 Review
Review

Affiliation:
Internal Medicine, Department of Gastroenterology-Hepatology, Faculty of Medicine, Abant Izzet Baysal University, Bolu 14030, Turkiye
Email: kosekli@gmail.com
ORCID: https://orcid.org/0000-0002-3172-8200
Explor Endocr Metab Dis. 2025;2:101446 DOI: https://doi.org/10.37349/eemd.2025.101446
Received: June 11, 2025 Accepted: September 22, 2025 Published: November 10, 2025
Academic Editor: Geltrude Mingrone, Catholic University, Italy, King’s College London, United Kingdom
The article belongs to the special issue Current Views on Pathogenesis, Diagnosis and Management of Type 2 Diabetes Mellitus and Its Complications and Related Conditions
Fatty liver, defined as lipid accumulation in more than 5% of hepatocytes, has become an increasingly important contributor to liver cirrhosis, particularly as viral hepatitis is being brought under control through vaccination and antiviral therapies. Abdominal obesity and insulin resistance play a central role in its pathogenesis. The global burden of metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD), continues to rise in parallel with the increasing prevalence of obesity and type 2 diabetes mellitus (T2DM). While meta-analyses indicate that the overall worldwide prevalence of MASLD is approximately 30%, higher rates have been reported in regions such as Latin America and North America. Among individuals with T2DM, the prevalence may reach up to 65%, and MASLD in these patients frequently progresses to metabolic dysfunction-associated steatohepatitis (MASH) and advanced fibrosis. Shared pathogenic mechanisms—most notably insulin resistance and chronic low-grade inflammation—drive disease progression, contributing to increased morbidity and mortality. The bidirectional relationship is further underscored by the observation that MASLD itself predisposes to the development of T2DM. In addition, the coexistence of MASLD and T2DM may exert a synergistic effect on cardiovascular disease (CVD) risk, and emerging evidence suggests that MASLD may represent an independent risk factor for CVD. Consequently, individuals with both MASLD and T2DM should be recognized as a particularly high-risk population requiring comprehensive monitoring of both hepatic and cardiovascular health. In this review, we aim to provide a concise overview of the etiopathogenesis, diagnostic approaches, and therapeutic strategies related to the MASLD-T2DM interface, a global health challenge that can be regarded as a pandemic of modern times.
The accumulation of fat in more than 5% of hepatocytes is defined as fatty liver. Abdominal obesity and insulin resistance are thought to play a central role. Fatty liver disease is increasingly playing a role in the etiology of hepatic cirrhosis. While the global rise in obesity and lifestyle changes contribute to the pathogenic cascade that facilitates hepatosteatosis, the partial control of viral hepatitis, which has been a leading cause of cirrhosis, through vaccination and antiviral treatment brings hepatosteatosis to the forefront [1]. In the coming years, hepatosteatosis appears likely to rank higher among the etiologies of cirrhosis. It is known that hepatosteatosis is more frequently observed in individuals with diabetes mellitus compared to the general population. This brief review will address the relationship between hepatosteatosis and diabetes mellitus, its etiopathogenesis, diagnosis, and treatment.
It is well-known that the global prevalence of nonalcoholic fatty liver disease (NAFLD) is increasing. In a meta-analysis analyzing 479 studies involving approximately 78 million participants, NAFLD was found to be a global health issue, despite showing regional differences in prevalence. Reported prevalence rates were 30.9% in Asia, 30.2% in Europe, 29% in North America, and 34% in South America, with a global prevalence reported as 30.2% [2].
An analysis of data from 92 studies involving over 9 million individuals found that the global prevalence of NAFLD increased from 25.3% during the 1990–2006 period to 38.2% in 2016–2019, marking a 50.4% rise over nearly three decades. In this study, the highest prevalence was observed in Latin America, Middle East, and North Africa [3]. Another meta-analysis, including 578 studies, reported the global prevalence of NAFLD as 29.4%. The study highlighted an increase in prevalence from 27.9% during the 2000–2010 period to 31.6% in the 2011–2021 period. The highest prevalence rates were found in North America (40.3%) and Europe (32.2%). Additionally, the incidence rate of NAFLD was estimated at 46.24 cases per 1,000 person-years [4]. Metabolic dysfunction-associated steatotic liver disease (MASLD) is responsible for 26% of all liver cirrhosis cases [5].
NAFLD, also known as MASLD, and type 2 diabetes mellitus (T2DM) are strongly associated, and both conditions increase the risk of cardiovascular disease (CVD). Recent findings suggest that MASLD may be an independent risk factor for CVD. Due to the strong relationship between MASLD and T2DM, it is difficult to evaluate the independent cardiovascular effects of each condition. However, patients with both T2DM and MASLD have a higher risk of CVD compared to those with T2DM alone, indicating a synergistic increase in risk supported by shared pathophysiological mechanisms. Some antidiabetic treatments have been shown to exert beneficial effects on both MASLD and CVD. Patients with both T2DM and MASLD should be considered a high-risk group for CVD and may benefit from more intensive cardiovascular protection strategies. Long-term follow-up studies are needed to determine whether treatment of MASLD can effectively reduce cardiovascular risk [6].
The prevalence of MASLD is high among individuals with T2DM. MASLD is the most common cause of chronic liver disease, and T2DM is a significant risk factor. Insulin resistance and chronic inflammation are the main mechanisms through which T2DM contributes to the development of MASLD. The coexistence of MASLD and T2DM further increases the risks of morbidity and mortality.
In an analysis of 156 studies involving 1,832,125 patients, the prevalence of NAFLD among individuals with T2DM was found to be 65.04% (95% CI: 61.79–68.15%, I2 = 99.90%), while the prevalence of nonalcoholic steatohepatitis (NASH) was 31.55% (95% CI: 17.12–50.70%, I2 = 97.70%). Among individuals diagnosed with both T2DM and NAFLD, 35.54% (95% CI: 19.56–55.56%, I2 = 100.00%) had clinically significant fibrosis (F2–F4), and 14.95% (95% CI: 11.03–19.95%, I2 = 99.00%) had advanced fibrosis (F3–F4) [7].
Hepatosteatosis occurs as a result of the accumulation of fat exceeding 5% in hepatocytes, due to the combined influence of factors such as insulin resistance, genetic predisposition, environmental factors, and excessive caloric intake. The increased fat content in the liver results from enhanced hepatic uptake, oxidation of fatty acids (FAs), very low-density lipoprotein (VLDL) export, and de novo lipogenesis (DNL), ultimately leading to excessive fat accumulation in hepatocytes. Common pathways play a role in the development of both hepatosteatosis and diabetes mellitus. While some individuals with hepatosteatosis go on to develop diabetes, fatty liver is present in approximately 70% of the diabetic population. Liver steatosis is present in 95% of the diabetic subjects with a body mass index of 35 kg/m2 or higher [8, 9].
Hepatic fat accumulation occurs when the balance between incoming FAs and their metabolism in the liver is disrupted. These FAs originate from dietary intake, DNL, and lipolysis of adipose tissue. In the early stages of MASLD, increased hepatic VLDL secretion and β-oxidation attempt to compensate for the excess influx of FAs. However, when β-oxidation becomes impaired and triglyceride (TG) removal via VLDL becomes insufficient, TG begins to accumulate in hepatocytes. The composition of dietary FAs also influences this process and can exacerbate hepatic fat accumulation.
As a result of insulin resistance, hyperinsulinemia develops, which further raises the fatty acyl-CoA pool in the liver [10]. Fructose is a substrate for both lipogenesis and gluconeogenesis. It is degraded to acetate in the gut and transported to the liver by portal circulation [11]. Fructose has a higher lipogenic potential compared to glucose due to this dual mechanism [12]. In hyperglycemia, SREBP1c stimulates DNL in the liver through carbohydrate-responsive element-binding protein (ChREBP) [13].
There is a bidirectional relationship between liver steatosis and T2DM. Insulin resistance causes progression in MASLD, while MASLD deteriorates insulin resistance further [14]. When insulin resistance develops in muscle, adipose tissue, and the liver, the pancreas compensates by increasing insulin secretion. During the period before the onset of T2DM, hyperinsulinemia, caused by excessive insulin secretion from pancreatic beta cells and reduced hepatic insulin clearance, may trigger the development of T2DM [15]. DNL is activated by insulin resistance through the SREBP1C pathway and by hyperglycemia through the ChREBP pathway [16].
Excess saturated fats consumed through the diet accumulate in hepatocytes. The increased fat content in hepatocytes triggers insulin resistance. The oxidation of free FAs leads to mitochondrial dysfunction, a decrease in adiponectin levels, and an elevation in pro-inflammatory markers [17].
Endoplasmic reticulum (ER) stress is a key factor in the development of insulin resistance. Hyperglycemia and excessive lipid accumulation lead to a high production rate of reactive oxygen species (ROS), which initiate ER stress. ER stress promotes the progression of fatty liver disease by activating NF-κB and Kupffer cells, thereby increasing the release of pro-inflammatory mediators [18]. NF-κB is a mediator that triggers insulin resistance. It influences the expression of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6). TNF-α activates the JNK pathway, and IL-6 stimulates the STAT3 pathway—both of which interfere with insulin signaling [19]. ER stress triggers an inflammatory response that enhances the production of pro-inflammatory cytokines, leads to immunogenic cell death, and disturbs metabolic balance and immune tolerance. These processes contribute to hepatocyte necrosis, steatosis, and the development of fibrosis [20].
Hepatic insulin resistance is a key component of systemic insulin resistance and is also associated with decreased insulin sensitivity in skeletal muscle and adipose tissue. Hepatic insulin resistance linked to NAFLD is triggered by the accumulation of hepatic diacylglycerol (DAG) and activation of protein kinase C epsilon (PKCε). DAG activates PKCε near the cell membrane, inhibiting insulin receptor kinase, which impairs the phosphorylation of IRS-1/2 and PI3K, weakening insulin signaling. As a result, glycogen synthesis in the liver decreases, while gluconeogenesis increases through FOXO1 activation, leading to excessive glucose release via GLUT2 [21].
Does hyperinsulinemia develop as a result of insulin resistance, or does the presence of hyperinsulinemia lead to insulin resistance? The traditional view is that insulin resistance occurs first. However, studies in T2DM suggest that the initial abnormality may be hyperinsulinemia, caused by excessive insulin secretion from beta cells and/or reduced hepatic insulin clearance, with insulin resistance developing subsequently [15]. It has been reported that insulin resistance may not always be associated with a high body mass index (BMI) and that T2DM may result more from impaired insulin secretion rather than insulin resistance [22].
There are studies suggesting that NAFLD develops before T2DM and that this condition is associated with insulin resistance. In a study where NAFLD was detected in 43% of patients with type 1 diabetes mellitus (T1DM), it was shown that these patients required higher doses of insulin due to weight gain linked to unhealthy lifestyle habits and increased insulin resistance. It has also been demonstrated that T1DM patients with NAFLD exhibit insulin resistance, being less sensitive to insulin, and have a higher visceral adiposity index [23].
In an interesting study examining the role of insulin resistance in liver fat accumulation and diabetes development, insulin secretion was found to increase while insulin sensitivity decreased in lean individuals with normal glucose tolerance, obese individuals, and obese individuals with fatty liver. Hepatic insulin extraction progressively increased across the lean, obese, and obese + fatty liver groups. Total hepatic insulin extraction remained stable even at high insulin uptake rates, while a linear correlation was observed between systemic insulin levels and total extrahepatic insulin extraction. This suggests that the elevated plasma insulin concentrations seen after oral glucose intake in overweight and obese individuals with fatty liver, compared to lean individuals with normal glucose tolerance, are primarily due to increased insulin secretion, rather than reduced insulin clearance by the liver or other tissues. Additionally, in both the obese group with normal glucose tolerance and the obese + NAFLD group, post-glucose load hyperinsulinemia was associated with increased insulin secretion. No decrease was detected in extrahepatic insulin extraction in these groups.
In summary, total hepatic insulin clearance remained stable at high insulin uptake rates, and a linear correlation existed between systemic insulin levels and total extrahepatic insulin clearance. Therefore, the increased plasma insulin concentrations following oral glucose intake in overweight and obese individuals with MASLD, compared to lean normal glucose-tolerant individuals, stem from enhanced insulin secretion rather than reduced insulin clearance by hepatic or extrahepatic tissues [24].
It has been demonstrated that patients with MASLD exhibit insulin resistance in adipose tissue, liver, and muscle. Insulin sensitivity in the muscle and liver was similarly impaired in MASLD patients, regardless of whether they had pre-diabetes or T2DM. Only insulin resistance in adipose tissue worsened further in T2DM, and this condition showed a correlation with the severity of insulin resistance in the muscle and liver, as well as with steatosis detected by magnetic resonance spectroscopy (MRS). In light of these findings, it has been concluded that insulin resistance in adipose tissue plays a significant role in the severity of MASLD in patients with T2DM. Lipotoxicity may impair beta-cell function in individuals predisposed to T2DM. Reducing metabolic stress may improve functional damage to beta cells [25].
It has been shown that fetuin-A, a glycoprotein secreted by the liver, placenta, and tongue, can induce insulin resistance by inhibiting the tyrosine kinase activity of the insulin receptor. Fetuin-A, which is secreted in excess by a fatty liver, has also been shown to inhibit glucose-stimulated insulin secretion (GSIS) from pancreatic islet cells [26].
Ceramides also play a role in the development of insulin resistance; inhibition of ceramide synthesis in NAFLD rat models has been shown to reduce hepatic steatosis and fibrosis. While ceramide-rich hepatic lipids are observed in metabolic NAFLD, this is not prominent in PNPLA3-related NAFLD. However, some NAFLD models may not exhibit insulin resistance despite increases in DAG and ceramides, suggesting that the effect of PKCε on glucose homeostasis may not be directly hepatic [27]. Intrahepatic fat accumulation leads to the buildup of ceramides and diacylglycerols in the liver, which inhibit insulin signaling and contribute to hepatic insulin resistance [28]. As a result, glycemic control becomes more difficult in individuals with both NAFLD and T2DM. Therefore, patients with T2DM who also have NAFLD require more intensive antidiabetic treatment compared to diabetic patients without NAFLD [29]. Hyperglycemia can facilitate the adhesion of macrophages and monocytes to the endothelium, promote vascular smooth muscle dysfunction, and activate pro-inflammatory macrophages, thereby contributing to the development of coronary artery disease [30].
AMPK increases glucose uptake, FA oxidation, and autophagy. It also inhibits protein and cholesterol synthesis. AMPK activation inhibits lipogenesis and increases insulin sensitivity [31].
The genetic basis of hepatic steatosis is closely associated with insulin resistance and hereditary susceptibility to MASLD [32]. Genome-wide association studies and genetic analyses have identified several variants that contribute to phenotypic heterogeneity in both disease development and progression risk. Among these, PNPLA3, TM6SF2, GCKR, MBOAT7, and HSD17B13 are considered the most relevant. The PNPLA3 I148M variant regulates TG mobilization from lipid droplets and thereby increases hepatic fat accumulation, while the TM6SF2 E167K variant appears to promote steatosis by impairing VLDL secretion. MBOAT7, which is involved in the remodeling of phosphatidylinositol FA chains, plays a critical role in hepatic lipid metabolism, and GCKR promotes hepatic fat accumulation by enhancing DNL. Furthermore, HSD17B13 has been implicated in the regulation of biologically active lipids and estrogen-related signaling pathways. Importantly, the PNPLA3 148Met and TM6SF2 167Lys variants have been strongly associated with liver fat accumulation, progression to NASH, cirrhosis, and hepatocellular carcinoma (HCC), although they do not appear to be directly related to metabolic syndrome (MS) or insulin resistance. Collectively, these variants represent important genetic determinants that may serve as predictors of disease progression in specific subgroups of patients with MASLD [33–35].
Chylomicrons and dietary intake contribute directly to liver fat accumulation. Dietary FAs are absorbed in the small intestine and released into the bloodstream in the form of chylomicrons. While most chylomicrons are stored in adipose tissue, a portion is taken up by the liver. Especially in the postprandial state, a significant share of the FAs entering the liver comes from chylomicrons and their remnants. Furthermore, the type of dietary FAs plays a role in promoting hepatic steatosis; for instance, saturated FAs and simple sugars can enhance DNL, intensifying fat accumulation in the liver.
The increase in liver fat occurs due to the disruption of the balance between FAs, lipolysis, and TGs. To reduce the influx of FAs into hepatocytes, VLDL secretion and beta-oxidation rates increase.
IL-1, IL-6, TNF-α, C-reactive protein, fibrinogen, and fetuin—these cytokines and acute-phase reactants—are elevated in individuals with NAFLD and T2DM. Therefore, a state of low-grade inflammation exists in both conditions, which contributes to an atherogenic environment.
Hepatokines like fetuin-A, fetuin-B, retinol-binding protein 4 (RBP4), and selenoprotein P act as mediators involved in the development of NAFLD and insulin resistance [10].
Adiponectin levels were found to be low in NAFLD patients, suggesting that this condition facilitates the disruption of FA metabolism and hepatocyte inflammation [36].
Incretins, which affect the enteropancreatic axis, may play a role in the relationship between diabetes and NAFLD. Healthy individuals have shown a higher incretin effect (55%) compared to non-diabetic NAFLD patients (39%), diabetic NAFLD patients (20%), and diabetic patients without fatty liver (2%) [37].
Evidence indicates strong associations between the gut microbiota, insulin sensitivity, and factors such as lipopolysaccharides (LPS), short-chain FAs (SCFAs), and bile acids [38]. The link between gut microbiota and the progression of NAFLD to T2DM remains poorly understood. Research has shown that individuals with NAFLD exhibit a distinct gut microbiota composition compared to those with both NAFLD and T2DM, as well as to patients with only NAFLD or only T2DM. These microbial differences may play a role in disease development. One study reported that Bacteroidetes and Firmicutes were significantly reduced in NAFLD patients with T2DM, whereas Proteobacteria and Actinobacteria levels were elevated. Additionally, no significant difference was found between patients in the NAFLD and T2DM groups. Moreover, the abundance of Firmicutes was lower in both the NAFLD and T2DM groups compared to healthy individuals [39].
It has been found that changes in the gut microbiota in NAFLD accurately reflect the presence of liver fibrosis and cirrhosis [40]. Common alterations in the gut microbiota are also observed in NAFLD, T2DM, and obesity [41]. Changes in the microbiota alter intestinal permeability, stimulating proinflammatory cytokines such as SCFAs and trimethylamine N-oxide, which in turn trigger ER stimuli [42, 43].
In fatty liver, TG-rich lipoproteins increase, HDL-C levels decrease, and small LDL particles increase. Fat accumulation in the liver and insulin resistance increase VLDL production, and this increase increases cholesterol ester transfer protein (CETP) activity [44, 45].
Plasminogen activator inhibitor type 1-PA-I, an inhibitor of the fibrinolytic pathway, is produced in the liver. PA-I levels increase in fatty liver [46]. In T2DM, procoagulant factors and platelet activation increase, while endogenous anticoagulant levels decrease. Therefore, fatty liver and T2DM pose a risk for thrombosis [47]. Table 1 shows etiological causes of MASLD.
Main factors responsible for the pathogenesis of steatotic liver.
| Factor | Description/Mechanism |
|---|---|
| Fat accumulation | > 5% fat in hepatocytes; due to an imbalance between input (diet, lipolysis, DNL) and output (VLDL secretion, β-oxidation) |
| Insulin resistance | Central to MASLD, it causes and worsens hepatic fat accumulation and glucose metabolism impairment |
| Hyperinsulinemia | Worsens MASLD by impairing glucose regulation and promoting fat storage |
| Genetic factors | Variants like PNPLA3 and TM6SF2 influence susceptibility and disease progression |
| Diet & environment | Excess caloric intake and environmental contributors promote steatosis |
| MASLD & T2DM coexistence | Linked with systemic inflammation, ER stress, mitochondrial dysfunction, and cytokine-mediated insulin signaling disruption |
| Gut microbiota | Alterations increase intestinal permeability and inflammation, contributing to MASLD and T2DM |
| Hepatokines | Low adiponectin and high fetuin-A levels contribute to insulin resistance |
DNL: de novo lipogenesis; VLDL: very low-density lipoprotein; MASLD: metabolic dysfunction-associated steatotic liver disease; ER: endoplasmic reticulum; T2DM: Type 2 diabetes mellitus.
In the diagnosis of NAFLD, both invasive and non-invasive methods are used. The invasive method is liver biopsy, which is generally the last choice for both patients and physicians due to its invasiveness. However, this does not prevent it from remaining the gold standard in diagnosis. A true-cut biopsy provides detailed information about liver tissue and is a unique method for evaluating fat accumulation in hepatocytes, hepatocyte inflammation, and liver fibrosis.
There is no single imaging method with high specificity for diagnosing hepatic steatosis. The diagnostic methods used vary in sensitivity, specificity, and have different advantages and disadvantages. Numerous guidelines advocate for screening diabetic patients for fatty liver disease, and vice versa, investigating the presence of fatty liver in individuals with diabetes. Likewise, the presence of either condition warrants an evaluation of other cardiometabolic risk factors.
Transabdominal ultrasonography (TAUS) is a sensitive method for diagnosing liver steatosis. It is widely available, relatively inexpensive, and easily repeatable—making it advantageous. Most asymptomatic cases are often first diagnosed using TAUS. In a meta-analysis of 49 studies, TAUS demonstrated a sensitivity of 84.8% and a specificity of 93.6% (AUROC 0.93) for detecting moderate-to-severe hepatic steatosis [48]. However, its accuracy in detecting low-stage hepatosteatosis drops to 20%. In the presence of obesity, sensitivity drops to 49% and specificity drops to 75% [49]. Variability between different operators or even within the same operator performing evaluations at different times is another weakness of this method.
Significant progress has been made in detecting hepatic steatosis using measurement techniques based on the speed of sound waves passing through liver tissue. The controlled attenuation parameter (CAP), provided by the vibration-controlled transient elastography (VCTE) platform, is currently the most widely used method. In a meta-analysis including 2,735 patients, the optimal cutoff values for CAP were determined as 248 dB/m for steatosis grade S0 and above, 268 dB/m for S1 and above, and 280 dB/m for S2 and above, with AUROC values of 0.823, 0.865, and 0.882, respectively [50].
There are some limitations to CAP measurement, such as significant overlap in CAP values across steatosis degrees and the existence of heterogeneous optimal threshold values across studies. Additionally, further studies are needed to assess its performance in obese individuals [51].
MRS has the ability to detect hepatic steatosis as low as 2% by directly measuring the chemical composition of liver fat [52]. In a meta-analysis of 46 studies, MRS had an average sensitivity of 73–89% and a specificity of 92–96%, outperforming ultrasound (US) and CT [53]. However, its widespread use in clinical practice is limited due to reasons such as limited accessibility, complexity in use, and interpretation [54].
MRI proton density fat fraction (PDFF) is another method with high accuracy in showing liver steatosis and the degree of steatosis [55]. The performance of this method is affected by diabetes and obesity [56].
Transient elastography and magnetic resonance elastography (MRE) are methods used to assess liver fibrosis. Due to fibrosis, the collagen content in liver tissue increases, making the liver stiffer compared to normal tissue, and it has become possible to measure this stiffness. Transient elastography (FibroScan) measures liver parenchymal stiffness by assessing the speed of sound waves traveling through the tissue, known as liver stiffness measurement (LSM). It is used to evaluate liver fibrosis and has been widely applied in chronic viral hepatitis. More recently, it has also gained acceptance for assessing hepatic fibrosis in MASLD [57]. A healthy liver is generally considered to have a liver stiffness value of less than 5.5 kPa [58]. It is more sensitive in detecting stage 3 and stage 4 (advanced) fibrosis [59]. Because it has a high negative predictive value, its success in excluding advanced-stage fibrosis is greater than its ability to definitively detect fibrosis [60].
It has been reported that a combination of blood biochemistry and anthropometric measurements can lead to more accurate results in the diagnosis of MASLD and hepatic fibrosis [61]. In this context, the most commonly used parameters are as follows:
where age is in years, ALT and AST are in U/L, and platelet count is in 109/L [61].
NFS (NAFLD fibrosis score) = –1.675 + 0.037 × age + 0.094 × BMI + 1.13 × impaired fasting glucose (yes = 1, no = 0) + 0.99 × AST/ALT – 0.013 × platelet count – 0.66 × albumin
where age in years, BMI in kg/m2, AST and ALT in U/L, platelet count in 109/L and albumin in g/dL [63].
FIB-4 is a commonly used and simple scoring system. However, its sensitivity is low in individuals with T2DM, in the elderly, and in detecting moderate levels of fibrosis [64]. FIB-4 and NAFLD fibrosis score (NFS) have moderate accuracy in predicting fibrosis at stage > F3 (AUROC ~0.77 for FIB-4 and ~0.75 for NFS). However, their sensitivity is low in individuals under the age of 35 [65].
Tests based on collagen components can also provide additional evidence regarding liver fibrosis.
Enhanced liver fibrosis (ELF) is derived from three markers related to extracellular collagen metabolism [66]. ELF = 2.278 + 0.851 × ln[hyaluronic acid (HA)] + 0.751 × ln[amino-terminal propeptide of type III procollagen (PIIINP)] + 0.394 × ln[tissue inhibitor of metalloproteinase 1 (TIMP 1)] as measured on the ADVIA Centaur XP/XPT system, Atellica IM Analyzer, and Atellica CI Analyzer.
A meta-analysis demonstrated that the ELF test showed particularly good performance in detecting significant and advanced fibrosis, or cirrhosis [67].
ADAPT, calculated using age, presence of diabetes, Pro-C3 levels, and platelet count, is a model that has recently shown relatively high performance in identifying advanced fibrosis in MASLD within tertiary hepatology care settings, as well as in excluding advanced fibrosis in low-risk populations [68, 69].
In recent years, combined scores that utilize blood tests and imaging modalities (such as elastography and assessment of steatosis) have been proposed and tested for the diagnosis of fibrosis. The diagnostic performance of these combined scores is still in the research phase [61].
In subjects with SLD or metabolic risk factors, fibrosis assessment using non-invasive methods is recommended, along with the following strategy (Figure 1) [61].
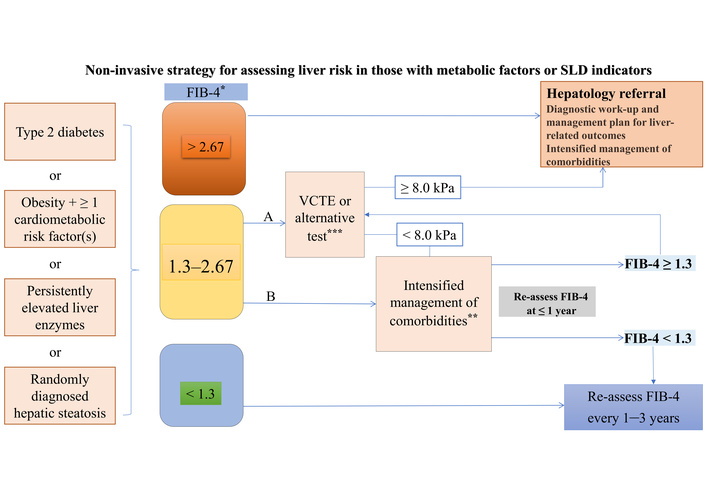
Fibrosis assessment using non-invasive methods in individuals with SLD or metabolic risk factors. Individuals with (1) T2DM or (2) abdominal obesity and ≥ 1 additional cardiometabolic risk factor(s) or (3) persistently elevated liver enzymes or (4) randomly diagnosed hepatic steatosis should undergo a multi-step diagnostic process, as indicated in the figure, to identify individuals with MASLD and advanced fibrosis. The algorithm can also be applied in the case of incident finding of steatosis. This strategy is intended to identify individuals at risk of developing liver-related outcomes. *: FIB-4 thresholds valid for age ≤ 65 years (for age > 65 years: lower FIB-4 cut-off is 2.0); **: e.g., lifestyle intervention, treatment of comorbidities (e.g., GLP1RA), bariatric procedures; ***: e.g., MRE, SWE, ELF, with adapted thresholds. A and B are options, depending on medical history, clinical context, and local resources. ELF: enhanced liver fibrosis; FIB-4: fibrosis-4 index; GLP1RA: glucagon-like peptide-1 receptor agonist; MRE: magnetic resonance elastography; SLD: steatotic liver disease; SWE: shear wave elastography; VCTE: vibration-controlled transient elastography. Adapted from [61]. © 2024, The Author(s). Licensed under a CC BY 4.0.
MASLD is a prevalent hepatic manifestation of MS. Its association with T2DM is particularly significant, as the presence of T2DM accelerates the progression of MASLD and heightens the risk of developing severe liver complications. Conversely, MASLD can impair glycemic control, creating a bidirectional relationship that complicates management strategies [70–74].
Approximately 70% of individuals with T2DM are affected by MASLD, and about 20% of these patients may develop advanced liver fibrosis. The coexistence of these conditions significantly elevates the risk of cirrhosis, HCC, and liver-related mortality [72]. The pathogenesis of MASLD in T2DM involves insulin resistance, chronic inflammation, and lipid metabolism dysregulation. These factors contribute to hepatic steatosis and can progress to steatohepatitis and fibrosis.
Given the high rate and potential severity of MASLD in patients with T2DM, early detection is crucial. Clinical guidelines recommend routine screening for MASLD in individuals with T2DM, utilizing non-invasive methods such as liver ultrasound and fibrosis scoring systems like FIB-4 and NFS. Advanced imaging techniques, including transient elastography and MRE, can further assess liver stiffness and fibrosis stage. However, liver biopsy remains the gold standard for definitive diagnosis, particularly in cases where non-invasive tests yield inconclusive results.
Lifestyle intervention is the cornerstone of MASLD management. Weight loss through dietary changes and increased physical activity has been shown to reduce hepatic steatosis and improve insulin sensitivity. A weight reduction of at least 5% can decrease liver fat, while a loss of 10% or more may resolve steatohepatitis and reverse fibrosis [73]. The Mediterranean diet, rich in monounsaturated fats and low in saturated fats, has demonstrated efficacy in improving liver histology and metabolic parameters. Regular aerobic exercise, combined with resistance training, enhances these benefits and supports long-term weight maintenance [75].
While no medications are specifically approved for MASLD, several antidiabetic agents show promise in managing the condition. Pioglitazone, a member of the thiazolidinedione class, enhances insulin sensitivity and has been linked to histological improvements in steatohepatitis. Glucagon-like peptide-1 receptor agonists (GLP1RAs) like liraglutide and semaglutide support weight loss and have shown effectiveness in reducing liver fat and inflammation. SGLT2 inhibitors, such as empagliflozin and dapagliflozin, help regulate blood glucose levels and may also decrease hepatic steatosis. Additionally, novel treatments like dual GIP and GLP1RA (e.g., tirzepatide) are currently being explored for their potential role in the management of MASLD [76].
In individuals with obesity, T2DM, and MASLD, bariatric surgery has been shown to produce substantial weight loss and improvements in liver histology. Techniques such as gastric bypass and sleeve gastrectomy are associated with reductions in hepatic steatosis, inflammation, and fibrosis. Nonetheless, surgical treatment should be individualized, with careful assessment of potential risks and expected benefits.
Managing MASLD in patients with T2DM effectively requires a multidisciplinary team approach, incorporating hepatologists, endocrinologists, dietitians, and primary care physicians. This coordinated model of care ensures a holistic strategy that addresses both liver-related and metabolic health needs, promoting better overall outcomes [77]. Ongoing evaluation of liver function, blood glucose levels, and cardiovascular risk factors is crucial in managing MASLD in patients with T2DM. Educating patients on the importance of lifestyle changes and ensuring adherence to treatment regimens are key components in slowing disease progression.
In addition to lifestyle modifications and antidiabetic agents, several emerging and investigational therapies are being explored for the management of MASLD, particularly in patients with T2DM.
Resmetirom, a selective THR-β agonist, received FDA approval in March 2024 for the treatment of metabolic dysfunction-associated steatohepatitis (MASH) in patients with moderate to severe fibrosis without cirrhosis. It acts by enhancing hepatic fat metabolism, leading to reductions in hepatic fat content and ameliorations in fibrosis [78–81].
Lanifibranor, a pan-PPAR agonist, is currently undergoing phase III clinical trials. It targets multiple pathways involved in MASLD pathogenesis, including lipid metabolism, inflammation, and fibrosis, showing promise in improving liver histology [82].
FXR agonists, such as obeticholic acid, have been investigated for their potential to reduce hepatic inflammation and fibrosis by modulating bile acid pathways. However, some agents in this class have faced challenges in clinical trials, including adverse effects and limited efficacy [81].
FGF21 analogues are being studied for their metabolic benefits, including improvements in insulin sensitivity, lipid profiles, and reductions in liver fat. These agents may offer therapeutic potential for MASLD, especially in patients with concurrent metabolic disorders [83].
Recent evidence in the literature indicates that alterations in the gut microbiota play a role in MASLD development. Therapeutic approaches targeting the gut-liver axis, including probiotics, prebiotics, and synbiotics, are under investigation for their potential to improve liver health by modulating intestinal flora [81].
Figure 2 summarizes the management of MASLD in T2DM.
Summary of the management of MASLD in T2DM. MASLD: metabolic dysfunction-associated steatotic liver disease; T2DM: type 2 diabetes mellitus; GLP1RAs: glucagon-like peptide-1 receptor agonists.
Inhibitors of key enzymes involved in DNL, including acetyl-CoA carboxylase (ACC), stearoyl-CoA desaturase-1 (SCD1), and diacylglycerol acyltransferase-2 (DGAT2), are currently under investigation [84, 85]. In addition, PPAR agonists have shown therapeutic potential [86]. Another promising area of research is the development of dual agonists targeting the FXR and the Takeda G-protein receptor 5 (TGR5) [87]. These compounds may exert beneficial effects through multiple mechanisms, including reduction of TG levels, downregulation of ChREBP expression [88], improvement of insulin sensitivity via FXR activation [89], enhancement of GLP-1 activity [90], and strong anti-inflammatory actions mediated through TGR5 [89]. Such developments hold promise for novel pharmacological approaches to complement lifestyle interventions in the management of MASLD.
In conclusion, MASLD represents a significant health concern in individuals with T2DM, necessitating early screening, accurate diagnosis, and comprehensive management. While lifestyle modification remains the cornerstone of therapy, pharmacological agents and, when appropriate, surgical interventions may provide effective adjunctive treatment strategies.
CAP: controlled attenuation parameter
ChREBP: carbohydrate-responsive element-binding protein
CVD: cardiovascular disease
DNL: de novo lipogenesis
ELF: enhanced liver fibrosis
ER: endoplasmic reticulum
FAs: fatty acids
FGF: fibroblast growth factor
FIB-4: fibrosis-4 index
FXR: farnesoid X receptor
GLP1RAs: glucagon-like peptide-1 receptor agonists
HCC: hepatocellular carcinoma
IL-6: interleukin-6
LSM: liver stiffness measurement
MASLD: metabolic dysfunction-associated steatotic liver disease
MRE: magnetic resonance elastography
MRS: magnetic resonance spectroscopy
MS: metabolic syndrome
NAFLD: nonalcoholic fatty liver disease
NASH: nonalcoholic steatohepatitis
NFS: nonalcoholic fatty liver disease fibrosis score
PDFF: proton density fat fraction
PKCε: protein kinase C epsilon
PPAR: peroxisome proliferator-activated receptor
SCFAs: short-chain fatty acids
T1DM: type 1 diabetes mellitus
T2DM: type 2 diabetes mellitus
TAUS: transabdominal ultrasonography
TG: triglyceride
THR-β: thyroid hormone receptor-β
TNF-α: tumor necrosis factor-alpha
VCTE: vibration-controlled transient elastography
VLDL: very low-density lipoprotein
MAK: Visualization, Writing—original draft, Writing—review & editing, Conceptualization. The author read and approved the submitted version.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3674
Download: 49
Times Cited: 0
Elif Basaran, Gulali Aktas
Roanne Lecky ... Catriona Kelly
Maher Monir Akl, Amr Ahmed
Yutang Wang ... Guang Yang
Ivonne G. Narváez-Ortiz ... Alberto Maceda-Serrano
Osama Mahmoud Mehanna ... Ahmad Shaban Abd El Monsef
Thewodros Leka, Hemda Garelick
Halima Babikir Eltahir ... Abdelrahim Osman Mohamed
