics. As seen in various such virus-host duality (reviewed in Kaján et al. [250], 2020), this interplay has led to genetic diversification in both parties, as natural selection favours individuals that can outpace their counterparts [250]. A study by Latinne et al. [251] also showed a correlation between Rhinolophus sinicus bats and SARS-CoV-2, thus providing the opportunity for coevolution with its hosts. This ongoing coevolutionary saga is also evident in the latest inextricable linkage between SARS-CoV-2 and humans, wherein the virus has been seen to mutate occasionally, creating a lineage of variants (as explained in Figure 3). Though it hasn’t yet been deduced concretely regarding their association, understanding their complex interplay is crucial for deciphering the mechanisms of pathogenesis, developing targeted therapies, and unveiling the mysteries of evolution.
Mostly involving respiratory manifestations, COVID-19 was also known to be involved in affecting other organs, followed by complications and dysregulation of host responses. Though the immune system plays the most vital role in fighting COVID-19, ironically it also turns out to be harmful and eventually becomes the primary cause of COVID-19-affiliated death. Thus, amidst the notion of acknowledging the reason behind the innumerable deaths due to COVID-19, it can be stated that the derangement of the immune system and post-infection is the culprit. Armed with this knowledge, therapeutic guidelines could be potentially updated, thereby allowing medical professionals to provide optimum care for patients with COVID-19.
AAb: autoantibody
ACE: angiotensin-converting enzyme
Ang: angiotensin
ARDS: acute respiratory distress syndrome
CCL2: C-C motif chemokine ligand 2
COVID-19: coronavirus disease 2019
CSF: cerebrospinal fluid
CXCL8: C-X-C motif chemokine ligand 8
DAMPs: damage-associated molecular patterns
GI: gastrointestinal
GM-CSF: granulocyte macrophage colony stimulating factor
IFN-β: interferon-β
IgG: immunoglobulin G
IL-1: interleukin-1
IRF3: interferon response factor 3
ISGs: interferon-stimulated genes
JAK: Janus kinase
LGP2: laboratory of genetics and physiology 2
mAbs: monoclonal antibodies
MCP-1: monocyte chemoattractant protein-1
MDA5: melanoma differentiation-associated protein 5
MERS: Middle East respiratory syndrome
MHC-I: major histocompatibility complex class I
mRNA: messenger RNA
NAbs: neutralizing antibodies
NETs: neutrophil extracellular traps
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NK: natural killer
NLRs: nucleotide-binding oligomerization domain-like receptors
NSP13: non-structural protein 13
NTD: N-terminal domain
ORFs: open reading frames
PAMPs: pathogen-associated molecular patterns
PANoptosis: pyroptosis, apoptosis, and necroptosis
PRRs: pattern recognition receptors
RAS: rat sarcoma
RBD: receptor-binding domain
RIG-1: retinoic acid inducible gene-1
S protein: spike protein
SARS: severe acute respiratory syndrome
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
STAT: signal transducers and activators of transcription
TBK-1: TANK-binding kinase-1
TGF-β1: transforming growth factor-beta 1
Th17: T helper 17
TLE1: transducin-like enhancer of split-1
TLRs: Toll-like receptors
TMPRSS2: transmembrane protease serine 2
TNF: tumor necrosis factor
VOCs: variants of concern
OSS: Writing—original draft, Writing—review & editing, Visualization. AN, RM, and KP: Writing—review & editing. RD: Conceptualization, Writing—review & editing, Supervision. SK: Conceptualization, Writing—review & editing, Supervision, Funding acquisition. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The study was financially supported by the IMRD grant (AIIMS Intramural Grant [COVID 45]) from AIIMS Delhi received by SK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Marc H.V. Van Regenmortel
Christine Jacomet
Vladimir N. Uversky
Brent Brown ... Ingo Fricke
Chittaranjan Baruah ... Bhabesh Deka
Brent Brown ... Enrique Chacon-Cruz
Mikolaj Raszek ... Alberto Rubio-Casillas
Ankit Kumar ... Vijay Mishra
Yulia Desheva ... Irina Isakova-Sivak
Kenneth Lundstrom
