Review
Review

Affiliation:
1University Institute of Pharmaceutical Sciences, UGC Centre of Advanced Studies (UGC-CAS), Panjab University, Chandigarh 160014, India
2Department of Pharmacy, School of Medical & Allied Sciences, GD Goenka University, Gurugram 122103, India
Email: monuyadav.pharmacology@gmail.com
ORCID: https://orcid.org/0000-0002-3003-4186
Affiliation:
1University Institute of Pharmaceutical Sciences, UGC Centre of Advanced Studies (UGC-CAS), Panjab University, Chandigarh 160014, India
ORCID: https://orcid.org/0000-0001-8887-7322
Affiliation:
2Department of Pharmacy, School of Medical & Allied Sciences, GD Goenka University, Gurugram 122103, India
Affiliation:
2Department of Pharmacy, School of Medical & Allied Sciences, GD Goenka University, Gurugram 122103, India
Affiliation:
3Dr. K.N. Modi Institute of Pharmaceutical Education and Research, Modinagar 201204, Uttar Pradesh, India
ORCID: https://orcid.org/0000-0002-5904-4185
Affiliation:
1University Institute of Pharmaceutical Sciences, UGC Centre of Advanced Studies (UGC-CAS), Panjab University, Chandigarh 160014, India
Affiliation:
2Department of Pharmacy, School of Medical & Allied Sciences, GD Goenka University, Gurugram 122103, India
Affiliation:
4School of Medical & Allied Sciences, K. R. Mangalam University, Gurugram 122103, Haryana, India
ORCID: https://orcid.org/0000-0002-0870-4972
Affiliation:
1University Institute of Pharmaceutical Sciences, UGC Centre of Advanced Studies (UGC-CAS), Panjab University, Chandigarh 160014, India
Explor Med. 2022;3:494–515 DOI: https://doi.org/10.37349/emed.2022.00110
Received: June 15, 2022 Accepted: September 10, 2022 Published: October 31, 2022
Academic Editor: Lindsay A. Farrer, Boston University School of Medicine, USA; Md Noushad Javed, K.R. Mangalam University, India
The article belongs to the special issue Techniques in Repurposing and Targeted Delivery: Bringing a New Life to Shelved Drugs
Mitochondria are important organelles for high energy synthesis, reactive oxygen species balancing, antiapoptotic molecule production, membrane stability, intracellular calcium buffering, neuroplasticity and neurotransmission. Dysfunction in mitochondria is considered to be involved in the pathophysiology of mental problems. It has been observed that several drug types used to treat brain illnesses can harm mitochondria by altering the oxidative phosphorylation system and the gene expression of mitochondria-related proteins. In some studies, it has been observed that mitochondrial biogenesis shows a therapeutic effect in the management of mitochondrial disorders. Many therapeutic compounds are effective in the activation of mitochondrial biogenesis. The comorbidity of mental problems observed in those with mitochondrial dysfunction and the change in the efficacy of the cellular respiratory system have attracted researchers to understand the pathways and possible therapeutic strategies in neurological disorders. This article has attempted to understand the impact of mitochondrial function and mitochondrial dysfunction in the pathogenesis of brain disorders to develop potential therapeutic drugs.
Around 200 to 2,000 mitochondria are found in a single cell to supply energy, whereas human germ cells like oocytes contain 10,000 and spermatozoa have 16 mitochondria. Almost all cells contain it, with the exception of adult erythrocytes. It primarily generates energy through two metabolic processes: the Krebs cycle of citric acid and the electron transport chain (ETC) [1]. Two molecules of pyruvate are produced by the cytosolic breakdown of glucose or carbohydrates, and these molecules are transported into the mitochondrial tricarboxylic acid (TCA) cycle. Pyruvate carboxylase and pyruvate dehydrogenase (PDH) come into contact with the pyruvate transported across the membranes (PDH). PDH is a mixture of the three enzymes: dihydrolipoyl dehydrogenase, transacetylase, and PDH. Coenzymes and substrates are required for the PDH to operate. Numerous nutrients are necessary for healthy mitochondrial activity (Table 1). A lot of neurodevelopmental processes also depend on mitochondria. It is essential for cellular stability, energy production through the metabolism of steroid hormones, proteins, lipids, reactive oxygen species (ROS) level control, and the apoptosis mechanism [2, 3]. The development of mouse embryos and the formation of synapses depend on mitochondrial fission and fusion [4]. Additionally, inappropriate cellular energy production caused by abnormalities in these pathways impairs neurodevelopment by interfering with neurotransmission, myelination, and neural connectivity [5]. Many understandings document that functional alteration of complex-I contributes to the ruin of cellular respiration and disturbs the mitochondrial factional system in brain disorders [6]. By modifying numerous cellular pathways, disturbed brain energy metabolism and mitochondrial malfunctioning oxidative stress have been linked to the etiology of brain diseases [7]. Molecular studies proposed a strong link between inflammation and mitochondrial impairment [8]. The combined effect of neuroinflammation and mitochondrial dysfunction can also activate apoptosis [9]. Neurogenesis, the process of making new neurons, is thought to be an important part of synaptic plasticity in natural settings and in the repair of neurons that have been damaged [10, 11]. Several studies have consistently found that different types of stress reduce hippocampal neurogenesis in the adult brain, and neurogenesis is also a pathophysiological hypothesis of brain disorders [12]. Long-term administration of various drugs used for neurological disorders is observed to affect mitochondrial gene expression [13]. Many of these drugs, which are reported in Table 2, may halt the mitochondrial respiratory chain. Clozapine is found to change mitochondrial morphology, its membrane potential, and stimulate inflammation [14]. Moreover, haloperidol also causes degeneration of ultrastructural alterations in mitochondria [15]. The importance of genetic variants in the mitochondrial nicotinamide adenine dinucleotide (NADH)-ubiquinone oxidoreductase 75 kDa subunit (NDUFS1) with antipsychotic drug-induced weight gain has been brought to light by the association between nuclear-encoded mitochondrial genes and antipsychotics [16]. The protective effect of estrogen on decreasing mitochondrial dysfunction and increasing the function of ETC while activating antioxidant defense systems has been reported in some studies [17, 18]. Furthermore, N-acetylcysteine has been shown to protect mitochondria from cadmium-induced oxidative damage and toxicity by increasing cytosolic cytochrome c release, decreasing mitochondrial membrane potential, decreasing B-cell lymphoma 2 (Bcl-2) expression, and increasing poly-polymerase cleavage, caspase cascades, and p53 expression [19, 20]. Chloroacetic acid significantly increased oxidative biomarkers, mitochondrial dysfunction and decreased intracellular glutathione (GSH) via increasing cytochrome c release and stimulating the mitochondria-dependent apoptosis system by initiating caspase cascades, which were inhibited with the treatment of N-acetylcysteine [21]. Many nutrients may alter the functioning of mitochondrial complexes as well as inflammatory states of neurons, including protective actions against drug-induced mitochondrial dysfunctions [21, 22]. A list of such nutrients is tabulated in Tables 1 and 2 respectively as below:
Nutrients for mitochondrial complexes function
| Mitochondrial complex | Nutrients |
|---|---|
| ETC complexes | Copper, iron, sulfur, riboflavin and ubiquinone coenzyme Q10 (CoQ10) |
| PDH complex | Riboflavin, niacin, thiamin, pantothenic acid and lipoic acid |
| TCA cycle |
|
| Shuttling electrons between ETC complexes | Copper, iron and ubiquinone |
Drug-induced mitochondrial dysfunction
| Drugs class | Drugs |
|---|---|
| Alcoholism medications | Disulfiram |
| Anti-anxiety | Alprazolam, diazepam |
| Anti-dementia | Tacrine, galantamine |
| Antidepressants | Amitriptyline, amoxapine, citalopram, fluoxetine, Symbyax, Sarafem, Fontex, Foxetin, ladose, Fluctin, prodep, Fludac, oxetin |
| Antiepileptic | Valproic acid, mood stabilizers lithium |
| Anti-Parkinson’s | Tolcapone |
| Antipsychotics | Chlorpromazine, fluphenazine, haloperidol, risperidone, quetiapine, clozapine, olanzapine |
| Barbiturates | Amobarbital, aprobarbital, butabarbital, butalbital, hexobarbital, methylphenobarbital, pentobarbital, phenobarbital, primidone, propofol, secobarbital, thiobarbital |
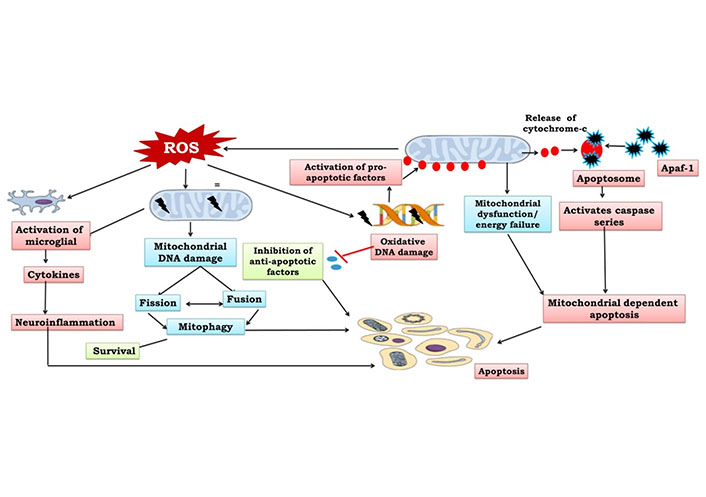
The primary energy source for cellular function is provided by the mitochondria, which make adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). Continuous mitochondrial fission and fusion, mitochondrial biogenesis, and mitophagy all work together to control the shape, quantity, quality, turnover, and heredity of mitochondria. ROS, fatty acid oxidation, amino acid metabolism, pyridine production, phospholipid alterations, calcium regulation, as well as cell viability, senescence, and death are all under the control of mitochondria [23]. The careful balancing of two opposing processes is necessary to maintain mitochondrial homeostasis—mitophagy, which removes harmed mitochondria and mitochondrial biogenesis which produces new mitochondria. Recent studies have identified a number of specific molecules involved in preserving the homeostasis of mitochondria, including the phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) Pakin, which activates protein synthesis in damaged mitochondria and the ligand-activated transcription factor aryl hydrocarbon receptor, also acts as a protector from oxidative stress [24]. In response to stress, mitophagy, a mitochondrial quality control system that allows the removal of damaged and unnecessary mitochondria, stops these negative effects and restores cellular balance. Permanent mitochondrial damage can either activate innate immunity by increasing ROS or releasing mitochondrial deoxyribonucleic acid (mtDNA), or cause cell death by releasing cytochrome c and other apoptogenic proteins [25, 26].
Apoptosis is a biologically regulated process of planned cell death that is managed by a few particular genes. It is an energy-dependent process to increase the growth of cancerous cells, while uncontrolled apoptosis can facilitate the neurodegeneration process [27, 28]. This process involves quick phagocytosis by surrounding cells, cytoplasmic shrinkage and chromatin condensation. The basic physiological principles are regulated by the apoptotic process which eliminates superfluous glial cells and neurons that are unable to create neural connections [1]. A large number of neurons are formed throughout adult neurogenesis in which neurons further compete to form neuronal connections, and those neurons not having neurotrophic or electrical activity undergo the apoptotic process. Biologically, the apoptotic system is activated by both extrinsic and intrinsic pathways via many factors. Tumor necrosis factor (TNF) and cluster differentiation 95 (CD95) recruit caspase-8 in the extrinsic pathway via Fas- (transmembrane proteins) (Figure 1). Further, leakage of caspase-8 into the cytosol entailed the cleavage and initiation of the BH3-interacting death agonist to trigger the caspase cascade [29, 30]. This finally facilitates an increase in outer mitochondrial membrane permeability to release apoptotic molecules. The intrinsic pathway is further divided into two pathways, including endoplasmic reticulum and mitochondrial apoptosis that are initiated by intracellular signaling molecules. Internal apoptosis activators, such as oxidative stress, glutamate’s hyperactivation of N-methyl-D-aspartate receptors (NMDARs) and elevated Ca2+ and ROS levels induce mitochondrial apoptosis. This releases cytochrome c into the cytoplasm which binds to apoptotic protease activating factor 1 (Apaf-1), activating a number of kinases that can reduce the mitochondrial membrane potential. Caspases-3 and -7, which are also activated by stimulated caspase-9, are involved in cell death. Bcl-2 prevents the release of cytochrome c into the cytoplasm by blocking the permeability transition pore [31]. Apoptosis factors, mitochondrial membrane permeabilization and endonuclease G transport to the nucleus and lead to chromatolysis and ultimately apoptosis by deactivating repair mechanisms, ending cell cycle development, disabling antiapoptotic molecules, facilitating cytoskeletal and disassembling of nuclear structures and triggering cell phagocytosis [32] (Figure 1). Certainly, apoptosis is necessary for the primary steps of development and adult neurogenesis and also mediates long-term potentiation and synaptic plasticity [1]. Therefore, mitochondrial dysfunction can modify mitochondrial membrane potential and release various apoptosis factors.
Neural stem cell proliferation promotes neuron differentiation, known as neurogenesis, is an extremely complicated mechanism. Furthermore, during neurogenesis, the number of mitochondria per cell increases, and the neurogenesis process is dependent on the number of mitochondria and total ATP production [33]. Moreover, mitochondria get to enter the developing axon and help in the process of axogenesis. There is evidence that axogenesis may stop as a result of mitochondrial malfunction. [3]. Mitochondria are thought to be involved in the traffic between multiple subcellular compartments, including axons, cell bodies, dendrites, and synaptic terminals, as part of the neuroplasticity mechanism [34]. Therefore, mitochondria support the development of the brain through forming dendrites and axons by providing energy at presynaptic as well as postsynaptic sites [1]. Hence, mitochondrial dysfunction can cause the cessation of differentiation and maturation of neurons. Furthermore, the mitochondrion controls neuroplasticity to maintain various neuronal functions such as neural growth, neural differentiation, dendritic remodelling, and neurotransmitter release [3, 35]. During the neuronal process, presynaptic activity increases the transmission of long-lasting synaptic formation, which is known as post-tetanic potentiation. It involves the processing of neural circuitry for the formation of memory. Additionally, mitochondria trap Ca2+ from the cytoplasm and accumulate it in their matrix while taking part in a variety of Ca2+-mediated signaling events [36]. Many mitochondrial genetic studies have also demonstrated that mitochondria influence various brain functions and behaviors.
Synaptic plasticity is a modification that happens at the synapse (a junction between the neurons that permits their communication) to help with functional and structural plasticity. Mitochondria play an important role in synaptic plasticity and neuronal development [37]. It has an important role at presynaptic axon terminals as well as at postsynaptic dendritic spines. The axons of hippocampal neurons have more mitochondria that can move around than their dendrites. Mitochondria are metabolically active and extremely charged in dendrites. In synapse formation, the total number of mitochondria in dendrites is very important for structural plasticity. dynamin-related protein 1 (Drp1) mutations can prevent mitochondrial division; however, optic atrophy 1 [OPA1, OPA1 mitochondrial dynamin-like guanosine triphosphate (GTPase)] facilitates mitochondrial fusion. The process of synaptogenesis is stopped when Drp1 (A38K) is turned on. This also decreases the number of mitochondria in dendrites. It has been reported that treatment with creatine can enhance the dendritic mitochondrial mass and increase their functions [38, 39]. The neuronal activity of presynaptic axons, as well as postsynaptic dendrites, is dependent upon intracellular Ca2+. Mitochondria function as Ca2+ sensors and play an important role in neuronal transduction [36, 37]. According to recent research, long-term potentiation (LTP) is also influenced by ROS [40]. Some studies suggest the role of ROS in synaptic plasticity of the hippocampal region by reducing the function of protein phosphatases 2A & 2B and increasing the activity of extracellular-regulated protein kinase C, protein tyrosine kinases, kinase 2 and ryanodine receptor type 3 [41, 42]. Excessive levels of Ca2+ can lead to initiation of pro-apoptotic elements and apoptotic mechanisms which are further involved in long-term depression (LTD). A balance between LTP and LTD is important for cerebral health. Synaptic plasticity may be impacted by alterations in mitochondrial function, including Ca2+ modulation, ROS generation, energy metabolism, and mitochondrial mobility within axons and dendrites [43].
Ca2+ controls the various cell functions by the initiation of ATP consumption (ion transport, movement, contraction, Ca2+ pumps) and by triggering ATP production via the activation of OXPHOS and the glycogen breakdown pathway [44]. The mitochondrial calcium uniporter (MCU) complex, which is made up of the calcium channel proteins MCU and MCU regulatory (MCUR) subunit (MCUb) as well as regulatory subunits such as essential MCUR (EMRE), MCUR, mitochondrial calcium uptake protein 1 (MICU1), and MICU2, is responsible for calcium entrance into mitochondria [45]. Ca2+ regulates various mitochondrial activities, from metabolism to cell death. Both the endoplasmic reticulum and the mitochondria contribute to calcium homeostasis which acts as the second messenger and is engaged in neuroplasticity and the release of neurotransmission. The inner membrane of mitochondria has Ca2+ uniporters for the influx of Ca2+, whereas H+/Ca2+ and Na+/Ca2+ are the antiporters for the efflux of Ca2+. On the other hand, the outer membrane of mitochondria is permeable to Ca2+ [46]. The citric acid cycle enzymes are activated when there is enough Ca2+ in the mitochondria. This helps make more ATP [47]. Both isocitrate dehydrogenase and oxoglutarate dehydrogenase are mitochondrial matrix enzymes that are also initiated by Ca2+. Additionally, PDH makes it easier for pyruvate to be converted into acetyl coenzyme A (acetyl-CoA), a crucial chemical involved in the Krebs cycle and another conversion into oxaloacetate. Moreover, relocation of Ca2+ from endoplasmic reticulum (ER) to mitochondria is essential to maintain cell energy and a decrease in transmission can lead to a decline in OXPHOS and energy crisis. High Ca2+ uptake into mitochondrion can increase the mitochondrial penetrability of transition pores while halting the formation of ATP [30]. The apoptosis system, mitochondrial swelling, and the release of cytochrome c can all be facilitated by increased mitochondrial membrane porousness for molecules less than 1,500 Daltons [1].
Many theories propose a strong link between inflammation and mitochondrial dysfunction by activating the redox-sensitive inflammatory response. However, excessive ROS generation could exhaust the antioxidant defense mechanism, leading to mitochondrial malfunction. Preclinical studies on transgenic mice revealed that increasing catalase levels in mitochondria reduce age-related pathological disorders [48]. Some investigations suggest mitochondrial dysfunction could initiate inflammatory responses by activating nuclear factor kappa B (NF-κB). Therefore, the influx of mitochondrial Ca2+ is involved in the development and regulation of cell signals and mitochondrial activity. However, through boosting NF-κB dependent pro-inflammatory signaling, mitochondrial Ca2+ excess contributes to the generation of ROS, leading to the release of pro-inflammatory mediators. Resveratrol, in combination with pyrrolidine dithiocarbamate (NF-κB inhibition), has been shown to reduce inflammatory gene expression in aged rats [49, 50]. Some studies suggest that upregulation of NF-κB may contribute to elevating the immune responses in the cortical region of the brain in schizophrenia [51, 52]. Moreover, prolonged immune activation is a critical mechanism involved in prenatal events such as infection exposure that can cause chronic changes in mitochondrial activity in leucocytes in adults. Serum thiobarbituric acid reactive substances (TBARS) and interleukin 6 (IL-6) levels are also elevated in the early and late stages of neurological disorders [53].
The possible sources of reactive species are dopamine metabolism, mitochondrial ETC and genetic factors. It is an extremely regulated mechanism, the passing out of molecular oxygen (O2) by mitochondrial ETC to form superoxide that gets transformed to H2O2 by superoxide dismutase. In a clinical trial, people with neurological brain problems had more TBARS. This shows that this compound can change ETC activity by changing how complex I works in mitochondria [54, 55]. Lipids, iron-sulfur complexes, and the sulfhydryl groups of mitochondrial enzymes are all oxidised when ROS is released in excess. A few examples of the neurological illnesses that can be caused by oxidative damage to membrane DNA, lipids, and proteins include Parkinson’s disease (PD), schizophrenia, bipolar disorder, epilepsy, migraine, Leigh syndrome, mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes [56, 57]. In vitro and in vivo research suggests that reactive nitrogen species (RNS) and ROS are responsible for teratogenesis, birth defects, and diseases of the nervous system [58]. As a result of constant exposure to reactive species, intramembrane mitochondrial proteins like cytochrome c, pro-caspases, and pro-apoptotic factors are released, which starts a chain of events that leads to apoptosis [59]. Additionally, oxidative stress also triggers deletions and point mutations of mtDNA that worsen the impairment of mitochondrial activity, which is also a reason for neurological disorders [60].
Other proteins are written by genes in the cell’s DNA in addition to the thirteen protein components of the ETC that are encoded by the mitochondria’s mtDNA [1]. They serve as the source of energy for the OXPHOS, citric acid cycle, and the production of ATP from glucose, pyruvate, and oxygen. ETC is made up of five protein complexes, each of which regularly produces ATP at complex V [61]. However, complexes I, III, and IV distribute proton ions to the mitochondrial intermembrane space to produce electrochemical for the production of ATP, which is a cellular energy fuel and necessary for the trafficking of ion channels and receptors, neuronal membrane, and neurotransmitters in vesicles. Additionally, in order to produce NADH phosphate (NADPH), glucose-6-phosphate dehydrogenase (G6PD) is necessary (G6P) [62, 63]. Its deficiency is reported to induce mental disorders. NADPH is required for the maintenance of cellular redox balance by reducing GSH [64–66]. G6PD is especially rich in the brain per unit protein and it gets enhanced in response to oxidative stress. The previous finding showed that hexokinase 1 (HK1) detachment from the outermost membrane of mitochondria (OMM) in the cortex region of the postmortem brain tissue of schizophrenic individuals. It is a rate-limiting enzyme and its attachment to mitochondria is important for the metabolism of brain energy [67, 68]. The enzyme attachment to mitochondria is also essential for the survival of brain cells and neurons by reducing oxidative damage. Furthermore, G6P is a key metabolite of HK1 and a substrate of G6PD, which is an important modulator for HK1 binding with OMM by activating the inhibition feedback pathway [69]. Moreover, reduced G6PD activity increases the cytosolic level of G6P, thus diminishing the attachment of HK1 to OMM. So, when HK1 gets separated from OMM, it makes it harder for mitochondria to work and leads to more free radicals and oxidative stress, which can turn on G6PD [70]. These reports suggested connections between HK1 mitochondrial detachment, mitochondrial dysfunction, G6PD activity, oxidative stress, and imbalanced pH with G6PD deficient neurological brains. By lowering oxidative stress, N-acetyl cysteine, a precursor to GSH, has shown therapeutic promise in easing the symptoms of G6PD deficiency in individuals with psychosis [71]. Furthermore, because mitochondria are the primary organelles needed for energy production, they are crucial for the brain’s highly metabolically active tissue. The genetic information needed to make the polypeptides that make up the mitochondrial respiratory chain is in the mitochondrial genome. Alterations in mtDNA and any mitochondrial dysfunction have been observed to be involved in the pathogenesis of brain disorders [1]. According to a study, platelet activity in complex I gets higher in schizophrenia patients, and this is associated with positive psychotic symptoms. Further understanding suggests that decreased activity of complex I and III enzymatics accumulates free electrons that increase mtDNA. Chronic accumulation of free redials is supposed to cause mtDNA depletion. Antipsychotic medications have also been seen to increase the mitochondrial respiratory chain complex I deficit and oxidative stress in schizophrenia patients, which further contributes to the development of extrapyramidal diseases [72, 73]. Further, evidence since 1934 demonstrates that decreased GSH and increased lactate are the possible hallmarks of impaired energy metabolism in schizophrenia. In a number of postmortem studies, the pathophysiology of schizophrenia has been related to abnormal energy metabolism brought on by the dissociation of the glycolytic enzyme HK1 from the outer mitochondrial membrane protein. The mechanism underlying this is a voltage-dependent anion channel [74]. Reduced mitochondrial ATP generation and an increase in non-mitochondrial anaerobic glucose metabolism result from the dissociation of HK1, therefore desisting excessive lactate synthesis, which has been seen as a cerebrospinal fluid (CSF) biomarker in the schizophrenic brain. Supporting studies like proteome analysis have shown dysregulation of energy metabolism in Wernicke’s area, alteration of calcium homeostasis in the dorsolateral region of the prefrontal cortex and dysfunction of glycolysis in the thalamus area of the schizophrenic brain. It has also been discovered that decreased glycolytic mechanisms and PDH enzyme levels increase cellular acidosis and lactate levels in the prefrontal cortex [3]. Reduced PDH activity and increased lactate concentration can cause anaerobic respiration, which indicates mitochondrial dysfunction.
Amyloid-β (Aβ) peptides are found in both neurons and mitochondria [75]. A peptide buildup in mitochondria inhibits complex II and IV of the mitochondrial enzyme system, increasing ROS and reducing ATP synthesis [76]. A peptide buildup also prevents proteins from entering the mitochondria, which could damage or mutate mtDNA [77]. Additionally, it has been noted that the development of A peptides reduces the activity of several enzymes, including isocitrate dehydrogenase, PDH, TCA cycle, and α-ketoglutarate dehydrogenase (αKGDH) [78]. Accumulation of the Aβ peptide results in abnormalities in the release of cytochrome c in excess, opening of the mitochondrial permeability transition pore (mPTP), and mitochondrial Ca2+ buffering [75]. Because Aβ peptides interact with the mitochondrial membrane protein Aβ binding alcohol dehydrogenase (ABAD), which results in aberrant mitochondrial trafficking, decreased mitochondrial mobility, and mitochondrial malfunction, synaptic degeneration occurs [79, 80]. It has been found that amyloid precursor protein (APP) mutations also lead to modifications in Ca2+ homeostasis and the apoptosis process [81]. Notably, Aβ peptide accumulation and tau hyperphosphorylation lead to accelerated DRP1 nitrosylation that may cause neurodegeneration and rapid mitochondrial fission. According to a study, Aβ peptides reduce proliferator-activated receptor-coactivator-1 (PGC-1) expressions, which in turn decrease mtDNA content and mitochondrial biogenesis, and ultimately enhance neurodegeneration and contribute to the onset of Alzheimer’s disease (AD) [82].
Neuroinflammation is suggested to be inculcated pathophysiologically in neurological disorders, which is confirmed by epidemiological, preclinical, and genome investigations [83]. Furthermore, clinical trials have also indicated the significance of anti-inflammatory potential agents in treating psychosis as a complementary therapy. Activation of microglia in brain macrophages is mainly responsible for the occurrence of neuroinflammation. Because of the increased translocator protein (TSPO) expression in the mitochondria of microglia, TSPO is used as a target in positron emission tomography (PET) imaging studies [84, 85]. Several PET studies on the expression of TSPO in neurological disorders have found no significant group effect; one found a reduced expression of TSPO in patients with the first episode of neurological disorders and four reported a high expression of TSPO in patients with chronic neurological disorders [86]. It has been reported that an increase in TSPO expression is seen in patients with treated neurological disorders but not in patients without prior treatment. According to a new meta-analysis of earlier PET investigations on individuals with psychosis utilising second-generation radioligands, psychosis increases the expression of the downstream TSPO gene [87, 88]. The connection between neurological disorders, brain anatomy and inflammation is not clear enough. Previous studies on morphometry and cerebral volumetry have revealed a decrease in grey matter volume, ventricular hypertrophy, and brain narrowing in schizophrenia-related brain abnormalities [87]. These neurodegenerative disorders are suggested, but they are suggested to reduce neuronal loss, synaptic stress, synaptic density, and the incidence of neurological disorders. The activation of microglia is hypothesized to influence brain volume. Preclinical and cell culture studies suggest that brain tissue can be destroyed by hyperactivation of microglia. It has been shown that AD, immunodeficiency syndrome, multiple sclerosis, and PD colocalize the density of TSPO and neurodegenerative disorders in the brain [89]. Recently, various observations have indicated a relationship between peripheral inflammation and grey matter deficits in the pathophysiology of neurological disorders. In people at high clinical risk of neurological disorders, the loss of grey matter in the frontal lobe has been associated with basal levels of pro-inflammatory cytokines in plasma. Also, IL-1β gene polymorphism has been observed to reduce grey matter volume in the prefrontal cortex of brains with neurological disorders [90].
Many in vivo and in vitro studies have shown that dopamine is also involved in oxidative stress, neuroinflammation, mitochondrial dysfunction, and apoptosis via ROS production, cytochrome c release, p38 kinase activation, followed by caspase-3 and caspase-9 [91, 92]. Moreover, in an in vitro study, it has been observed that chronic activation of dopamine in cortical neurons can cause dysfunction in calcium homeostasis and mitochondrial impairment can lead to apoptosis. Dopamine is also seen to reduce the cellular ATP by inhibiting the activity of mitochondrial complex I [61]. The important relationship between dopamine and mitochondrial respiration is thought to play a role in neurological diseases. Glutamate causes excitotoxicity in the central nervous system (CNS) by activating the NMDARs. Glutamatergic dysfunction contributes to brain volume reduction, which leads to disrupted synaptic plasticity, cortical microcircuitry, and NMDARs signaling [93]. Glutamatergic neurotransmission is potentially dependent upon calcium homeostasis and appropriate mitochondrial function. Recent findings suggest that the influx of Ca2+ by NMDARs causes an overload of mitochondrial calcium, which may further lead to mitochondrial impairment and trigger the signals for death. This excitatory neurotransmitter is also involved in the synthesis of nitric oxide in neurons by forming peroxynitrite by reacting with superoxide and may inhibit the complexes I, II, and IV of mitochondria [94]. Continuous glutamate exposure may saturate the mitochondrial matrix, which inhibits the synthesis of ATP [3].
Through OXPHOS, mitochondria produce the ATP required to power cellular activity. A scaffold protein called disrupted-in-schizophrenia-1 (DISC-1) participates in a number of neurodevelopmental processes, including synaptogenesis, intracellular cyclic adenosine 3, 5’-monophosphate (cAMP) signaling, neurite outgrowth, neurogenesis, neuronal migration in the developing cortex, and mitochondrial fusion and fission in the neurons [95–97]. Mitofilin, a mitochondrial inner-membrane protein, is how this protein functions in mitochondria [97–99]. The possible function of DISC-1 in controlling mitochondrial activity via mitochondrial interacting molecules was shown by molecular pharmacological investigation. In a study, it was found that the genetic process known as t(1;11) translocation causes abnormal DISC-1 proteins to lack the terminal amino acids necessary for interaction with binding molecules, suggesting mitochondrial dysfunction and stifling various neuronal processes involved in the onset of schizophrenia. D-amino acid oxidase activator (DAOA)/G72, another gene linked to neurological illness susceptibility, has been linked to regulating mitochondrial morphology [100, 101]. An increase in endogenous/exogenous ratio and a mitochondrial protein are both encoded by the gene known as LG72. G72 stimulates mitochondrial fission in primary neurons [101]. Also, it has been thought that the G72/G30 transgenic mouse model leads to a problem with the mitochondria, which can cause a lack of GSH and problems with thinking in psychosis.
Stroke, which is characterized by a sudden reduction in blood flow to the affected area of the brain, resulting in hypoxia and glucose deprivation, is one of the major causes of mortality and adult disability worldwide. More and more evidence points to the importance of mitochondrial dysfunction in the apoptotic and necrotic neuronal cell death that results from cerebral ischemia. Mitochondrial dysfunction is another sign of neuronal cell death brought on by ischemia and reperfusion. The infract core, which is irreparably destroyed within a few minutes of stroke and cannot be saved by reperfusion, and the penumbra, which has diminished function but retains structural integrity and can be saved by reperfusion, are the two primary zones of ischemia injury. The penumbra is an important pharmacological target for treating ischemia because it can be healed. Cell death in the infarct penumbra is mediated by a separate molecular mechanism from that in the infarct core. There is strong evidence from the experimental model, particularly with middle cerebral artery occlusion (MCAO), that the death of neurons in the penumbra is caused by mitochondrial malfunction. Ischemic neuronal death which results from ischemia, depolarization of the mitochondrial membrane potential (m), reduction of ATP synthesis, accumulation of PINK1, increased production of ROS by Parkin, increases in matrix calcium, and opening of the mPTP, all cause neuronal death [102–104]. Mitochondria play a significant role in this process. The mPTP opens when the amount of mitochondrial Ca2+ rises, which is well known to result in an excess production of ROS. As a result, targeting the mitochondria may be an effective strategy for treating ischemia- related damage.
Key areas of the brain affected by AD, a progressive neurodegenerative disorder caused by synaptic dysfunction and cell loss, include the hippocampus, frontal cortices, and related structures. The disease is characterized histopathologically by extracellular Aβ deposition and neurofibrillary degeneration, both of which result from the neurodegenerative process known as amyloidogenesis. Given that the mechanisms of neuronal degeneration downstream of Aβ are similar to those of mitochondrial dysfunction, including membrane-associated oxidative stress, disturbed Ca2+ homeostasis, impaired energy metabolism, and possibly apoptosis, it is possible that mitochondrial changes contribute to the development of the illness. Aβ stimulates the development of mitochondrial dysfunction in AD neurons by increasing mitochondrial O2 synthesis, lowering ATP production, and increasing mitochondrial Ca2+ absorption, which can lead to the opening of the permeability transition pores (mPTP) and apoptosis [105]. “Ubiquinone” is the main component of mitoquinone mesylate (Mito Q), an antioxidant that specifically targets the mitochondria. Aβ accumulates and exerts its antioxidant properties against oxidative stress by being selectively absorbed into mitochondria in a membrane potential-dependent manner. This shows a potential AD treatment drug.
The substantia nigra pars compacta (SNpc), which is primarily responsible for motor movement disorders like rigidity, tremor, bradykinesia, and postural insufficiency, is the site of a selective loss of dopaminergic neurons that leads to PD, the second most common progressive neurodegenerative disease. Early reports of complex I impairment in the postmortem SNpc of PD patients and dopaminergic neuron degeneration brought on by environmental toxins rotenone and MPTP (1-methyl-4-Phenyl-1,2,3,6-tetrahydropyridine) were the first lines of evidence connecting mitochondrial dysfunction to PD [106]. When mitochondrial complex I is absent, ROS is produced, DNA is damaged, and poly adenosine di-phosphate (ADP)-ribose polymerase (PARP) is activated. This is one of the mechanisms that cause PD (PARP). In turn, PARP worsens the initial mitochondrial malfunction by reducing intracellular nicotinamide adenine dinucleotide (NAD+) reserves, which interferes with the mitochondrial respiratory chain and ATP generation. Motor, brainstem, and neocortical neurons in mice with the synuclein A53T mutation die, and mtDNA is damaged. These facts demonstrate that one of the most promising avenues for treating PD is a mitochondrial issue [107].
Huntington’s disease (HD), a neurological condition, can be inherited from one generation to the next. There is presently no cure for HD, which harms a person’s physical, mental, and emotional health. It is caused by a single faulty gene called “Huntington” on chromosome 4, which primarily affects the caudate and later the cortex in the striatum. It is well known that the nucleotides A, G, C, and T make up genes. In the case of HD, the letters C-A-G are repeated 20–40 times. One of the main signs of this disorder is uncontrollable movement of the arms, legs, face, and upper body, as well as issues with thinking and reasoning skills, including judgement and focus. Mitochondrial dysfunction has a crucial role in the etiology of HD. The cleaved rodent mutant gene (mtHtt) interacts with p53, Sp1, TAFII130, and CREB (cAMP response element) binding protein, among other transcription regulators. The activity of mitochondria is regulated by p53. As a result of mtHtt’s interaction with p53, p53-dependent transcription is brought about by an increase in p53 level. Neuroprotection against the mutant Htt gene may be achieved by small interfering RNA (siRNA)-mediated inhibition of p53 function or gene deletion. By knocking down the p53 gene, it is possible to protect striatal cells against harm and mitochondrial dysfunction, such as a drop in cytochrome c oxidase (COX) activity and mitochondrial membrane potential. It has also been demonstrated that the activity of the mitochondrial complexes II (succinate dehydrogenase), III (cytochrome c reeducates), and IV has decreased in postmortem investigations of HD patients (COX). As a result, the level of calcium in the mitochondria decreased, which in turn caused the production of ROS, which in turn affected the mitochondrial potential and ultimately caused the mitochondrial process to be disrupted. The nuclear coactivator in mitochondrial biogenesis is PGC-1α. In cell culture and in vivo experiments, increased PGC-1 expression is neuroprotective against mutant Htt gene toxicity; its deletion is particularly susceptible to mutant Htt toxicity [108–111]. Resveratrol, an antioxidant that activates sirtuin 1 (SIRT1), causes induction of OXPHOS, and may increase activation of the PGC-1-peroxisome proliferator-activated receptors (PPAR) signaling pathway, which further improves mitochondrial function, has been shown to improve mitochondrial function in mice with HD in several studies.
No matter the cause, all types of seizures exhibit an acute, high energy demand in certain brain regions, which is satisfied by mitochondria, the main source of ATP needed for regular neuronal activity and synaptic transmission [112]. Neurons are more vulnerable to mitochondrial dysfunction due to their high energy requirements and lack of regeneration ability. One of the possible causes of epileptic seizures has been discovered as mitochondrial malfunction, which is brought on by an imbalance in calcium homeostasis and the production of ROS. Mitochondrial dysfunction further causes epilepsy by altering the energy demand, normal neuronal excitability and synaptic transmission; it also causes acquired epilepsy, which accounts for 60% of all epileptic cases [113]. Most of the pathogenic DNA mutations causing epilepsy are mtDNA mutations. There are 169 classes of mitochondrial gene mutations that have been identified to affect the mitochondrial function [8344A–G (mutation associated with myoclonic seizures), mitochondrially encoded transfer RNA (tRNA) lysine (MT-TK+), mitochondrially encoded tRNA leucine (MT-TL1), mitochondrially encoded tRNA phenylalanine (MT-TF+), mitochondrially encoded ATP synthase 6 (MT-ATP6+)]. For these reasons, mitochondria are contemplated as a propitious target against epileptic seizures [114].
Glycolysis gives neurons their energy, but since the brain cannot store glycogen, glucose needs to be continuously supplied. When this OXPHOS is impaired, the neuron cannot meet its energy needs, which results in numerous physiological changes and a reduction in ATP generation. According to a study, mild chronic stress impairs animal mitochondrial OXPHOS, which lowers mitochondrial membrane potential, alters the mitochondrial structure in numerous mouse brain regions, including the hippocampus, cortex, and hypothalamus, and causes depression [115, 116]. In addition to lifestyle, diet, and environmental factors, genetics also leads to an increase in mitochondrial susceptibility. The hypothalamic-pituitary-adrenal (HPA) axis is hyperactive in depression and is associated with increased levels of CNS glucocorticoids. However, long-term exposure to excessive glucocorticoids disrupts the respiratory chain, increases the production of ROS and changes the structure of the mitochondria, all of which contribute to the death of hippocampus neurons [117]. The inhibition of mitochondrial 2-oxoglutarate dehydrogenase by nitric oxide and ROS results in glutamate excitotoxicity because the NMDARs are activated. Additionally, this changes the polarity of the mitochondrial membrane, which has been linked to symptoms of depression [23].
It has been demonstrated in in vitro and in vivo investigations that mitochondrial biogenesis may benefit mitochondrial diseases. A number of therapeutic medications are utilized to trigger mitochondrial biogenesis. A biomarker of mitochondrial malfunction is energy shortage. However, these substances are ineffective in treating the underlying cause of mitochondrial dysfunction [118]. Nuclear respiratory factors 1 and 2 (NRF1 and NRF2), PGC-1 and other transcription factors such as PPAR interact to control the complex pathway that controls mitochondrial biogenesis [119]. Once activated, PPARs and NRFs both boost the expression of genes involved in fatty acid oxidation and the transcription of OXPHOS genes (FAO). PGC-1, PGC-1α can be deacetylated and phosphorylated by Sirt1 and adenosine monophosphate-activated protein kinase (AMPK), respectively, to boost its activity [120]. Sirt1 is a nuclear deacetylase that deacetylates acetyl-lysine residues in proteins using oxidized NAD+. Increased cellular NAD+ levels cause Sirt1 to become active. Nicotinamide riboside and PARP 1 can help with this rise [121]. In animal models of COX deficiency, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), an AMPK agonist, has been shown to improve respiratory chain complex activity. A pan-PPAR activator called bezafibrate has been studied in the fibroblasts of people with mitochondrial disorders, stimulated PGC-1 levels, and increased mitochondrial respiratory chain deficits [122]. Intriguingly, human fibroblasts and animal models have both been shown to respond favorably to resveratrol as a stimulant of mitochondrial biogenesis [123, 124]. Retinoic acid may also activate the retinoid X receptor-alpha (RXRalpha) in cybrid containing the m.3243A>G mutation, improving the respiratory chain deficit, according to data [125].
Preclinical studies reported that the protective functions of mitochondria may be increased by endogenous antioxidant defense mechanisms and respiration rate coenzyme with the treatment of CoQ10, vitamins, acetyl, L-carnitine and α-lipoic acid, resveratrol for the management of neurological disorders [50, 65, 75]. Clinical trials have been conducted on some therapeutic compounds for the treatment of mitochondrial dysfunction-induced disorders (Tables 3 and 4).
Clinically approved treatments for neurological disorders induced by mitochondrial dysfunction
| Drug name | Activity | Clinical trial number |
|---|---|---|
| EPI-743 | Antioxidant | NCT01721733 |
| KH176 | Antioxidant | NCT02544217 |
| RP103 (cysteamine bitartrate delayed release) | Cystine-depleting agent | NCT02023866 |
| Idebenone | Antioxidant | NCT00887562 |
| RP103 (cysteamine bitartrate delayed release) | Cystine-depleting agent | NCT02473445 |
| Medium chain triglycerides | Shift heteroplasmy | NCT01252979 |
| L-arginine | NO precursor | NCT01603446 |
| L-arginine (IV) | NO precursor | JMA-IIA00023 |
| L-arginine (PO) | NO precursor | JMA-IIA00025 |
| Arginine and citrulline | NO precursor | NCT01339494 |
| Taurine | Taurine modification | UMIN000011908 |
| Pyruvate | NAD donor | JMA-IIA00093 |
| DCA | Lowering lactate levels | NCT00068913 |
DCA: dichloroacetate; NO: nitric oxide
List molecules that regulate several mitochondrial functions impaired in neurodegenerative disorder examined under preclinical as well as in clinical trials
| Drugs/molecule | Disease | Type of studies | Outcomes | References |
|---|---|---|---|---|
| α-lipoic acid | AD | Preclinical and clinical | Neuroprotective effect, learning and memory improvement | [126] |
| Inosine | ALS | Preclinical and clinical | Safety, tolerability, and effective in increasing urate serum levels | [127] |
| Inosine/urate | PD | Preclinical and clinical | Safety, tolerability, and effective in increasing urate serum levels | [128, 129] |
| Melatonin | ALS, PD, AD | Preclinical and clinical | Safety, better sleep, and a decrease in oxidative stress indicators; no advantages for motor activity; no ameliorations in cognitive functions | [130, 131] |
| Mito-Apocynin | AD, PD | Preclinical | Motor deficit & neuroinflammation attenuation (neuroprotection) | [132–134] |
| Mito Q | ALS, AD, HD, PD | Preclinical | Lengthening lifespan, mitigating cognitive decline, and increasing hindlimb strength; preventing the loss of dopaminergic neurons in a 6-OHDA PD mice model and decreasing ROS-induced autophagy while fostering mitochondrial fusion | [135, 136] |
| N-acetylcysteine | AD, PD, HD | Preclinical | An improvement in cognitive and motor impairments; an increase in brain connections, GSH levels, TH and complex 1 activity; protection against neuroinflammation | [137] |
| SKQ1 | AD | Preclinical | Improvements in cognition and behavior (reduction of ROS generation) | [138] |
| Szeto-Schiller tetrapeptides | AD, PD, ALS | Preclinical | Improved anterograde axonal transport and synaptic activity, increased survival and improved behavior in SOD1G93A mice, as well as increased lifespan and improved motor ability | [138, 139] |
| Vitamin C | AD | Preclinical | Protection of mitochondrial morphology (reduction of oxidative stress damage) and prevention of apoptosis | [140] |
| Carotenoids (astaxanthin) | AD | Preclinical | Hippocampal neurons treated with A 1–42 oligomers and astaxanthin are protected from the formation of ROS; decreasing neuroinflammation and synaptotoxic events | [141] |
| Vitamin E | AD | Preclinical, clinical | Vitamin E on glutamate-treated astrocytes: healing of mitochondrial damage (MMP stabilization and decreased lipid peroxidation); additional research on AD sufferers is required | [142–144] |
| Vitamin E | ALS | Preclinical, clinical | Several clinical investigations have revealed contradictory results in terms of delaying the onset and course of ALS, but more research is required | [145] |
| PGC-1α | PD | Preclinical | Synuclein oligomerization in a cell culture model with PGC-1 restoration; animals with the A30P synuclein gene: decreased synuclein oligomerization | [146] |
| Olesoxime | PD, HD | Preclinical | Enhancing mitochondrial function and preventing apoptosis; stabilizing the mitochondrial membrane improvement in behavior and cognition | [147] |
| Olesoxime | SMA | Preclinical, clinical | Efficacy in motor improvement and safety have been confirmed; it could be administered in combinatorial therapy | [148] |
| Curcumin | ALS | Preclinical, clinical | Increased lifespan and slowed illness progression, although more research on various delivery techniques is required | [149] |
| Flavonoids | ALS | Preclinical | Motor performances improvement [prevention of MN (motor neurons) loss] | [145] |
| Quercetin | AD | Preclinical | Improvements in cognitive capabilities, antioxidant activity, MMP and mitochondrial morphological restoration, a reduction in ROS, a rise in ATP levels, and suppression of apoptosis | [149] |
| Resveratrol | AD | Preclinical, clinical | Cognitive decline mitigation | [149] |
| Pramipexol | PD | Preclinical | Neuroprotection (mPTP opening prevention and a decrease in ROS production) | [150] |
ALS: amyotrophic lateral sclerosis; 6-OHDA: 6-hydroxydopamine; MMP: matrix metalloproteinases; MN: motor neurons; SKQ1: Visomitin; SMA: spinal muscular atrophy; TH: thyroid hormone
In addition to producing ATP, mitochondria also maintain Ca2+ homeostasis, activate apoptosis, and control the body’s antioxidant defense system. Understanding the pathogenesis of neurological illnesses requires an understanding of how mitochondrial function, neural activity, and energy metabolism interact biologically. There is strong evidence that the pathophysiology of numerous neurodegenerative disorders is heavily influenced by oxidative damage, DNA mutations, mitobiogenesis, mitophagy, mitochondrial dynamics, metabolism, and mitochondrial interactions with other organelles. Redox dysregulation is a result of genetic and environmental risk factors in neuropsychiatric diseases. Emerging targets for the creation of novel medications for neuropsychiatric diseases include genes that control mitochondrial dynamics. Preclinical research has also shown that increasing endogenous antioxidant defense mechanisms and respiration rates, such as CoQ10, vitamins, acetyl L-carnitine, and L-lipoic acid, can protect mitochondria against inflammation and apoptosis and is being investigated for the treatment of neurological disorders. Preclinical investigations deepen our understanding of the relationship between mitochondrial dysfunction and neurological illnesses, but more clinical evidence is required to fully comprehend the pathophysiology of brain disorders. Clinical trials that looked at novel drugs for the treatment of neurological conditions brought on by mitochondrial malfunction are now complete. There is a critical need for greater research in the area of treating mitochondrial dysfunction and the creation of more potent treatments. The goal of this paper was to create a conversation on the main ways that mitochondria affect neurodegenerative disorders. Currently, a few of these systems are thought to be key targets for pharmaceutical therapies to prevent neurodegeneration. A wider window for intervention will be provided by research into other factors that affect how mitochondrial functions are controlled in neural cells.
AD: Alzheimer’s disease
ALS: amyotrophic lateral sclerosis
ATP: adenosine triphosphate
Aβ: amyloid-β
CoQ10: coenzyme Q10
COX: cytochrome c oxidase
DISC-1: disrupted-in-schizophrenia-1
Drp1: dynamin-related protein 1
ETC: electron transport chain
G6P: glucose-6-phosphate
G6PD: glucose-6-phosphate dehydrogenase
GSH: glutathione
HD: Huntington’s disease
HK1: hexokinase 1
MCU: mitochondrial calcium uniporter
MCUR: mitochondrial calcium uniporter regulator
mPTP: mitochondrial permeability transition pore
mtDNA: mitochondrial deoxyribonucleic acid
NAD+: nicotinamide adenine dinucleotide
NF-κB: nuclear factor kappa B
NMDARs: N-methyl-D-aspartate receptors
NRF1: nuclear respiratory factor 1
OMM: outermost membrane of mitochondria
OXPHOS: oxidative phosphorylation
PARP: poly adenosine di-phosphate-ribose polymerase
PD: Parkinson’s disease
PDH: pyruvate dehydrogenase
PET: positron emission tomography
PGC-1: proliferator-activated receptor-coactivator-1
PPAR: peroxisome proliferator-activated receptors
ROS: reactive oxygen species
SIRT1: sirtuin 1
TCA: tricarboxylic acid
TSPO: translocator protein
MY and AK contributed conception and design of the study; N Singh, MD and NR organized the paper; N Sharma performed the reference setting and formating; MY wrote the first draft of the manuscript; JD, N Sharma, PD and NM wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The authors are thankful to Science and Engineering Research Board-Department of Science and Technology (SERB-DST, New Delhi, Letter number PDF/2018/002605) and GD Goenka University, Gurgaon, 122103, India [Seed Grant (R&D.12/21/01)] for the financial support to get the access to the journals which are paid and the data included in this paper are from these journals and provided funds to attend conferences and workshops to collect data for this paper.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Neha Minocha ... Parijat Pandey
Sangeet Kumar Mall ... Md Sabir Alam
Rouchan Ali ... Pooja A. Chawla
Rosy Yesela Mancilla Santa Cruz ... María Segunda Aurora Prado
Tausif Alam
Abhishek Chaurasiya ... Pooja A. Chawla
Dilpreet Singh ... Pooja A. Chawla
Shikha Choudhary ... Md Sabir Alam