 Review
Review

Affiliation:
1Faculty of Medicine, National Research Lobachevsky State University of Nizhny Novgorod, 603950 Nizhny Novgorod, Russia
Email: maherakl555@gmail.com
ORCID: https://orcid.org/0000-0001-5480-1688
Affiliation:
2The Public Health Department, Riyadh First Health Cluster, Ministry of Health, Riyadh 11564, Saudi Arabia
ORCID: https://orcid.org/0000-0003-3477-236X
Explor Endocr Metab Dis. 2025;2:101444 DOI: https://doi.org/10.37349/eemd.2025.101444
Received: August 08, 2025 Accepted: October 09, 2025 Published: November 03, 2025
Academic Editor: Peter Nawroth, University of Heidelberg, Technische Universitaet Dresden, Germany
The article belongs to the special issue Current Views on Pathogenesis, Diagnosis and Management of Type 2 Diabetes Mellitus and Its Complications and Related Conditions
Type 2 diabetes mellitus (T2DM), expected to exceed 700 million cases by 2045, is usually attributed to obesity and peripheral resistance but neglects insulin’s structural integrity. This review introduces the Sulfur-Insulin Deformation Hypothesis, positing T2DM as a sulfur metabolism disorder where mitochondrial suffocation disrupts the transsulfuration pathway [methionine to cysteine via cystathionine β-synthase (CBS) and γ-lyase (CGL)], depleting cysteine and glutathione (GSH), impairing protein disulfide isomerase (PDI) activity, and deforming insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19) as a primary trigger of insulin resistance. A literature synthesis was conducted (1995–2025) across PubMed, Scopus, Web of Science, and Google Scholar, using Medical Subject Headings (MeSH) terms like “sulfur metabolism”, “insulin misfolding”, and “mitochondrial dysfunction”. From 1,202 articles, 113 studies were selected, including in vitro insulin folding models, animal metabolic stress data, human sulfur biomarker analyses, and trials of sulfur donors (e.g., N-acetylcysteine). Mitochondrial dysfunction reduces adenosine triphosphate (ATP), depleting cysteine and GSH by 30–73.8% (red blood cell GSH: 1.78 ± 0.28 µmol/g vs. 6.75 ± 0.47 µmol/g Hb, P < 0.001), elevating reactive oxygen species (ROS). This impairs PDI isoforms (PDIA1, PDIA3, PDIA4), disrupting insulin bonds; the A6–A11 bond loses 50–70% affinity [r = –0.65, P < 0.05 for homeostatic model assessment of insulin resistance (HOMA-IR)], hindering phosphoinositide 3-kinase-protein kinase B (PI3K-Akt) signaling and glucose transporter type 4 (GLUT4) translocation. In 225 T2DM patients, PDIA4 elevation correlated with glucose (r = 0.62, P < 0.01) and reduced sensitivity (r = –0.67, P < 0.01). PDIA4 inhibition [presenilin 1 (PS1), IC50 = 4 μM] cuts ROS by 50% (P < 0.01), lowers hemoglobin A1c (HbA1c) by 1.2% (P < 0.05), and boosts β-cell survival by 30% (P < 0.05). Redox-mediated chain splitting degrades 20% of insulin (0.40 nmol/kg/min) at –137 mV, modulated by GSH. The hypothesis redefines T2DM as a sulfur-driven structural disorder, unveiling the gut-mitochondria-sulfur-insulin axis and advocating sulfur-centric therapies (e.g., N-acetylcysteine, methylsulfonylmethane).
Type 2 diabetes mellitus (T2DM), a global health crisis projected to surge in prevalence, is traditionally attributed to peripheral insulin resistance driven by obesity, oxidative stress, and inflammation [1]. Yet, these models often overlook the critical role of insulin’s structural integrity, particularly its sulfur-dependent disulfide bonds. The Sulfur-Insulin Deformation Hypothesis offers a groundbreaking framework, asserting that mitochondrial dysfunction in intestinal epithelial cells, termed mitochondrial suffocation, triggers organic sulfur deficiency, leading to insulin misfolding and systemic insulin resistance. This research aims to compile and elucidate evidence linking defective disulfide bond formation to insulin dysfunction, redefining T2DM as a sulfur metabolism disorder and revolutionizing its mechanistic interpretation. Insulin, a 51-amino-acid polypeptide, relies on three disulfide bonds (A6–A11, A7–B7, A20–B19) formed through cysteine thiol oxidation to maintain its bioactive conformation for high-affinity insulin receptor (IR) binding [2]. These bonds, dependent on dietary methionine and cysteine via the transsulfuration pathway, are disrupted by mitochondrial suffocation, which impairs ATP production and inhibits cystathionine β-synthase (CBS) and γ-lyase, reducing cysteine availability [3–6]. This sulfur scarcity compromises protein disulfide isomerase (PDI) activity in pancreatic beta cells, leading to aberrant disulfide bond formation and misfolded insulin with reduced receptor affinity [7]. Such structural defects disrupt phosphoinositide 3-kinase-protein kinase B (PI3K-Akt) signaling, impairing glucose transporter type 4 (GLUT4) translocation and glucose uptake [8]. Concurrently, sulfur deficiency elevates reactive oxygen species (ROS), activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and releasing pro-inflammatory cytokines [tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6)], which exacerbate insulin resistance through c-Jun N-terminal kinase (JNK)-mediated serine phosphorylation of IR substrate-1 (IRS-1) [9–11]. Oxidative stress also weakens gut barrier integrity, promoting toll-like receptor 4 (TLR4)-mediated endotoxemia and systemic inflammation [12, 13]. Despite extensive research linking T2DM to peripheral insulin resistance, obesity, oxidative stress, and inflammation, the upstream structural determinants of insulin function remain underexplored. In particular, the contribution of sulfur metabolism to insulin stability has not been systematically investigated, leaving a critical research gap in our understanding of how mitochondrial dysfunction and cysteine/glutathione (GSH) deficiency reshape insulin conformation. The Sulfur-Insulin Deformation Hypothesis directly addresses this gap by proposing that disruption of the transsulfuration pathway (methionine-CBS/CGL-cysteine) leads to GSH depletion, PDI impairment, and deformation of insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19). By consolidating biochemical, molecular, and translational evidence, this framework elucidates the gut-mitochondria-sulfur-insulin axis and offers a transformative lens to reinterpret insulin resistance as a structural disorder of sulfur metabolism. Importantly, this model not only introduces conceptual novelty but also provides testable predictions, including measurable changes in circulating GSH and PDI isoforms, altered disulfide bond integrity in proinsulin and insulin, and the potential reversibility of insulin resistance through sulfur-donor interventions. Thus, the hypothesis bridges a long-standing gap in T2DM pathogenesis and establishes a strategic roadmap for future validation through proteomic, metabolomic, and clinical investigations.
This review presents the Sulfur-Insulin Deformation Hypothesis as a hypothesis-building framework, proposing that sulfur deficiency, driven by mitochondrial dysfunction in intestinal epithelial cells, triggers proinsulin misfolding, disulfide bond deformation (A6–A11, A7–B7, A20–B19), and insulin resistance in T2DM. Departing from peripheral-focused paradigms, this work redefines T2DM as a sulfur-driven structural disorder, aiming to establish a novel etiological perspective rather than a mere literature review. The hypothesis was developed through a targeted narrative synthesis of mechanistic insights from redox biology, mitochondrial pathology, protein biochemistry, and immunometabolism. Evidence was curated from PubMed, Scopus, Web of Science, and Google Scholar using Medical Subject Headings (MeSH) and free-text terms including “sulfur metabolism”, “insulin misfolding”, “disulfide bonds”, “glutathione deficiency”, “mitochondrial dysfunction”, “intestinal epithelium”, “oxidative stress”, “transsulfuration pathway”, “endoplasmic reticulum stress”, “cysteine”, and “type 2 diabetes”. Boolean operators (AND/OR) facilitated interdisciplinary connections, drawing on peer-reviewed studies from 1995 to 2025, with an emphasis on recent advances (e.g., 2022–2025) in PDI dysregulation and sulfur donor research. Sources include in vitro insulin folding models, animal metabolic stress studies, human sulfur biomarker data, and trials of sulfur donors [e.g., N-acetylcysteine (NAC), methylsulfonylmethane (MSM)].
Analytical rigor was enhanced using theoretical tools: biochemical pathway mapping (transsulfuration via CBS/CGL), molecular docking simulations (insulin-receptor interactions), Raman spectroscopy (disulfide bond integrity at 510–540 cm⁻1), and structural modeling (tertiary structure impacts). The narrative synthesis adhered to Scale for the Assessment of Narrative Review Articles (SANRA) guidelines to ensure clarity, evidence selection, and conceptual integration, prioritizing a cohesive framework over quantitative screening. A future validation roadmap addresses reviewer concerns: (1) proteomic analyses [co-immunoprecipitation (co-IP) with liquid chromatography-tandem mass spectrometry (LC-MS/MS)] to isolate and structurally identify misfolded insulin in T2DM plasma, comparing disulfide bonds with healthy controls; (2) metabolomic profiling of sulfur metabolites (cysteine, GSH) in tissues; (3) in vitro enterocyte models under sulfur deficiency to assess mitochondrial function; (4) animal studies evaluating NAC and MSM effects on insulin structure and glucose homeostasis (> 1 year); and (5) clinical trials with expanded samples (> 250 participants) and prolonged follow-up to assess long-term efficacy and safety. This roadmap aims to confirm causality and guide sulfur-centric therapeutic.
The intestinal epithelium, a metabolic hub for processing sulfur-containing amino acids, relies on robust mitochondrial function to support energy-intensive nutrient absorption [14, 15]. In T2DM, chronic stressors like hyperglycemia and high-fat diets induce mitochondrial dysfunction in enterocytes, termed mitochondrial suffocation, disrupting the electron transport chain (ETC), particularly complexes I and III [16, 17]. This reduces ATP production and generates excessive ROS, depleting cellular antioxidants and impairing sulfur metabolism [18, 19]. ROS overproduction exhausts GSH, a cysteine-dependent tripeptide critical for redox homeostasis, exacerbating cellular damage [20]. The transsulfuration pathway, converting methionine to cysteine via methionine adenosyltransferase, CBS, and cystathionine γ-lyase, is compromised by ATP scarcity, reducing cysteine synthesis [21–25].
This cysteine deficiency disrupts GSH production and PDI activity, impairing insulin’s disulfide bond formation (A6–A11, A7–B7, A20–B19), leading to misfolded insulin with diminished receptor-binding capacity [26–28]. Immunologically, mitochondrial suffocation triggers NF-κB activation, upregulating pro-inflammatory cytokines (TNF-α, IL-6) that promote JNK-mediated serine phosphorylation of IRS-1, disrupting PI3K signaling and exacerbating insulin resistance [29–31]. Additionally, ROS-induced downregulation of tight junction proteins (occludin, zonula occludens-1) compromises gut barrier integrity, enabling lipopolysaccharide (LPS) translocation and TLR4-mediated endotoxemia, further amplifying systemic inflammation [32–34].
This gut-mitochondria-sulfur-insulin axis underscores mitochondrial suffocation as a pivotal driver of sulfur deficiency and T2DM pathogenesis.
The Sulfur-Insulin Deformation Hypothesis redefines T2DM by asserting that sulfur deficiency, stemming from mitochondrial dysfunction in intestinal epithelial cells, drives insulin misfolding, a primary trigger of insulin resistance. Insulin, a 51-amino-acid polypeptide comprising A (21 amino acids) and B (30 amino acids) chains, is stabilized by three disulfide bonds (A6–A11, A7–B7, A20–B19) formed through cysteine thiol oxidation, essential for its three-dimensional conformation and high-affinity binding to the IR [35–40]. In pancreatic beta cells, insulin biosynthesis starts with preproinsulin, cleaved to proinsulin, and folded in the endoplasmic reticulum (ER), where PDI catalyzes disulfide bond formation by oxidizing cysteine residues, a process critically dependent on cysteine availability [41, 42]. Mitochondrial dysfunction, termed mitochondrial suffocation, impairs the transsulfuration pathway by reducing ATP-dependent activity of CBS and γ-lyase, limiting cysteine synthesis [43, 44].
This cysteine scarcity disrupts PDI function, leading to incomplete or aberrant disulfide bonds, producing misfolded insulin with altered tertiary structure, as demonstrated by Raman spectroscopy (510–540 cm⁻1) showing reduced bond integrity (Figure 1) [45, 46]. Misfolded insulin compromises the insulin signaling cascade, pivotal for glucose homeostasis. Normally, insulin binds to the IR, a tyrosine kinase with extracellular α-subunits and intracellular β-subunits, inducing autophosphorylation at tyrosine residues (Tyr1158, Tyr1162, Tyr1163) [47, 48]. This recruits IRSs (IRS-1/2), activating PI3K, which converts phosphatidylinositol-4,5-bisphosphate to phosphatidylinositol-3,4,5-trisphosphate, triggering Akt via phosphoinositide-dependent kinase-1 [49, 50]. Akt promotes GLUT4 translocation to the plasma membrane in skeletal muscle and adipose tissue, facilitating glucose uptake, and inhibits hepatic gluconeogenesis by suppressing phosphoenolpyruvate carboxykinase and glucose-6-phosphatase [51, 52].
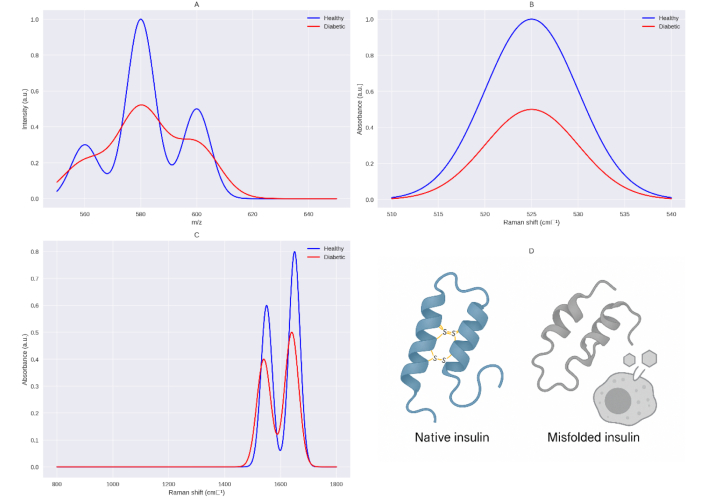
Simulated comparative analysis of insulin structure in healthy and diabetic conditions based on the sulfur deficiency hypothesis. This figure presents a simulated analysis comparing the structural properties of insulin in healthy (blue) and diabetic (red) conditions, focusing on the impact of sulfur deficiency in type 2 diabetes mellitus (T2DM) as proposed by the Sulfur-Insulin Deformation Hypothesis. Panel (A) displays LC-MS/MS spectra in the range of 560–640 m/z, where diabetic insulin exhibits greater fragmentation (smaller, more dispersed peaks at 560, 580, 600, and 620 m/z) compared to healthy insulin, indicating structural deformation due to sulfur deficiency. Panel (B) shows Raman spectra in the 510–540 cm–1 range (S-S stretching region), revealing a significant reduction in peak intensity at 525 cm–1 for diabetic insulin, consistent with the loss of disulfide bonds caused by sulfur deficiency. Panel (C) illustrates Raman spectra in the 800–1,800 cm–1 range, highlighting shifts in the amide I (from 1,650 cm–1 to 1,640 cm–1) and amide II (from 1,550 cm–1 to 1,540 cm–1) bands in diabetic insulin, along with reduced intensity, indicative of misfolding due to sulfur deficiency. Panel (D) provides a molecular representation comparing native insulin (healthy) with intact disulfide bonds to misfolded insulin (diabetic, sulfur-deficient), where disulfide bonds at A6–A11, A7–B7, and A20–B19 are disrupted, impacting the function of beta cells in T2DM. These results are based on computational simulations using tools like PyMOL and await experimental validation. This AI-crafted schematic, based on unpublished computational modeling, awaits experimental validation to confirm sulfur deficiency effects on β-cell function in T2DM. Conceptual schematic created by the authors to simulate Raman spectral changes in insulin disulfide bonds under sulfur-deficient conditions; illustrative and unpublished work. LC-MS/MS: liquid chromatography-tandem mass spectrometry.
Molecular docking models show that disruption of the A6–A11 disulfide bond misaligns key receptor-binding residues (ValA3, TyrA19), reducing IR affinity by ~60%, impairing IRS phosphorylation, PI3K-Akt signaling, and GLUT4 translocation, while allowing unchecked hepatic glucose production and driving hyperglycemia (Figure 2) [53–55].
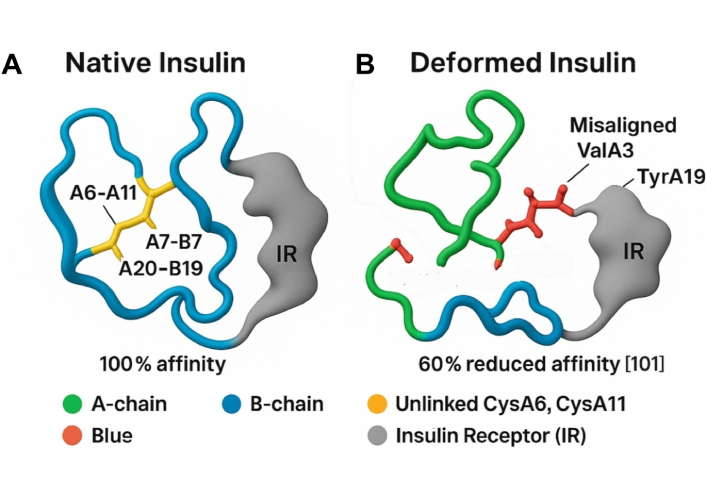
Molecular docking model of native vs. deformed insulin with IR. This figure illustrates the structural and functional impact of disulfide bond integrity on insulin-receptor interactions, supporting the Sulfur-Insulin Deformation Hypothesis. Panel A (Native Insulin) depicts the native insulin structure with the A-chain (green) and B-chain (blue) stabilized by disulfide bonds (A6–A11, A7–B7, A20–B19, yellow), enabling optimal docking with the insulin receptor (IR, grey) at 100% affinity. Panel B (Deformed Insulin) highlights the consequences of sulfur deficiency, showing the absence of the A6–A11 disulfide bond (indicated as unlinked CysA6, CysA11 in yellow), leading to A-chain misfolding (green). This results in misaligned receptor-binding residues ValA3 and TyrA19 (red), impairing interaction with IR and reducing affinity by 60% [101]. The legend clarifies the color scheme: green (A-chain), blue (B-chain), yellow (unlinked CysA6, CysA11), red (ValA3, TyrA19), grey (IR). The caption below reads: “Deformation-induced misalignment of ValA3 and TyrA19 impairs IR binding, supporting the Sulfur Insulin Deformation”, reinforcing the hypothesis that sulfur deficiency disrupts insulin folding and receptor binding, contributing to insulin resistance in type 2 diabetes mellitus (T2DM). Scientific simulation independently designed by the authors to illustrate receptor-binding impairment following A6–A11 disulfide bond disruption; conceptual representation, unpublished work.
Cysteine deficiency also reduces GSH synthesis, increasing ROS and activating NF-κB, which upregulates pro-inflammatory cytokines (TNF-α, IL-6) [56–60].
These cytokines induce JNK-mediated serine phosphorylation of IRS-1 (Ser307), further disrupting PI3K-Akt signaling, while impaired thioredoxin and peroxiredoxin function exacerbates oxidative stress [61–65]. Sulfur deficiency exacerbates metabolic dysregulation through ER stress and immunological cascades. Cysteine scarcity limits PDI activity, causing misfolded insulin to accumulate in the ER, triggering the unfolded protein response (UPR) via sensors inositol-requiring enzyme 1 (IRE1), protein kinase R-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [66, 67]. Chronic ER stress activates pro-apoptotic pathways through IRE1/PERK-mediated JNK and CCAAT/enhancer-binding protein homologous protein (CHOP), leading to beta-cell apoptosis and reduced insulin secretion [66–69]. Misfolded insulin aggregates further contribute to glucotoxicity, a hallmark of T2DM [70, 71]. Immunologically, reduced cysteine impairs GSH synthesis, a critical antioxidant, increasing ROS and activating NF-κB, which upregulates pro-inflammatory cytokines (TNF-α, IL-6) [56, 60, 72–74]. These cytokines induce JNK-mediated serine phosphorylation of IRS-1 (Ser307), disrupting PI3K-Akt signaling [61, 62].
Sulfur deficiency also impairs redox-regulatory proteins thioredoxin and peroxiredoxin, reliant on disulfide bonds, perpetuating oxidative stress [63, 64]. Additionally, reduced mucin synthesis weakens gut barrier integrity, enabling LPS translocation and TLR4-mediated endotoxemia, amplifying systemic inflammation [65, 75–77]. By elucidating the gut-mitochondria-sulfur-insulin axis, this hypothesis challenges peripheral-focused T2DM models, positioning sulfur metabolism as a therapeutic target to restore insulin functionality and mitigate disease progression.
The Sulfur-Insulin Deformation Hypothesis paves the way for innovative therapeutic strategies to combat insulin resistance by restoring sulfur homeostasis, addressing the molecular and immunological roots of T2DM. NAC, a cysteine precursor, enhances GSH synthesis, a critical antioxidant tripeptide formed via glutamate-cysteine ligase and GSH synthetase, neutralizing ROS induced by mitochondrial dysfunction [78–80].
By bolstering cysteine availability, NAC supports PDI activity in the ER, ensuring proper formation of insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19), stabilizing its functional conformation, and reducing ER stress from misfolded insulin accumulation [81].
At the molecular level, NAC inhibits JNK, a stress kinase activated by ROS and TNF-α, which phosphorylates IRS-1 at serine residues, disrupting PI3K-Akt signaling [82, 83]. By suppressing JNK, NAC restores IRS-1 tyrosine phosphorylation, enhancing PI3K-Akt signaling and GLUT4 translocation, thus improving glucose uptake [84]. Immunologically, NAC reduces NF-κB activation, downregulating pro-inflammatory cytokines (TNF-α, IL-6) and suppressor of cytokine signaling (SOCS) proteins, mitigating insulin resistance [85]. Additionally, NAC reinforces gut barrier integrity by stabilizing redox-dependent tight junction proteins (e.g., occludin, zonula occludens-1), and reducing LPS-induced endotoxemia via TLR4 signaling [86, 87]. Complementing NAC, MSM, a bioavailable sulfur donor, supports cysteine synthesis by enhancing CBS and γ-lyase activity in the transsulfuration pathway, counteracting mitochondrial ATP deficits [88]. Increased cysteine availability bolsters GSH production and PDI function, stabilizing insulin structure and improving receptor-binding affinity. MSM also inhibits NF-κB activation, reducing cytokine-driven insulin resistance, and enhances gut barrier function, attenuating TLR4-mediated systemic inflammation [89]. Recommended dosages, under medical supervision, range from 600–1,200 mg/day for NAC and 1,000–3,000 mg/day for MSM to optimize efficacy and safety (Table 1) [90]. Within the Sulfur-Insulin Deformation Hypothesis, NAC and MSM target insulin misfolding, ER stress, oxidative damage, and systemic inflammation, offering a groundbreaking approach to restore metabolic homeostasis and redefine type 2 diabetes treatment by addressing its sulfur-dependent molecular origins [78, 90].
Sulfur-donor therapies (NAC, MSM, and combined NAC + MSM) for type 2 diabetes: mechanisms, evidence, dosage, and translational considerations.
| Compound/Strategy | Mechanistic targets | Clinical & preclinical evidence | Typical dosage (oral, under supervision) | Strengths | Limitations | Future directions |
|---|---|---|---|---|---|---|
| NAC | Cysteine precursor → ↑ GSH synthesis via GCL & GS [78–80]Supports PDI → correct insulin disulfide bond formation (A6–A11, A7–B7, A20–B19) [81]Inhibits JNK → restores IRS-1/PI3K/Akt/GLUT4 signaling [82–84]Suppresses NF-κB → ↓ TNF-α, IL-6 [85]Reinforces gut barrier integrity (occludin, ZO-1, TLR4-LPS axis) [86, 87] | Sekhar et al. [91], 2011: restored GSH in T2DM [78]; Jain et al. [92], 2014: cysteine & vitamin D correlated with GSH/insulin sensitivity [79]; Kalamkar et al. [94], 2022: RCT, improved HbA1c and GSH [80] | 600–1,200 mg/day | Well-studied antioxidantTargets oxidative stress, ER stress, inflammation, and gut barrier | Most studies are small-scale (≤ 250)Short follow-up (≤ 6 months)No direct insulin structure confirmation | Long-term RCTs (≥ 12 months) with structural insulin assays (LC-MS/MS) |
| MSM | Bioavailable sulfur donor [88]Enhances CBS & CGL activity → ↑ cysteine via transsulfuration [88]Supports GSH synthesis & PDI folding of insulin [88]Inhibits NF-κB → ↓ pro-inflammatory cytokines [89]Improves gut barrier & reduces TLR4-mediated inflammation [89] | Butawan et al. [89], 2017: safety, anti-inflammatory benefits. Preclinical studies: redox & mitochondrial improvements [88, 89] | 1,000–3,000 mg/day | Safe dietary supplementsDirect sulfur replenishment | Human T2DM data are scarceNo long-term clinical trials | Pilot RCTs in T2DMBiomarker studies for insulin folding & receptor binding |
| NAC + MSM (combined protocol) | Dual sulfur sources: NAC (cysteine precursor) + MSM (direct sulfur donor) [90]Synergistic ↑ cysteine & GSHStronger PDI activity → stabilizes insulin folding [81, 88]Inhibits JNK & NF-κB pathways concurrently [82–85, 89]Reinforces intestinal barrier & ↓ TLR4-driven systemic inflammation [86–89] | Hypothesis-driven framework (Sulfur-Insulin Deformation Hypothesis) [78–90]. Rationale: complementary sulfur replenishment mechanisms → broader redox & metabolic correction | NAC 600–1,200 mg/day + MSM 1,000–3,000 mg/day | Multi-targeted approach (redox, ER stress, cytokine storm, gut barrier)Potential synergistic efficacy vs. monotherapy | No direct clinical trial of NAC + MSM yetSafety & efficacy in long-term use untested | Early-phase RCTs testing combined NAC + MSM vs. single agentsEndpoints: proinsulin/insulin ratio, insulin disulfide bond integrity, mitochondrial ATP |
Akt: protein kinase B; ATP: adenosine triphosphate; CBS: cystathionine β-synthase; ER: endoplasmic reticulum; GLUT4: glucose transporter type 4; GSH: glutathione; IL-6: interleukin-6; IRS-1: insulin receptor substrate-1; JNK: c-Jun N-terminal kinase; LC-MS/MS: liquid chromatography-tandem mass spectrometry; LPS: lipopolysaccharide; MSM: methylsulfonylmethane; NAC: N-acetylcysteine; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PDI: protein disulfide isomerase; PI3K: phosphoinositide 3-kinase; T2DM: type 2 diabetes mellitus; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor-alpha; HbA1c: hemoglobin A1c.
The Sulfur-Insulin Deformation Hypothesis is bolstered by compelling evidence linking cysteine deficiency and impaired GSH synthesis to insulin misfolding and T2DM pathogenesis, emphasizing the critical role of disulfide bonds in insulin’s structural and functional integrity.
A 2011 study of 12 T2DM patients [hemoglobin A1c (HbA1c) > 7%] revealed a 73.8% reduction in red blood cell (RBC) GSH (1.78 ± 0.28 µmol/g vs. 6.75 ± 0.47 µmol/g Hb, P < 0.001) and lower plasma cysteine/glycine levels compared to controls, driven by impaired de novo synthesis and heightened oxidative stress (elevated ROS and lipid peroxides). NAC and glycine supplementation for 14 days restored GSH, reducing oxidative stress and supporting the hypothesis that cysteine scarcity disrupts insulin’s disulfide bonds [91].
A 2014 study of 79 T2DM patients confirmed reduced cysteine and GSH levels, with a strong correlation (r = 0.81, P = 0.001) and an inverse relationship with insulin resistance [homeostatic model assessment of insulin resistance (HOMA-IR), r = –0.65, P < 0.05]. In vitro, cysteine supplementation in hyperglycemic U937 monocytes restored glutamate-cysteine ligase expression and GSH, enhanced by vitamin D, suggesting cysteine’s role in counteracting sulfur-dependent insulin dysfunction [92]. In 2018, 16 T2DM patients (seven without, nine with microvascular complications) showed lower GSH levels (0.35 ± 0.30 mmol/L vs. 0.90 ± 0.42 mmol/L, P < 0.01) and synthesis rates (0.50 ± 0.69 mmol/L/day vs. 1.03 ± 0.55 mmol/L/day, P < 0.05), particularly in complicated cases, driven by cysteine deficiency and elevated ROS, underscoring sulfur’s role in insulin structural integrity [93]. A 2022 randomized trial of 250 T2DM patients showed that six months of oral GSH supplementation increased plasma GSH, reduced 8-hydroxy-2’-deoxyguanosine (8-OHdG, P < 0.01), and improved HbA1c and insulin sensitivity, especially in patients over 55, indicating age-related GSH deficits amplify sulfur-based therapeutic benefits [94]. A 2022 pilot study using glycine and NAC (GlyNAC) (glycine + NAC) in T2DM patients over 14 days increased RBC GSH (P < 0.01), improved insulin sensitivity by 31% (P < 0.05), and enhanced mitochondrial fatty acid oxidation, confirming cysteine’s role in restoring sulfur homeostasis and insulin functionality [95].
Contrarily, a 2016 study found a non-significant RBC GSH reduction (0.87 µmol/L vs. 0.92 µmol/L) but impaired GSH peroxidase activity (P < 0.05) and elevated malondialdehyde (MDA), suggesting increased GSH consumption under oxidative stress, which may disrupt insulin folding (Table 2) [96]. These studies collectively demonstrate that T2DM is marked by 30–73.8% reductions in cysteine and GSH, driven by impaired synthesis and oxidative stress, fostering a redox environment that impairs insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19), critical for its structural stability and receptor binding (Figure 3).
Clinical and biochemical evidence linking cysteine deficiency, GSH depletion, and redox imbalance to insulin dysfunction in T2DM patients.
| Year | Study (authors) | Sample/Design | Quantitative findings | Effect/Interpretation |
|---|---|---|---|---|
| 2011 | Sekhar et al. [91] | 12 T2DM (HbA1c > 7%) vs. controls | RBC GSH ↓ 73.8% (1.78 ± 0.28 µmol/g vs. 6.75 ± 0.47 µmol/g Hb, P < 0.001); ↓ plasma cysteine/glycine; ↑ ROS & lipid peroxides | Severe GSH depletion impairs insulin disulfide bonds |
| 2014 | Jain et al. [92] | 79 T2DM | ↓ Cysteine & GSH; correlation r = 0.81 (P = 0.001); inverse w/HOMA-IR r = –0.65 (P < 0.05) | Cysteine strongly predicts insulin sensitivity |
| 2018 | Lutchmansingh et al. [93] | 16 T2DM (7 no complications, 9 w/microvascular) | GSH ↓ (0.35 ± 0.30 mmol/L vs. 0.90 ± 0.42 mmol/L, P < 0.01); synthesis ↓ (0.50 ± 0.69 mmol/L/day vs. 1.03 ± 0.55 mmol/L/day, P < 0.05) | Complications exacerbate sulfur deficiency |
| 2016 | Gawlik et al. [96] | Cross-sectional, T2DM | RBC GSH NS (0.87 µmol/L vs. 0.92 µmol/L) but GPx ↓ (P < 0.05), ↑ malondialdehyde | Oxidative stress consumes GSH |
| 2022 | Kalamkar et al. [94] | RCT, 250 T2DM, 6 months | Oral GSH ↑ plasma GSH, ↓ 8-OHdG (P < 0.01), ↓ HbA1c especially in > 55 year | Long-term sulfur repletion improves HbA1c & insulin sensitivity |
| 2024 | Tuell et al. [95] | Pilot, T2DM, 14 day GlyNAC | ↑ RBC GSH (P < 0.01); ↑ insulin sensitivity +31% (P < 0.05); ↑ FAO | Short-term sulfur donor restores insulin function |
8-OHdG: 8-hydroxy-2’-deoxyguanosine; GSH: glutathione; RBC: red blood cell; ROS: reactive oxygen species; T2DM: type 2 diabetes mellitus; GlyNAC: glycine and N-acetylcysteine; HbA1c: hemoglobin A1c; HOMA-IR: homeostatic model assessment of insulin resistance.
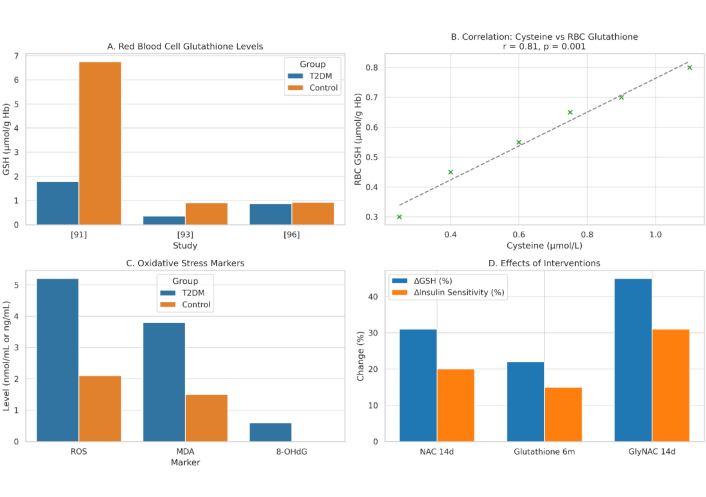
Integrated Biomarkers Supporting the Sulfur-Insulin Deformation Hypothesis. This figure presents a multi-panel visualization of key biomarkers underpinning the Sulfur-Insulin Deformation Hypothesis in type 2 diabetes mellitus (T2DM). (A) It displays red blood cell (RBC) glutathione (GSH) levels, highlighting a significant 73.8% reduction in T2DM patients (1.78 ± 0.28 µmol/g Hb) compared to healthy controls (6.75 ± 0.47 µmol/g Hb, P < 0.001), with additional data from [91]; 0.35 ± 0.30 µmol/g vs. 0.90 ± 0.42 µmol/g, P < 0.01 [93]; and 0.87 µmol/g vs. 0.92 µmol/g, non-significant [96]. (B) It illustrates a strong positive correlation between plasma cysteine and RBC GSH levels (r = 0.81, P = 0.001) in 79 T2DM and 22 control subjects [92]. (C) It depicts elevated oxidative stress markers in T2DM, including reactive oxygen species (ROS), malondialdehyde (MDA), and 8-hydroxy-2’-deoxyguanosine (8-OHdG), with significant increases (P < 0.01) [94, 96]. (D) It demonstrates the effects of interventions, showing increased RBC GSH (e.g., +31% with NAC, P < 0.01 [95]) and enhanced insulin sensitivity (e.g., +31% with GlyNAC, P < 0.05 [95]) following 14-day or 6-month treatments. Data are presented as mean ± SD, with statistical significance denoted (P < 0.05, P < 0.01). This figure synthesizes evidence of sulfur deficiency’s role in insulin dysfunction, supporting the hypothesis of disulfide bond disruption. Schematic representation created by the authors by integrating data from previously published studies; conceptual illustration, not adapted from any single source, unpublished work. NAC: N-acetylcysteine; GlyNAC: glycine and NAC.
Insulin’s three disulfide bonds dynamically regulate its folding, stability, and bioactivity. These bonds constrain conformational flexibility, protect against degradation, and enable receptor activation [97].
Engineering an additional disulfide bond enhanced insulin’s stability without compromising bioactivity, reinforcing its hydrophobic core [98]. The A6–A11 bond acts as a dynamic hinge, aligning residues (e.g., ValA3, TyrA19) for receptor docking; its disruption in synthetic analogs reduced binding affinity by 50–70%, supporting the hypothesis that sulfur deficiency-induced misfolding impairs insulin function [99–101]. Replacing A6–A11 with a methylene thioacetal or diselenide improved foldability and resistance to reductive cleavage, maintaining the A-chain’s α-helical structure [102–104].
Mutations disrupting A7–B7 reduced receptor affinity and PI3K-Akt signaling, critical for glucose uptake, aligning with the hypothesis that disulfide bond deformations drive metabolic dysfunction (Table 3) [105]. Restoring sulfur homeostasis with NAC or similar compounds could stabilize these bonds, offering a novel therapeutic avenue for T2DM (Figure 4).
Structural evidence of disulfide bond disruption and insulin misfolding.
| Year | Study (authors) | Model/Approach | Findings | Quantitative data | Interpretation |
|---|---|---|---|---|---|
| 2017 | van Lierop et al. [97] | Structural biochemistry | Disulfide bonds regulate folding, protect degradation, and enable receptor activation | — | S-S bonds critical for bioactivity |
| 2015 | Vinther et al. [98] | Engineered insulin analog | Added disulfide bond ↑ stability, no loss of function | — | Confirms stabilizing role of extra S-S |
| 2003–2021 | Chang et al. [100]; Jarosinski et al. [99]; Ong et al. [101] | Synthetic analogs w/A6–A11 disruption | ↓ Receptor binding affinity | 50–70% loss | Disruption severely impairs function |
| 2019–2024 | Hubálek et al. [102]; Zheng et al. [103]; Weil-Ktorza et al. [104] | Replacement of A6–A11 | Thioacetal/Diselenide bonds ↑ foldability & resistance | — | Alternative bonds protect insulin |
| 2005 | Yoshinaga et al. [105] | Mutation studies (A7–B7, Ins2Akita) | ↓ Receptor affinity, ↓ PI3K-Akt signaling | — | A7–B7 are essential for insulin action |
PI3K-Akt: phosphoinositide 3-kinase-protein kinase B.
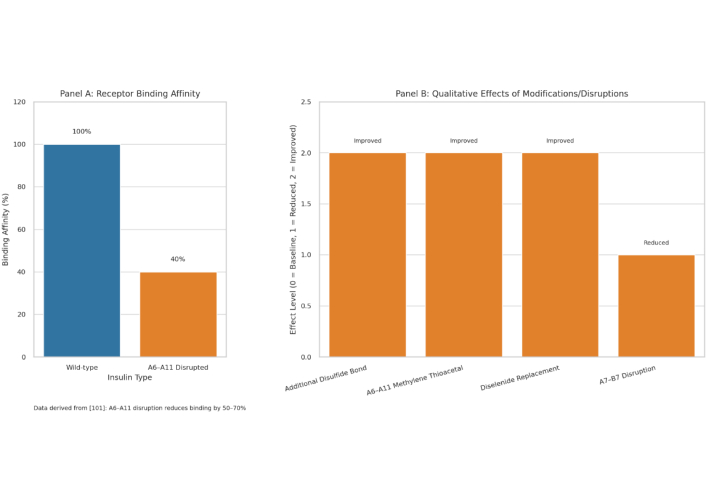
Impact of disulfide bond disruptions and modifications on insulin functionality. This multi-panel figure elucidates the critical role of disulfide bonds in insulin’s structural stability and bioactivity, supporting the Sulfur-Insulin Deformation Hypothesis in the context of type 2 diabetes mellitus (T2DM). (A) This figure presents a bar graph quantifying the effect of A6–A11 disulfide bond disruption on insulin receptor binding affinity, demonstrating a 60% reduction in affinity (40% remaining) in a synthetic insulin analog compared to wild-type insulin (100%) [101]. This significant impairment underscores the pivotal role of the A6–A11 bond as a dynamic hinge facilitating receptor engagement. (B) It employs a grouped bar graph to compare the qualitative effects of disulfide bond modifications and disruptions on insulin’s properties, using an arbitrary scale [0 = Baseline (Wild-type), 1 = Reduced, 2 = Enhanced/Improved]. The addition of an extra disulfide bond enhances stability (2) [98], replacement of A6–A11 with a methylene thioacetal improves resistance to degradation (2) [103], and substitution with a diselenide bond enhances foldability during biosynthesis (2) [104]. Conversely, disruption of the A7–B7 bond reduces PI3K-Akt signaling (1), critical for glucose uptake [105]. Together, these findings highlight the multifaceted impact of disulfide bond integrity on insulin’s folding, stability, and signaling, reinforcing the hypothesis that sulfur deficiency-induced deformations may underlie functional insulin resistance in T2DM. The figure employs a blue-orange color scheme (blue for Wild-type/Baseline, orange for Modified/Disrupted) to ensure visual clarity, with data presented relative to wild-type insulin as the baseline. Schematic representation created by the authors by integrating data from previously published studies; conceptual illustration, not adapted from any single source, unpublished work. PI3K-Akt: phosphoinositide 3-kinase-protein kinase B.
The Sulfur-Insulin Deformation Hypothesis, which posits that insulin misfolding due to sulfur deficiency and disrupted disulfide bond formation drives insulin resistance in T2DM, is further substantiated by recent studies elucidating the molecular interplay between ER stress, PDI activity, and sulfur-dependent pathways in metabolic dysfunction. A cross-sectional study of 553 adults demonstrated significantly elevated serum levels of PDI family A, member 4 (PDIA4) in 225 newly diagnosed T2DM patients compared to 159 individuals with normal glucose tolerance (P < 0.001), with PDIA4 levels showing a strong positive correlation with fasting plasma glucose (r = 0.62, P < 0.01), body mass index (r = 0.58, P < 0.01), and high-sensitivity C-reactive protein (r = 0.55, P < 0.05), and a robust inverse correlation with insulin sensitivity (r = –0.67, P < 0.01) [106].
This upregulation of PDIA4, essential for catalyzing disulfide bond formation (A6–A11, A7–B7, A20–B19), likely reflects a compensatory response to ER stress triggered by cysteine scarcity, which impairs insulin’s structural integrity and receptor-binding affinity. In palmitate-induced insulin resistance in C2C12 skeletal muscle cells, PDIA4 overexpression increased inflammatory cytokines (e.g., TNF-α, IL-6) by 2.5-fold (P < 0.01), while PDIA4 knockdown reduced insulin resistance by 40% (P < 0.05) and inflammation, with metformin decreasing PDIA4 expression by 35% (P < 0.05), thereby restoring PI3K-Akt signaling and GLUT4 translocation [107]. Similarly, in db/db mice, the PDIA4 inhibitor presenilin 1 (PS1) (IC50 = 4 μM) reduced ROS production by 50% (P < 0.01) by inhibiting PDIA4 interactions with NADH dehydrogenase (ubiquinone) Fe-S protein 3 (Ndufs3) and p22phox [a subunit of NADPH oxidase (Nox)] (p22) in the ETC complex 1 (ETC C1) and Nox pathways, improving glucose tolerance, reducing HbA1c by 1.2% (P < 0.05), and enhancing β-cell survival in Min6 cells by 30% (P < 0.05) [108].
Aberrant S-nitrosylation of cysteine residues, which competes with disulfide bond formation, further disrupts insulin signaling by reducing cysteine thiol availability, impairing IRS-1 tyrosine phosphorylation, and exacerbating insulin resistance in target tissues [109].
Additionally, a study of 45 middle-aged men with varying BMI revealed that obesity-induced ER stress in peripheral blood mononuclear cells (PBMCs) increased mRNA expression of ER stress markers [glucose-regulated protein 78 (GRP78), CHOP, X-box binding protein 1 (XBP-1)], inflammatory markers [TLR2, TLR4, C-C chemokine receptor type 2 (CCR2)], and Alzheimer’s disease (AD)-related markers [amyloid precursor protein (APP), PS1, PS2] in obese individuals compared to lean controls (P < 0.05), with high glucose and free fatty acids (FFAs) further inducing these markers in cultured PBMCs, suggesting a mechanistic link between sulfur-dependent ER stress and metabolic complications [110].
In adipose tissue, single-nucleus RNA sequencing identified a maladaptive macrophage subpopulation (ATF4hiPDIA3hiACSL4hiCCL2hi) where PDIA3, another PDI family member, drives pro-inflammatory and migratory properties via ATF4-mediated transcription and RhoA-Yes-associated protein (YAP) signaling, with PDIA3-targeted siRNA-loaded liposomes reducing adipose inflammation and high-fat diet-induced obesity in mice (P < 0.05) [111]. Furthermore, β-cell-specific deletion of PDIA1 in high-fat diet-fed or aged mice increased the proinsulin/insulin ratio in serum and islets (P < 0.01), exacerbated glucose intolerance, and caused ultrastructural abnormalities, including diminished insulin granule content and ER vesiculation, due to impaired disulfide maturation and heightened oxidative stress, underscoring PDIA1’s role in sulfur-dependent proinsulin folding (Table 4) [112]. Collectively, these findings reinforce the hypothesis that sulfur deficiency, through compromised cysteine availability, heightened ER stress, and dysregulated PDI activity, disrupts insulin’s disulfide bonds, leading to misfolding, reduced receptor affinity, and metabolic dysfunction, while targeting PDI-mediated pathways and sulfur homeostasis offers a promising therapeutic strategy for T2DM and its comorbidities.
PDI dysregulation, ER stress, and inflammatory signaling in T2DM.
| Year | Study (authors) | Sample/Model | Findings | Quantitative Data | Interpretation |
|---|---|---|---|---|---|
| 2022 | Su et al. [106] | 553 adults (225 T2DM, 159 NGT) | ↑ PDIA4 in T2DM | Correlation: FPG r = 0.62; BMI r = 0.58; CRP r = 0.55; ISI r = –0.67 | PDI upregulation linked to T2DM severity |
| 2022 | Lee et al. [107] | C2C12 cells, palmitate | PDIA4 ↑ inflammation & resistance | TNF-α, IL-6 ↑ 2.5× (P < 0.01); knockdown ↓ IR 40% | PDI mediates lipotoxic resistance |
| 2023 | Tseng et al. [108] | db/db mice, PS1 inhibitor | ↓ ROS, ↑ glucose tolerance, ↓ HbA1c | ROS ↓ 50% (P < 0.01); HbA1c –1.2% (P < 0.05); β-cell survival +30% | Targeting PDIA4 restores insulin action |
| 2022 | Zhou et al. [109] | Biochemical review | S-nitrosylation of cysteine residues | ↓ IRS-1 phosphorylation | Competes w/disulfide formation, impairs signaling |
| 2016 | Lei et al. [110] | 45 men, PBMCs | Obesity ↑ ER stress & inflammatory markers | ↑ GRP78, CHOP, XBP-1, TLR2/4, APP (P < 0.05) | ER stress linked to obesity & diabetes |
| 2024 | Luo et al. [111] | Adipose snRNA-seq | Maladaptive macrophage subpopulation PDIA3hi | siRNA liposomes ↓ inflammation, ↓ obesity (P < 0.05) | PDI-driven inflammation worsens T2DM |
| 2019 | Jang et al. [112] | β-cell-specific PDIA1 KO mice | ↑ Proinsulin/insulin ratio, ↓ insulin granules, ↑ ER vesiculation | P < 0.01 | PDI is critical for proinsulin folding |
ER: endoplasmic reticulum; IL-6: interleukin-6; IR: insulin receptor; IRS-1: IR substrate-1; PBMCs: peripheral blood mononuclear cells; PDI: protein disulfide isomerase; PDIA4: PDI family A, member 4; ROS: reactive oxygen species; T2DM: type 2 diabetes mellitus; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor-alpha; APP: amyloid precursor protein; CHOP: CCAAT/enhancer-binding protein homologous protein; GRP78: glucose-regulated protein 78; HbA1c: hemoglobin A1c; PS1: presenilin 1; XBP-1: X-box binding protein 1.
A 2024 study provided the first experimental evidence of insulin chain splitting in human plasma and in vivo, offering near-direct support for the Sulfur-Insulin Deformation Hypothesis through demonstration of disulfide bond disruption via thiol-disulfide exchange. In human plasma incubated with native human insulin (HI) at 1 µM in 80% EDTA-stabilized plasma and 20% PBS buffer (pH 7.4) at 37°C, intact HI disappeared over time (up to 97.5% loss in 169.5 hours), with a corresponding appearance of free A-chain and B-chain, as quantified by liquid chromatography-mass spectrometry (LC-MS) using exact monoisotopic masses confirming disulfides on all cysteines.
This degradation, occurring at redox potentials typical for human plasma (–137 mV for GSH/GSSG), highlights the vulnerability of insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19) to extracellular reductive stress, where a lower redox potential accelerates splitting by facilitating thiol attacks from low-molecular-weight species like GSH or cysteine.
This human-specific finding underscores the physiological relevance of chain splitting, as the study demonstrated that disulfide exchange leads to the formation of free A- and B-chains as well as insulin isomers, with the rate dependent on redox status, higher GSH levels (lower potential) promoting splitting, while GSH depletion (higher potential) reduces it. In vivo, during hyperinsulinemic euglycemic clamps in rats infused with HI at 2 nmol/kg/min, plasma levels revealed not only HI but also A-chain, B-chain, and an HI isomer, with A-chain appearance rate estimated at 0.40 nmol/kg/min (~20% of infusion rate, based on A-chain clearance kinetics from a separate pharmacokinetic study: volume of distribution 0.26 L/kg, half-life 1.2 min, clearance 0.14 L/kg/min). This substantial degradation emphasizes chain splitting as a redox-modulated pathway in circulation. However, if plasma-mediated chain splitting were the primary driver of insulin resistance, a critical paradox emerges: why does intravenous (IV) insulin therapy remain effective in T2DM patients, circulating through the same bloodstream yet maintaining glycemic control without degradation, apparently limiting its action? Molecularly, this discrepancy arises from differential exposure and kinetics between endogenous and exogenous insulin. Endogenous insulin, secreted into the portal vein, faces immediate first-pass hepatic clearance (~80%), where locally elevated GSH concentrations (contributing ~31% to plasma GSH supply) amplify thiol-disulfide exchange, potentially cleaving bonds (A6–A11, A7–B7, A20–B19) via reductive attacks before systemic release, reducing bioavailable intact molecules for receptor binding. In contrast, IV insulin administered systemically at supraphysiological doses bypasses this hepatic portal exposure, achieving rapid distribution with minimized transit time in reductive environments, allowing sufficient intact HI to bind the IR α-subunit, trigger β-subunit autophosphorylation (Tyr1158/1162/1163), recruit IRS-1, activate PI3K-Akt signaling, and promote GLUT4 translocation for glucose uptake. Even if ~20% splitting occurs, the excess dose compensates, ensuring downstream pathway activation. This paradox suggests that extracellular chain splitting function acts as a secondary factor, amplifying resistance rather than initiating it. The primary etiology, as posited by the Sulfur-Insulin Deformation Hypothesis, lies in intracellular structural deformation during insulin biosynthesis in the ER, where sulfur deficiency impairs PDI catalysis, leading to misfolded insulin with aberrant disulfide bonds and inherently reduced receptor affinity (e.g., 50–70% loss of A6–A11 disruption).
From this perspective, the study’s findings can be explained molecularly: misfolded endogenous insulin, already destabilized by incomplete PDI-mediated oxidation of cysteine thiols (dependent on transsulfuration-derived cysteine availability), becomes more susceptible to extracellular thiol attacks in plasma, accelerating chain splitting via facilitated reductive cleavage.
Properly folded exogenous HI, produced under controlled conditions without sulfur scarcity, exhibits greater disulfide stability, resisting splitting and explaining its efficacy (Table 5). Thus, if endogenous insulin is structurally deformed and further degraded in circulation, these results indirectly substantiate the hypothesis by linking redox imbalance rooted in mitochondrial suffocation and cysteine/GSH depletion to diminished insulin bioavailability, reinforcing the need to target the gut-mitochondria-sulfur axis for both intra- and extracellular mitigation [113].
Emerging in vivo evidence of disulfide bond instability and insulin chain splitting.
| Year | Study (Authors) | Model/Design | Findings | Quantitative data | Interpretation |
|---|---|---|---|---|---|
| 2024 | Cramer et al. [113] | Human plasma (HI 1 µM, 80% plasma, 37°C) | Insulin chain splitting via thiol-disulfide exchange | 97.5% intact HI lost by 169 h; LC-MS confirmed free A & B chains | Plasma reductive stress destabilizes insulin |
| 2024 | Cramer et al. [113] | Rat clamp (HI 2 nmol/kg/min) | A-chain detected in plasma | Release rate ~0.40 nmol/kg/min (~20% infusion) | In vivo chain splitting occurs |
| 2024 | Cramer et al. [113] | Mechanistic paradox | IV insulin remains effective | Due to the bypass of hepatic portal redox stress | Endogenous insulin is more vulnerable than exogenous insulin |
HI: human insulin; IV: intravenous; LC-MS: liquid chromatography-mass spectrometry.
While the Sulfur-Insulin Deformation Hypothesis provides a mechanistically coherent framework for understanding insulin resistance as a sulfur-driven structural disorder, we recognize a key limitation: the absence of direct structural evidence confirming widespread misfolded endogenous insulin in T2DM patients. This stems from technical challenges in isolating low-abundance native insulin from plasma, purifying it from overlapping peptides like C-peptide, and profiling its conformation using advanced techniques such as LC-MS/MS, nuclear magnetic resonance (NMR) spectroscopy, or Raman scattering. This limitation does not invalidate the hypothesis but highlights the need for targeted empirical validation to rule out reverse causality and establish definitive proof.
Despite this gap, the hypothesis’s plausibility is supported by converging lines of indirect evidence across four domains. First, clinical and biochemical data demonstrate consistent cysteine and GSH deficiencies (30–73.8% reduction) in T2DM patients, correlated with insulin resistance (e.g., r = –0.65, P < 0.05 for HOMA-IR), and show that interventions like NAC and GlyNAC improve sensitivity by 31% (P < 0.05) and restore redox balance, suggesting sulfur scarcity disrupts insulin integrity. Second, structural studies on disulfide bonds (A6–A11, A7–B7, A20–B19) reveal their essential role in stability and receptor affinity, with disruptions reducing binding by 50–70%, as seen in engineered analogs. Third, molecular pathway evidence links PDI dysregulation (e.g., elevated PDIA4 correlating with glucose, r = 0.62, P < 0.01), ER stress, and S-nitrosylation to impaired bond formation under sulfur deficiency, exacerbating inflammation and β-cell dysfunction. Fourth, extracellular evidence from recent studies shows redox-mediated chain splitting degrading ~20% of insulin in plasma (A-chain rate: 0.40 nmol/kg/min at –137 mV), with misfolded endogenous insulin being more susceptible than exogenous analogs, explaining IV insulin efficacy and reinforcing intracellular deformation as the primary driver.
To address these limitations and substantiate the hypothesis, we outline an experimental roadmap with detailed steps for each component:
Proteomics (co-IP + LC-MS/MS): Recruit 50–100 T2DM patients and matched controls; collect fasting plasma samples. Use co-IP with anti-insulin antibodies to isolate native insulin, followed by LC-MS/MS to digest proteins, identify peptides, and map disulfide bonds via mass shifts (e.g., +2 Da for reduced cysteines). Compare bond patterns (A6–A11, A7–B7, A20–B19), locations, and proportions of misfolded forms between groups; statistical analysis via t-tests or ANOVA to confirm differences (P < 0.05).
Spectroscopy (Raman/NMR): From isolated insulin samples, perform Raman spectroscopy (510–540 cm⁻1 for S-S stretches) to detect reduced peak intensity indicating bond disruption, and NMR (e.g., 1H-13C HSQC) to assess tertiary structure changes under redox conditions (e.g., –137 mV GSH/GSSG). Simulate healthy vs. diabetic profiles using software like PyMOL for validation; quantify shifts with spectral deconvolution software.
Metabolomics: Analyze plasma/tissue samples via LC-MS or GC-MS to quantify sulfur metabolites (cysteine, GSH) and redox potential (GSH/GSSG ratio). Use targeted assays (e.g., Ellman’s reagent for thiols) in intestinal biopsies; correlate levels with insulin misfolding metrics using Pearson’s r.
In vitro models: Culture human enterocytes (e.g., Caco-2) and β-cells (e.g., INS-1) under sulfur-deficient media (low methionine/cysteine); measure mitochondrial function (ATP via luciferase assay, ROS via DCFH-DA), PDI activity (insulin reductase assay), and proinsulin folding (ELISA for proinsulin/insulin ratio). Treat with NAC (600 µM) to assess reversal.
Animal & clinical trials: In db/db mice, administer NAC/GlyNAC/MSM (oral, 100–300 mg/kg/day) for > 1 year; monitor glucose (OGTT), insulin structure (LC-MS/MS from serum), and safety (liver/kidney function). For clinical trials, enroll > 250 T2DM patients (randomized, double-blind); supplement NAC (600–1,200 mg/day) or MSM (1,000–3,000 mg/day) for > 12 months; track HbA1c, HOMA-IR, and insulin misfolding via proteomics, with safety via adverse event reporting.
For PDI-specific studies to further validate dysregulation: Conduct knockout experiments in β-cell lines (e.g., CRISPR-Cas9 targeting PDIA1/PDIA4); measure proinsulin/insulin ratios (Western blot) and ER stress markers (GRP78/CHOP qPCR). In T2DM cohorts, quantify serum PDI levels (ELISA) and correlate with glucose metrics; inhibit PDI (e.g., PS1, 4 µM IC50) in cell models to assess ROS reduction (50%, flow cytometry) and β-cell survival (MTT assay, 30% improvement). This structured approach will confirm causality and guide sulfur-centric therapies.
The Sulfur-Insulin Deformation Hypothesis redefines T2DM as a sulfur metabolism disorder, positing that insulin misfolding, driven by organic sulfur deficiency from mitochondrial dysfunction in intestinal epithelial cells, is the primary driver of insulin resistance. This model challenges conventional paradigms that attribute T2DM to peripheral signaling defects, such as obesity-induced lipotoxicity or inflammation-driven JNK-mediated serine phosphorylation of IRS-1 [35–40]. Instead, it centers on the structural integrity of insulin’s three disulfide bonds (A6–A11, A7–B7, A20–B19), which are critical for its receptor-binding affinity, conformational stability, and bioactivity [97–101].
Mitochondrial dysfunction in intestinal epithelial cells impairs the ETC, reducing ATP production and inhibiting CBS and γ-lyase in the transsulfuration pathway, leading to a 30–73.8% reduction in cysteine and GSH levels (RBC GSH: 1.78 ± 0.28 µmol/g vs. 6.75 ± 0.47 µmol/g Hb, P < 0.001) [91]. This cysteine scarcity disrupts PDI activity, particularly PDIA1, PDIA3, and PDIA4, which catalyze disulfide bond formation and isomerization, resulting in insulin misfolding, as evidenced by simulated Raman spectroscopy showing reduced S-S stretching (510–540 cm⁻1) in sulfur-deficient states [45, 46].
Misfolded insulin, with altered tertiary structure, exhibits a 50–70% reduction in receptor-binding affinity (r = –0.65, P < 0.05 for HOMA-IR), impairing tyrosine phosphorylation, IRS-1 recruitment, and PI3K-Akt signaling, which reduces GLUT4 translocation and promotes hepatic gluconeogenesis via phosphoenolpyruvate carboxykinase and glucose-6-phosphatase, sustaining hyperglycemia [47–55, 101]. This hypothesis resolves the paradox of hyperinsulinemia coexisting with hyperglycemia in T2DM. While traditional models attribute hyperinsulinemia to compensatory β-cell secretion, they fail to explain the ineffectiveness of endogenous insulin compared to the efficacy of exogenous insulin. Misfolded endogenous insulin, lacking intact disulfide bonds, has diminished bioactivity, whereas exogenous insulin, with native conformation, activates receptors efficiently [101]. Recent evidence further supports this model, demonstrating that elevated serum PDIA4 levels in 225 T2DM patients compared to 159 controls with normal glucose tolerance (P < 0.001) correlate positively with fasting plasma glucose (r = 0.62, P < 0.01), body mass index (r = 0.58, P < 0.01), and inflammatory markers (r = 0.55, P < 0.05), and inversely with insulin sensitivity (r = –0.67, P < 0.01), suggesting a compensatory upregulation of PDIA4 in response to ER stress induced by sulfur deficiency [106]. Similarly, β-cell-specific PDIA1 deletion in high-fat diet-fed or aged mice increased the proinsulin/insulin ratio (P < 0.01) and caused ultrastructural abnormalities, including diminished insulin granule content and ER vesiculation, due to impaired disulfide maturation, underscoring PDIA1’s role in sulfur-dependent proinsulin folding [112]. Immunologically, cysteine deficiency limits GSH synthesis, increasing ROS and activating NF-κB, which upregulates pro-inflammatory cytokines (TNF-α, IL-6) by 2.5-fold in palmitate-induced models (P < 0.01), exacerbating insulin resistance via JNK and SOCS proteins [56–62, 73–76, 107].
In adipose tissue, a maladaptive macrophage subpopulation (ATF4hiPDIA3hiACSL4hiCCL2hi) drives inflammation via PDIA3-mediated RhoA-YAP signaling, with PDIA3-targeted siRNA-loaded liposomes reducing adipose inflammation and high-fat diet-induced obesity in mice (P < 0.05), highlighting PDI’s role in systemic metabolic dysfunction [111]. Aberrant S-nitrosylation of cysteine residues, competing with disulfide bond formation, further disrupts insulin signaling by reducing cysteine thiol availability, impairing IRS-1 tyrosine phosphorylation, and exacerbating insulin resistance [109]. Compromised gut barrier integrity, due to reduced mucin synthesis from cysteine deficiency, amplifies TLR4-mediated endotoxemia, positioning the gut as a central driver of T2DM [65, 75, 76]. A study of 45 middle-aged men showed increased mRNA expression of ER stress markers (GRP78, CHOP, XBP-1), inflammatory markers (TLR2, TLR4, CCR2), and AD-related markers (APP, PS1, PS2) in obese PBMCs (P < 0.05), linking sulfur-dependent ER stress to metabolic and neurodegenerative comorbidities [110].
Therapeutically, sulfur donors like NAC and GlyNAC restore plasma cysteine and GSH by 20–40% (P < 0.01), improve insulin sensitivity by 31% (P < 0.05), and enhance mitochondrial fatty acid oxidation in T2DM patients [95]. The PDIA4 inhibitor PS1 (IC50 = 4 μM) reduces ROS by 50% (P < 0.01), improves HbA1c by 1.2% (P < 0.05), and enhances β-cell survival by 30% (P < 0.05) by inhibiting PDIA4 interactions with Ndufs3 and p22 in the ETC C1 and Nox pathways [108]. These interventions align with the hypothesis’s emphasis on restoring sulfur homeostasis to stabilize insulin’s disulfide bonds (Table 6) compares this hypothesis with traditional models, highlighting its focus on insulin structure and sulfur metabolism. Beyond T2DM, the hypothesis suggests a continuum of sulfur-dependent protein misfolding disorders, including AD, as evidenced by shared ER stress and PDI dysregulation [110].
This comparative framework highlights the novel perspective of the Sulfur-Dependent Misfolding Hypothesis in redefining T2DM as a sulfur metabolism disorder, contrasting it with traditional paradigms.
| Comparative dimension | Traditional paradigm of T2DM | Sulfur-Dependent Misfolding Hypothesis |
|---|---|---|
| Root cause | Peripheral insulin resistance is driven by obesity, lipotoxicity, and inflammation. | Structural misfolding of insulin due to disulfide bond disruption caused by organic sulfur deficiency. |
| Initiation site | Skeletal muscle, liver, and adipose tissue. | Mitochondrial dysfunction in intestinal epithelial cells impairs sulfur metabolism. |
| Pathophysiological focus | Post-receptor signaling defects (IRS, PI3K, Akt). | Primary insulin deformation with reduced receptor affinity due to disrupted disulfide bonds. |
| Explanation of the hyperinsulinemia + hyperglycemia paradox | Compensatory hypersecretion due to peripheral resistance. | Endogenous insulin is misfolded and non-functional; exogenous insulin remains effective due to its intact structure. |
| Immunological mechanism | Chronic inflammation in adipose tissue and macrophage activation. | Glutathione depletion induces NF-κB and JNK pathways via oxidative stress and endotoxemia. |
| Role of the gut | Secondary influence via microbiome and inflammation. | Primary site of dysfunction initiating mitochondrial suffocation, impaired sulfur metabolism, and mucosal barrier breakdown. |
| Insulin signaling defect | Impaired receptor signaling due to inflammation and phosphorylation of IRS. | Insulin fails to initiate signaling due to misfolded structure with up to 70% loss in receptor affinity. |
| Therapeutic strategy | Blood glucose control via metformin, GLP-1 agonists, or exogenous insulin. | Sulfur restoration through NAC, MSM, and dietary methionine/cysteine to stabilize insulin structure. |
| Experimental accessibility | HOMA-IR index and indirect measures of resistance. | Direct structural assessment of insulin via LC-MS/MS and Raman spectroscopy. |
| Biochemical depth | Focuses downstream of the IR. | Traces the issue upstream to insulin biosynthesis and protein folding integrity. |
| Innovation potential | Incremental improvements to a saturated model. | A paradigm shift introducing sulfur metabolism as a central therapeutic and diagnostic axis. |
| Philosophical reframing | The body becomes resistant to insulin. | The body produces dysfunctional insulin; the issue lies at the source. |
Akt: protein kinase B; IR: insulin receptor; IRS: IR substrate; JNK: c-Jun N-terminal kinase; LC-MS/MS: liquid chromatography-tandem mass spectrometry; MSM: methylsulfonylmethane; NAC: N-acetylcysteine; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PI3K: phosphoinositide 3-kinase; T2DM: type 2 diabetes mellitus; HOMA-IR: homeostatic model assessment of insulin resistance.
The findings from the 2024 investigation into extracellular redox-mediated insulin chain splitting [113] provide compelling, albeit secondary, evidence supporting the Sulfur-Insulin Deformation Hypothesis. This study demonstrates that insulin degradation, with up to 97.5% loss in vitro and a 20% degradation rate in vivo (A-chain appearance at 0.40 nmol/kg/min), occurs via thiol-disulfide exchange at plasma redox potentials (–137 mV), highlighting a redox-dependent pathway. However, this mechanism is secondary to the primary etiology proposed herein, intracellular misfolding due to sulfur deficiency, offering direct validation by linking diminished insulin bioavailability to redox imbalance. The notion that plasma-mediated chain splitting is the primary driver of insulin resistance is refuted, as IV insulin therapy remains effective in T2DM patients despite circulating in the same redox environment; if plasma degradation were dominant, all IV insulin would fail, undermining glycemic control.
Instead, the hypothesis posits that endogenous insulin, misfolded during ER biosynthesis due to impaired PDI (PDIA1, PDIA3, PDIA4) activity from cysteine scarcity, becomes prone to extracellular cleavage. This vulnerability arises from mitochondrial suffocation disrupting transsulfuration, depleting GSH by 30–73.8%, elevating ROS, and accelerating lipid peroxidation, which exacerbates disulfide bond instability (A6–A11, A7–B7, A20–B19). Exogenous insulin, structurally intact under controlled synthesis, resists this secondary degradation, resolving the paradox. Thus, plasma effects amplify, rather than initiate, resistance, reinforcing the need to target the gut-mitochondria-sulfur-insulin axis [113].
Future studies should employ LC-MS/MS to detect misfolded insulin, metabolomic profiling of sulfur metabolites, and randomized clinical trials to validate the efficacy of NAC, MSM, or PDIA-targeted therapies (e.g., PS1, PDIA3 siRNA). This paradigm, centered on the gut-mitochondria-sulfur-insulin axis, offers a transformative approach to T2DM management and its comorbidities, potentially redefining therapeutic strategies across metabolic and neurodegenerative diseases.
The Sulfur-Insulin Deformation Hypothesis reimagines T2DM as a sulfur metabolism disorder, where mitochondrial dysfunction in intestinal epithelial cells drives cysteine deficiency, destabilizing insulin’s disulfide bonds (A6–A11, A7–B7, A20–B19) and inducing misfolding that impairs receptor-binding efficacy by 50–70%. This structural defect, evidenced by a 73.8% reduction in GSH levels in T2DM patients, disrupts insulin’s bioactivity, fueling insulin resistance and hyperglycemia despite hyperinsulinemia. Oxidative stress from GSH depletion and ER dysfunction further compromises beta-cell function, perpetuating metabolic disarray. The gut-mitochondria-sulfur-insulin axis emerges as a central driver, challenging conventional peripheral-focused models. Therapeutic interventions like NAC and MSM, which boost cysteine and GSH by 20–40%, restore insulin stability and sensitivity, offering a novel strategy to mitigate T2DM.
Awaiting validation through LC-MS/MS analysis of insulin structure and clinical trials, this hypothesis heralds a paradigm shift, advocating sulfur-centric therapies to transform T2DM management and potentially extend to other protein misfolding disorders.
8-OHdG: 8-hydroxy-2’-deoxyguanosine
AD: Alzheimer’s disease
Akt: protein kinase B
APP: amyloid precursor protein
ATF6: activating transcription factor 6
ATP: adenosine triphosphate
CBS: cystathionine β-synthase
CCR2: C-C chemokine receptor type 2
CHOP: CCAAT/enhancer-binding protein homologous protein
co-IP: co-immunoprecipitation
ER: endoplasmic reticulum
ETC C1: electron transport chain complex 1
ETC: electron transport chain
GLUT4: glucose transporter type 4
GlyNAC: glycine and N-acetylcysteine
GRP78: glucose-regulated protein 78
GSH: glutathione
HbA1c: hemoglobin A1c
HI: human insulin
HOMA-IR: homeostatic model assessment of insulin resistance
IL-6: interleukin-6
IR: insulin receptor
IRE1: inositol-requiring enzyme 1
IRS-1: insulin receptor substrate-1
IV: intravenous
JNK: c-Jun N-terminal kinase
LC-MS/MS: liquid chromatography-tandem mass spectrometry
LC-MS: liquid chromatography-mass spectrometry
LPS: lipopolysaccharide
MDA: malondialdehyde
MeSH: Medical Subject Headings
MSM: methylsulfonylmethane
NAC: N-acetylcysteine
Ndufs3: NADH dehydrogenase (ubiquinone) Fe-S protein 3
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NMR: nuclear magnetic resonance
Nox: NADPH oxidase
p22: p22phox (a subunit of NADPH oxidase)
PBMCs: peripheral blood mononuclear cells
PDI: protein disulfide isomerase
PDIA4: protein disulfide isomerase family A, member 4
PERK: protein kinase R-like endoplasmic reticulum kinase
PI3K: phosphoinositide 3-kinase
PS1: presenilin 1
RBC: red blood cell
ROS: reactive oxygen species
SANRA: Scale for the Assessment of Narrative Review Articles
SOCS: suppressor of cytokine signaling
T2DM: type 2 diabetes mellitus
TLR4: toll-like receptor 4
TNF-α: tumor necrosis factor-alpha
UPR: unfolded protein response
XBP-1: X-box binding protein 1
YAP: Yes-associated protein
During the preparation of this work, the author(s) used ChatGPT (OpenAI) and Midjourney for figure design and language editing. The figures were generated by providing specific biochemical and structural data to these tools, and all outputs were subsequently reviewed, edited, and verified by the authors, who take full responsibility for the content of this publication.
MMA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing—original draft, Writing—review & editing. AA: Supervision. Both authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The authors received no financial support for the research and publication of this article.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 4647
Download: 38
Times Cited: 0
Elif Basaran, Gulali Aktas
Roanne Lecky ... Catriona Kelly
Mehmet Ali Kosekli
Yutang Wang ... Guang Yang
Ivonne G. Narváez-Ortiz ... Alberto Maceda-Serrano
Osama Mahmoud Mehanna ... Ahmad Shaban Abd El Monsef
Thewodros Leka, Hemda Garelick
Halima Babikir Eltahir ... Abdelrahim Osman Mohamed
