 Review
Review

Affiliation:
Shanghai Frontiers Science Center of Genome Editing and Cell Therapy, Shanghai Key Laboratory of Regulatory Biology, School of Life Sciences, East China Normal University, Shanghai 200241, China
Affiliation:
Shanghai Frontiers Science Center of Genome Editing and Cell Therapy, Shanghai Key Laboratory of Regulatory Biology, School of Life Sciences, East China Normal University, Shanghai 200241, China
Email: yplai@bio.ecnu.edu.cn
ORCID: https://orcid.org/0000-0001-7445-6738
Explor Immunol. 2021;1:418–431 DOI: https://doi.org/10.37349/ei.2021.00028
Received: August 12, 2021 Accepted: December 01, 2021 Published: December 31, 2021
Academic Editor: Masutaka Furue, Kyushu University, Japan
The article belongs to the special issue Cross Talk Among Skin Cells and Immune Cells
Cutaneous homeostasis is maintained by dynamic cellular communications between different cell types in the skin through interactions with various mediators, including cytokines, chemokines and antimicrobial peptides/proteins (AMPs). Keratinocytes, as the major cell type of the epidermis, not only form a passive physical barrier, but also actively participate in the pathogenesis of many, if not all, inflammatory skin diseases. Keratinocytes highly interact with immune cells to shape, amplify or regulate inflammatory responses, thus triggering and/or sustaining these inflammatory skin diseases. In this review, crosstalk between keratinocytes and immune cells is summarized, and its contributions to two major inflammatory skin disorders including psoriasis and atopic dermatitis are highlighted.
Skin is the first line of host defense against danger signals from the environment. Cutaneous homeostasis and skin defense are maintained by dynamic cellular communications between different cell types in the skin through interactions with various mediators, including cytokines, chemokines and antimicrobial peptides/proteins (AMPs) [1]. Keratinocytes, as a major cell type of the epidermis in the skin, are derived from skin epithelial stem cells in the basal layer, and then differentiate into different keratinocyte layers above the basal zone, such as spinous, granular, and cornified layers [2]. To balance proliferation and epidermal thickness, keratinocytes undergo apoptosis and apoptotic keratinocytes contribute to stratum corneum (SC) formation [3]. Keratinocytes not only passively form a physical barrier against pathogens, but also can sense danger signals and initiate a primary inflammation to protect hosts. Moreover, keratinocytes are capable of responding to inflammatory mediators produced by immune cells to amplify or maintain inflammatory responses in the skin. Accumulating evidence has suggested that keratinocytes are actively involved in the pathogenesis of many, if not all, inflammatory skin diseases. We herein summarize the crosstalk between keratinocytes and immune cells, and highlight its contributions to two major inflammatory skin disorders including psoriasis and atopic dermatitis (AD).
Psoriasis is a common chronic inflammatory skin disease characterized by a considerable thick epidermis and a dense dermal infiltration of immune cells [4]. The debate about which cells, keratinocytes or immune cells, trigger psoriasis lasts over two decades. Undoubtedly, no matter who drives psoriasis, the crosstalk between keratinocytes and immune cells plays essential roles during the development of psoriasis [5]. Here we will discuss two different hypotheses about how the crosstalk between keratinocytes and immune cells drives psoriasis.
Early in 1991, Nickoloff [6] proposed the idea of the “cytokine network” in psoriasis. In this “cytokine network” hypothesis, keratinocytes and inflammatory cells, such as T cells and dendritic antigen-presenting cells, interact with each other to drive cutaneous inflammation, and keratinocytes were indicated to act as “signal transducers” to convert exogenous stimuli into the production of cytokines, adhesion molecules, and chemotactic factors responsible for initiation of “antigen-independent” cutaneous inflammation [7, 8]. The observation from Haase group that mice with the specific ablation of inhibitor kappa B kinase 2 (IKK2) in keratinocytes (K14-Cre/Ikk2FL/FL) spontaneously developed psoriasis-like pathologies in αβ T-cell-independent way by upregulating of tumor necrosis factor (TNF) receptor 1 (TNFR1)-dependent interleukin 24 (IL-24) and activating signal transducer and activator of transcription 3 (STAT3) in keratinocytes [9, 10] also suggests that the keratinocytes can initiate inflammatory responses in psoriasis. The latter discovery that inducible epidermal deletion of JunB/c-Jun in adult mice and Rag2-deficient JunB/c-Jun double-mutant mice developed a phenotype resembling the pathology of psoriasis [11] confirmed this hypothesis. Moreover, several groups demonstrated that genetic defects of caspase recruitment domain family member 14 (CARD14) [12–15] in keratinocytes can directly cause psoriasis via regulating IL-17A-induced mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-kB) activation to produce pro-inflammatory factors [16]. These observations suggest that keratinocytes can be considered as a trigger of cutaneous inflammation in psoriasis, and cytokines produced by keratinocytes play key roles in the initiation of inflammation in psoriasis.
In 2007, Gilliet and colleagues [17, 18] confirmed that keratinocytes initiated the onset of psoriasis via secreting the AMPs LL-37 (also called hCAP18) conjugated with self-DNA or RNA to activate Toll-like receptor 9 (TLR9) or TLR7/8 in plasmacytoid dendritic cells (pDCs) after skin injury. Activated pDCs released interferon alpha (IFNα)/IFNβ to trigger initial activation of myeloid dendritic cells (mDCs) and IL-17-producing T cells [18, 19]. Activated IL-17-producing T cells were then recruited into lesional skin and produced multiple cytokines such as IL-17 and IL-22 to establish a cytokine milieu that dictates specific gene signatures in keratinocytes, which, thus, overexpress several inflammatory mediators to amplify local immune reactions [20–22].
After that, our group also found that skin injury activated TLR3 in keratinocytes to produce multiple pro-inflammatory cytokines, such as TNF, IL-6 and IL-36 [23, 24]. IL-36 in turn induced keratinocytes to produce the antimicrobial protein regenerating islet-derived protein 3 alpha (REG3A). REG3A acted back on keratinocytes to inhibit keratinocyte differentiation, thus inducing epidermal hyperplasia [25]. Furthermore, IL-36 induced the pathological IL-23/IL-17 axis to mediate the crosstalk between dendritic cells (DCs) and IL-17-producing cells [26]. Besides IL-36, IL-6, together with transforming growth factor beta (TGFβ), have been shown to induce naive CD4+ T cells differentiation into T helper 17 (Th17) cells, while TNF activates DCs to produce IL-23 or induces keratinocytes to constantly produce chemokines such as chemokine (C-C-motif) ligand 20 (CCL20) and chemokine (C-X-C-motif) ligand 1 (CXCL1) to recruit immune cells to the site of injury [27–31], thus triggering skin inflammation in psoriasis (Figure 1). Therefore, the pathogenesis of psoriasis has once been conceptualized into an initiation by trauma (Koebner response) [17, 32].
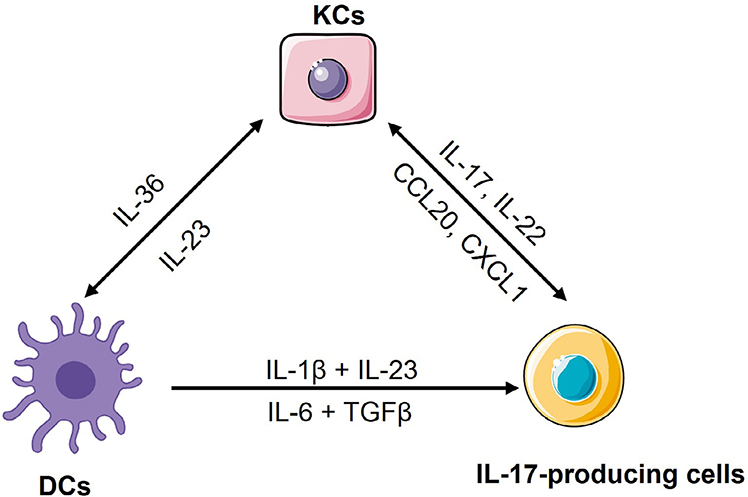
The crosstalk among keratinocytes, DCs and IL-17-producing cells. Upon skin injury, keratinocytes produce IL-36 to activate mDCs for releasing IL-1β, IL-6 and IL-23. In mice, IL-6, together with TGFβ, induce naive CD4+ T cells differentiation into Th17 cells, while in human, IL-1β together with IL-23, induce naive CD4+ T cells differentiation into Th17 cells. These cytokines also induce and/or activate other IL-17-producing cells to produce IL-17, IL-22 and other Th17 cytokines. Among these Th17 cytokines, IL-17 and IL-22 induce keratinocytes to produce chemokines such as CCL20 and CXCL1 for recruiting neutrophils and more IL-17-producing T cells into sites of inflammation in the skin, or induce keratinocytes to produce more IL-36, thus forming a feed-forward inflammatory loop. KCs: keratinocytes
Taken together, the above observations demonstrate that keratinocytes are capable of initiating local inflammation by recruiting and activating immune cells, and the crosstalk between keratinocytes and immune cells is critical for the initiation and progression of psoriasis.
Although our group and other groups have shown that the overactivation of keratinocytes in response to skin injury can trigger psoriasis, several mechanisms indicated that hyperproliferation and aberrant differentiation of keratinocytes are secondary phenomenon induced by immune activation [33], especially in a “feed-forward” mechanism of psoriasis [34]. In this mechanism, keratinocytes have been thought as a responder of inflammatory mediators released by DCs and/or IL-17-producing cells [35]. Mechanically, in lesional skin, activated mDCs by IL-36 produce high amount of IL-1β, IL-6 and IL-23 via unclear mechanisms [36–38]. IL-1β and IL-23 drive human naive CD4+ T cells differentiation into Th17 cell [39], while IL-6, together with TGFβ, promotes murine naive CD4+ T cells development into Th17 cell [40], and then IL-23 activates Th17 cells to produce IL-17A, IL-17F, IL-22 and other cytokines [17, 18, 41–43]. Among these cytokines, IL-17 cytokines induce keratinocytes or other innate immune cells to produce a plethora of cytokines [44–46], chemokines [47] and AMPs [25, 48, 49] to promote the expansion of Th17 cells, γδT cells and group 3 innate lymphoid cells (ILC3) or recruit neutrophils and more IL-17-producing T cells into sites of inflammation in the skin [26, 50–53] (Figure 1). In addition to inflammation, IL-17, IL-22 or IL-23 can maintain the acanthosis that is a hallmark of psoriasis. For example, IL-17A directly acts on keratinocytes to induce the expression of REG3A to promote keratinocyte hyperproliferation and epidermal hypoplasia [25]. Thus, the crosstalk between keratinocytes and the IL-17-producing T cells facilitates both immune and non-immune functions that drive cutaneous inflammation and epidermal hyperproliferation for developing the full-blown psoriasiform phenotype, and unceasing crosstalk between keratinocytes and immune cells comprises a positive feed-forward mechanism and further amplifies local inflammatory responses [5, 53]. The recent finding that mice with a specific deletion of IL-17 receptor A (IL-17RA) in keratinocytes, but not in T cells, neutrophils, or macrophages, were largely protected from the inflammatory response [54, 55] further confirms that this feed-forward inflammatory loop is critically sustained by the crosstalk between keratinocytes and immune cells via IL-17RA. All these data also suggest that targeting IL-17A/IL-17RA or IL-23 can cease the crosstalk between keratinocytes and immune cells, and would be effective treatment strategy for psoriasis patients [53]. Mounting clinical trials support the hypothesis, and show that antibodies targeting IL-17A/IL-17RA or IL-23, such as secukinumab, ixekizumab, brodalumab, tildrakizumab, guselkumab and risankizumab, have achieved tremendous success in the treatment of psoriasis [34, 53, 56].
Although psoriasis can be successfully treated by using the above biologics to cease the crosstalk between keratinocytes and immune cells, skin lesions often recur after withdrawal of these biologics [57–60]. Memory T cells residing in the skin have been considered as a major driver of psoriasis relapse [61, 62]. However, how skin resident memory T cells (TRM) are activated to reinitiate inflammation in psoriasis remains largely unknown. In addition to TRM, keratinocytes with stemness from the basal layer have recently been found to gain inflammatory memory during inflammation [63, 64]. Would keratinocytes with inflammatory memory be involved in psoriasis relapse? Can “memory” keratinocytes crosstalk with TRM to trigger psoriasis relapse? More efforts are required to address these questions.
AD is another chronic, relapsing and Th2 cell-driven inflammatory skin disorder characterized by barrier disruption, intense pruritus and eczematous lesions [65]. Therefore, Th2-type inflammation and barrier dysfunction are thought to be essential for the pathogenesis of AD [4]. So far, there are two views of AD pathogenesis, i.e. “inside-out” versus “outside-in” views [66]. In both views, keratinocytes are the key cell type that link barrier defects and the Th2 response in AD. Here we will summarize these two views in details.
Increasing evidence shows that keratinocytes from patients with AD produce increased amounts of multiple chemokines and cytokines compared with healthy cells, which mediate the crosstalk between keratinocytes and multiple immune cells. For example, compared to keratinocytes in normal patients, keratinocytes from lesional skin of AD patients produce the much higher level of thymic stromal lymphopoietin (TSLP) to potently activate CD11c+ DCs [67]. TSLP-activated DCs, on one hand, produce the Th2-attracting chemokines, thymus and activation-regulated chemokine (TARC, also known as CCL17) and macrophage-derived chemokine (MDC, also known as CCL22) to recruit Th2 cells into lesional skin of AD; on the other hand, prime Th2 cells to produce Th2 cytokines such as IL-4, IL-5 and IL-13 [68, 69]. These Th2 cytokines either induce and amplify the inflammation in eosinophils [70] or directly induce an immunoglobulin isotype switch to immunoglobulin E (IgE) in B cells [71], and then IgE triggers mast cells and basophils to release a diverse group of biologically active products including histamine and leukotriene C4 (LTC4) [72]. Moreover, these Th2 cytokines can directly drive keratinocytes to further produce more TSLP [73] or inhibit the expression of filaggrin (FLG), a protein involved in keratinocyte differentiation [74–76], to impair barrier formation of the skin [77, 78]. In addition to TSLP, keratinocytes from lesional skin of AD highly express IL-33 and IL-25 [79, 80]. IL-33 can activate Th2 cells and ILC2s to produce type 2 cytokines through a receptor complex consisting of IL-33-specific receptor ST2 and a co-receptor IL-1 receptor accessory protein (IL-1RAcP), or actives basophils to release IL-4 and then activates ILC2 [81]. IL-25 produced by keratinocytes also has a dual role in both inducing the Th2 response and inhibiting FLG synthesis in keratinocytes [82]. Moreover, both IL-33 and IL-4 can induce Th2 cells or mast cells to release IL-31 [83, 84], and then IL-31 promotes pruritus and scratching behavior in AD [85]. Scratching the skin, in turns, further induces the expression and/or release of IL-33 and IL-25 from keratinocytes [81, 86, 87]. Therefore, the crosstalk between keratinocytes and immune cells forms a positive feedback loop to drive the pathogenesis of AD (Figure 2). This is historically referred to as “the inside-out” model of AD [88].
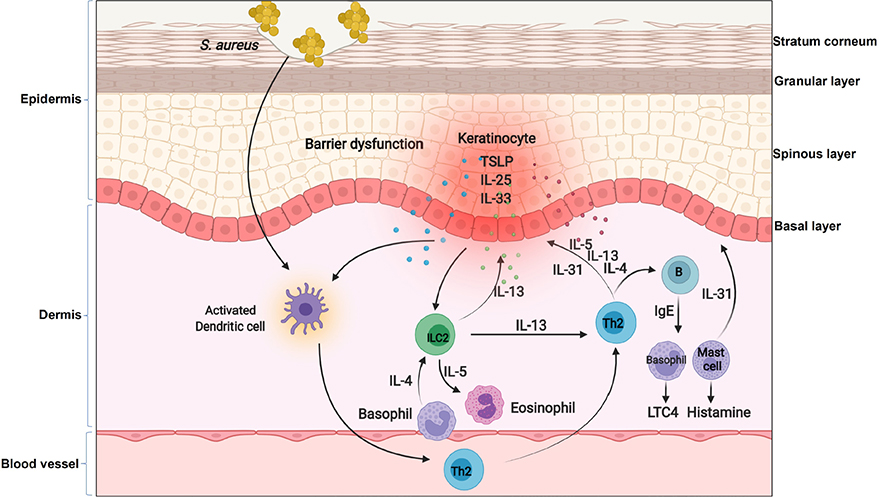
The inflammatory loop in AD. In the context of AD, keratinocytes produce cytokines such as TSLP, IL-25 and IL-33. These cytokines on one hand activate DCs to produce chemokines for recruiting Th2 cells and ILC2 into lesional skin, on the other hand directly active basophils to release IL-4 and then activate ILC2. Th2 cells and ILC2 produce IL-4, IL-5 and IL-13 to induce an immunoglobulin isotype switch to IgE in B cells, and then IgE triggers mast cells and basophils to release a diverse group of biologically active products including histamine and LTC4. Moreover, these Th2 cytokines can inhibit FLG expression in keratinocytes or further induce keratinocytes to produce TSLP. Furthermore, the genetic defects in keratinocytes impair skin barrier and facilitates Staphylococcus aureus (S. aureus) entry through the epidermal barrier. Penetrated S. aureus enables them to come direct contact with viable immune cells and enhances the expression of Th2 cytokines such as IL-4
Besides dysregulated immune responses, a defective barrier function is also a well-recognized feature of AD. Individuals with inherited aberrant production either in serine protease (SP)/antiprotease expression or FLG usually develop moderate to severe AD [66]. The strongest evidence for a primary structural defect of SC involved in the pathogenesis of AD derives from the link between loss-of-function mutations in the gene encoding keratinocyte FLG and AD [66, 74, 89]. Mutations in FLG are common in Northern European patients with AD [66, 90]. These patients have permeability barrier abnormality, as loss of intracellular protein FLG alters keratinocyte differentiation to disrupt the organization of the extracellular lamellar bilayers and decreases SC hydration [66]. This disrupted skin barrier leads to increased penetration of cutaneous antigens [91] or dysbiosis [92] in AD, which is involved in the subsequent inflammatory loop of the skin, regardless of its being a cause or a consequence in AD. For example, a loss-of-function mutation in FLG facilitates S. aureus entry through the epidermal barrier. Penetrated S. aureus enables them to come direct contact with viable immune cells and enhances the expression of Th2 cytokines such as IL-4 [93] (Figure 2). IL-4 inhibits the expression of FLG as well as multiple AMPs, such as human beta defensin 2 (HBD2), HBD3 and LL-37 [94], thus further impairing skin barrier and defense. Moreover, S. aureus releases phenol-soluble modulin alpha (PSMα) to induce the expression of IL-1α and IL-36α in keratinocytes [95], and this in turn stimulates γδT cells and ILC3 to produce IL-17, and then promoting skin damage and inflammation resembling AD [96, 97]. Therefore, the converging pathogenic features caused by FLG mutations lead to the development of specific strategies to restore barrier function in patients with AD, and topical application of moisturizers or barrier repair products has been shown to correct both the permeability barrier abnormality and normalize immune dysfunction induced by the S. aureus penetration [66, 93]. Altogether, these data suggest that primary inherited barrier abnormalities caused by the aberrant differentiation of keratinocytes not only stimulate downstream paracrine mechanisms that could further compromise permeability barrier function, but also cause dysregulated immune responses to drive the inflammatory loop in AD.
From the above statements, both views of AD pathogenesis highlight the crosstalk between keratinocytes and immune cells (including basophils, mast cells, ILC2s, DCs and Th2 cells) plays a definitive role in the induction and propagation of AD inflammation. Multiple cytokines, such as IL-4 and IL-13, link this crosstalk. Therefore, targeting these cytokines and their signaling pathway would effectively treat AD. Dupilumab, an IL-4Rα antagonist that inhibits both IL-4 and IL-13 signaling, and tralokinumab, an IL-13 inhibitor, have been approved for the treatment of AD [98, 99]. However, AD is a more complexed inflammatory skin disease with a wide spectrum of clinical phenotypes. Therefore, treatment of AD is still under great challenge. Moreover, relapse is common in AD patients, and the underlying mechanism of AD relapse needs further exploration. Whether the crosstalk between “memory” keratinocytes and resident TRM would be involved in AD relapse warrants further investigation.
Besides psoriasis and AD, there are several other forms of inflammatory skin diseases, such as cutaneous lupus erythematosus (CLE), acne vulgaris, pemphigus and pyoderma gangrenosum (PG). In these skin disorders, the crosstalk between keratinocytes and immune cells also plays critical roles in the pathogenesis of diseases. Here we summarize how the crosstalk between keratinocytes and immune cells are involved in other inflammatory skin disorders.
It is known that type I IFNs-mediated inflammation plays a critical role in the development of CLE. Ultraviolet is a common trigger for CLE and induces keratinocytes to produce type I IFNκ or induces keratinocyte apoptosis and necrosis [100]. The necrotic keratinocytes release DNAs, and DNAs complex with LL37 to stimulate the production of IFNα/β by pDCs. These type I IFNs further upregulate CLE-typical autoimmune nuclear autoantigens such as Sjogren’s-symdrome-related antigen A (SSA, also called Ro)/Ro52 [101], and then autoantigen-specific T cells recognize these autoantigens to promote autoreactive B-cell activation [102].
The commensal bacterium Cutibacterium acnes (C. acnes, formerly known as Propionibacterium acnes) has been thought as one of triggers of acne vulgaris. This bacterium can activate TLRs 2 and 4 in keratinocytes, and keratinocytes release pro-inflammatory cytokines such as IL-6 and TNF to induce innate immune and inflammatory events [103–105]. The innate immune activation of keratinocytes and the inflammatory milieu they generate in acne environment favor the activation of other cell types, including sebocytes, DCs, macrophages [106] and Th1/Th17 cells in lesions [107]. These events thus lead to the appearance of macroscopic inflammatory acne lesions in puberty.
Pemphigus vulgaris (PV) is an autoimmune blistering skin disease characterized by the loss of epidermal cell-cell adhesion caused by autoantibodies targeting keratinocyte surface antigens such as desmoglein (Dsg). In this disease setting, genetic factors facilitate auto-reactive T cells to induce autoantibody production against desmosomal cadherins, thus leading to keratinocyte dissociation and blister formation [108, 109]. Moreover, PV-derived autoantibodies stimulate keratinocytes to secrete cytokines such as TNF, IL-6 and IL-1α [110, 111], which contribute to the activation of antigen-specific Th cells such as Th2 cells [111]. The activated antigen-specific T cells interact with the B cell to induce the production of autoantibodies, thus triggering the development of PV phenotype.
PG is generally considered as an autoinflammatory skin disease with obvious neutrophil accumulation in skin lesions, although the cause of PG is not well understood. Keratinocytes play key roles in neutrophil accumulation. Because keratinocytes in PG produce high levels of the pro-inflammatory cytokines such as IL-1α, IL-1β, IL-8, IL-36, or chemokines including CXCL9, CXCL10 and CXCL11. IL-8, a major neutrophil chemotactic factor, promotes neutrophil recruitment, while other chemokines attract Th1 and Th17 cells infiltration into the wound periphery or around adnexal structures. These T cells produce IL-17A or other cytokines that in turn act on keratinocytes to induce the expression of various chemokines to recruit neutrophil accumulation or act in concert with TNF and IL-1β to further drive inflammatory pathways [112].
In past decades, there have been enormous progresses in the understanding of the crosstalk between keratinocytes and immune cells in the pathogenesis of inflammatory skin disorders. Keratinocytes express and secrete a variety of cytokines, chemokines, and AMPs to recruit and activate almost any type of immune cells, including DCs, Th1, Th2, and Th17 cells. Activated immune cells express inflammatory mediators to enforce immune responses of keratinocytes. Thus, the crosstalk between keratinocytes and immune cells forms a “feed-forward” or “feedback” loop to sustain the chronicity of inflammatory skin diseases. The recognition of the importance of the crosstalk between keratinocytes and immune cells leads to multiple successful drugs in the treatment of inflammatory skin diseases, such as neutralizing antibodies targeting the IL-23/IL-17 axis for psoriasis treatment [113] and the neutralizing antibody inhibiting IL-4R in the treatment of AD [99, 114]. However, the relapse of skin inflammatory diseases is still out of control. Future efforts are needed to explore the underlying intricate mechanism of the relapse of skin inflammatory diseases. Since both “memory” keratinocytes and TRM contribute to psoriasis relapse and the crosstalk between keratinocytes and immune cells plays key roles in multiple inflammatory skin diseases, whether the crosstalk between “memory” keratinocytes and TRM would be a general mechanism of the relapse of inflammatory skin diseases definitely warrant further investigation.
AD: atopic dermatitis
AMPs: antimicrobial peptides/proteins
CCL20: chemokine (C-C-motif) ligand 20
CLE: cutaneous lupus erythematosus
CXCL1: chemokine (C-X-C-motif) ligand 1
DCs: dendritic cells
FLG: filaggrin
IFNα: interferon alpha
IgE: immunoglobulin E
IL-24: interleukin 24
IL-17RA: interleukin 17 receptor A
ILC3: group 3 innate lymphoid cells
LTC4: leukotriene C4
mDCs: myeloid dendritic cells
pDCs: plasmacytoid dendritic cells
PG: pyoderma gangrenosum
PV: pemphigus vulgaris
REG3A: regenerating islet-derived protein 3 alpha
S. aureus: Staphylococcus aureus
SC: stratum corneum
TGFβ: transforming growth factor beta
Th17: T helper 17
TLR9: Toll-like receptor 9
TNF: tumor necrosis factor
TRM: resident memory T cells
TSLP: thymic stromal lymphopoietin
YL conceived the manuscript. YL and XN wrote the manuscript, and YL and XN edited the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The work in Yuping Lai’s group is supported by National Natural Science Foundation of China (82071785 & 31670925), National Key Research and Development Program of China (2016YFC0906200 & 2016YFC0906202 to YL), and ECNU Multifunctional Platform for Innovation (011). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Yen Hai Vu ... Gaku Tsuji
Masutaka Furue, Mihoko Furue
Mariko Seishima ... Kuniaki Saito
Ichiro Katayama ... Mari Wataya-Kaneda
Kanami Orihara
