 Review
Review

Affiliation:
1Department of Dermatology, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan
ORCID: https://orcid.org/0000-0001-5469-021X
Affiliation:
1Department of Dermatology, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan
2Research and Clinical Center for Yusho and Dioxin, Kyushu University, Fukuoka 812-8582, Japan
Email: furue@dermatol.med.kyushu-u.ac.jp; furuemasutaka00@yahoo.co.jp
ORCID: https://orcid.org/0000-0002-2967-1073
Affiliation:
1Department of Dermatology, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan
2Research and Clinical Center for Yusho and Dioxin, Kyushu University, Fukuoka 812-8582, Japan
ORCID: https://orcid.org/0000-0003-1472-1864
Explor Immunol. 2021;1:4–15 DOI: https://doi.org/10.37349/ei.2021.00002
Received: January 05, 2021 Accepted: January 26, 2021 Published: April 30, 2021
Academic Editor: Dominique J Charron, Hôpital Saint-Louis, France; Reem Al-Daccak, Hôpital Saint-Louis, France
The article belongs to the special issue Cross Talk Among Skin Cells and Immune Cells
 A Correction to this article was published on 7 November 2024
A Correction to this article was published on 7 November 2024
Atopic dermatitis (AD) is characterized by skin barrier disruption, type 2 immune dysregulation, chronic pruritus, and abnormal colonization by Staphylococcus aureus (S. aureus). Tapinarof, an aryl hydrocarbon receptor modulator, has been demonstrated to attenuate the development of AD in clinical studies. Recently, we found that tapinarof upregulated the expression of filaggrin and loricrin, which are essential proteins in skin barrier functions. Paradoxically, tapinarof induced interleukin (IL)-24 secretion by normal human keratinocytes. IL-24 is produced by T helper 2 lymphocytes and keratinocytes following stimulation by type 2 cytokines, and IL-24 is upregulated in the skin of patients with AD. Furthermore, IL-24 contributes to skin barrier disruption and hyperplasia in AD, and it may exacerbate skin inflammatory responses, itch, and S. aureus infection. In this review, we summarized the current findings regarding the detrimental role of IL-24 in AD, thereby suggesting that co-treatment of tapinarof with therapeutics that block IL-24 signaling may represent a promising strategy for managing AD.
Atopic dermatitis (AD), a common inflammatory skin disease, is associated with significant physio-psychological and socioeconomic burdens in affected patients [1]. AD is characterized by skin barrier disruption, type 2 immune dysregulation, chronic pruritus, and abnormal colonization by Staphylococcus aureus (S. aureus) [2, 3]. Among the emerging therapeutic agents for AD, topical tapinarof, an aryl hydrocarbon receptor (AHR) modulator, has been revealed to attenuate disease activity clinically [4–6]. Recently, we demonstrated that tapinarof upregulated the expression of filaggrin (FLG) and loricrin (LOR), which are important skin barrier-related proteins. Paradoxically, tapinarof stimulated human keratinocytes to secrete interleukin (IL)-24 [7]. Although IL-24 expression is upregulated in the epidermis in patients with AD [8, 9], its implications in the pathogenesis of AD remain poorly investigated. To date, IL-24 has been extensively studied in cancer and demonstrated to induce apoptosis in several different cancer cells without harming normal cells [10]. In addition, IL-24 was implicated to play a pivotal pathogenic role in inflammatory diseases such as psoriasis, arthritis, and inflammatory bowel disease. Furthermore, IL-24 was shown to have a potential role in type 2 inflammation-related allergic diseases, such as allergic rhinitis, asthma, and AD [11]. IL-24 contributes to skin barrier disruption and hyperplasia in AD and may promote skin inflammatory responses, itch, and S. aureus infection. Therefore, IL-24 may exacerbate AD lesions. In this review, we summarized the importance of IL-24 in the pathogenesis and treatment of AD.
IL-24 gene overexpression was detected in the lesional skin of patients with AD [12]. IL-24 protein expression was upregulated from the basal layer to the spinous cell layer, but not in the granular and cornified layers, in the epidermis of skin tissues from patients with AD [9]. In murine AD models, IL-24 expression was enhanced in the epidermis of mite-treated mice [9] and IL-4 transgenic mice [8]. No data have been presented regarding IL-24 serum levels in patients with AD; however, the serum levels of this cytokine did not differ between IL-4 transgenic and wild-type mice, suggesting the local role of IL-24 in AD pathophysiology [8].
IL-24 can be produced by immune cells, including monocytes, macrophages, mast cells, natural killer cells, and T and B lymphocytes, as well as non-immune cells, such as keratinocytes and melanocytes, in response to certain stimuli [10, 11] (Figure 1).
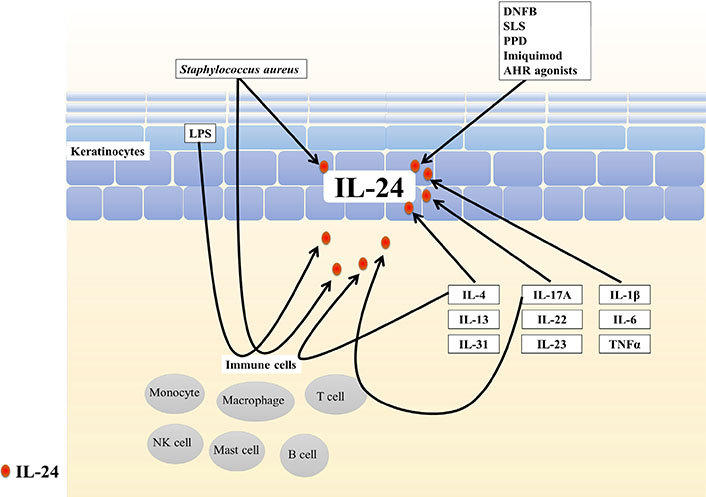
Sources of IL-24 in the skin. IL-24 can be produced by immune cells, including monocytes, macrophages, mast cells, natural killer cells, and T and B lymphocytes, and non-immune cells such as keratinocytes in response to certain stimuli. DNFB: 2,4-dinitrofluorobenzene; SLS: sodium lauryl sulfate; PPD: paraphenylenediamine; LPS: lipopolysaccharide
2,4-Dinitrofluorobenzene, which was revealed to induce AD-like pathology in mice [13], upregulates IL-24 expression in murine skin [14]. Sodium lauryl sulfate (a chemical irritant in human skin) stimulates IL-24 production in normal human keratinocytes [15]. IL-24 was overexpressed in an allergic contact dermatitis model induced by paraphenylenediamine exposure, in human and mouse skin [16]. IL-24 expression was also increased in imiquimod-stimulated HaCaT cells, an immortalized keratinocyte cell line [17]. Phorbol myristate acetate and ionomycin also induced IL-24 in T helper 2 (Th2) lymphocytes [10].
Some studies determined that IL-24 is the target cytokine of some oxidative environmental AHR agonists, such as 2, 3, 7, 8-tetrachloro-dibenzo-p-dioxin (TCDD), benzo(a)pyrene, particulate matter, and ultraviolet B irradiation, in primary human chorionic stromal cells, human lung adenocarcinoma cells, normal human bronchial epithelial cells, and normal keratinocytes, respectively [15, 18–21]. In line with these findings, we recently demonstrated that tapinarof, an antioxidative AHR modulator [22], stimulated normal human epidermal keratinocytes to generate IL-24 protein, whereas AHR depletion significantly attenuated the upregulation of IL-24 induced by tapinarof [7]. The AHR-binding sequence (GCGTG) is present in the promoter region of the IL-24 gene [18, 23]. These results indicated that AHR activation plays a vital role in the mechanism of AHR ligand-mediated IL-24 induction.
Staphylococcal strains, both pathogenic (S. aureus) and commensal strains (S. epidermidis and S. saprophyticus), induce IL-24 mRNA and protein expression in human peripheral blood mononuclear cells [24]. The expression of IL-24 was also upregulated by S. aureus infection in human keratinocytes [25].
Lipopolysaccharide, a microbial product and Toll-like receptor activator, stimulates monocytes [26], macrophages [24, 27], and activated T cells [28] to produce IL-24. Pellino 1 ubiquitin E3 ligase is activated by innate pattern-recognition receptors such as Toll-like receptors. Overexpression of Pellino 1 induced a significant increase in the expression of IL-24 in HaCaT cells [29].
It is widely accepted that the Th2 cytokines IL-4 and IL-13 are driving factors in AD pathogenesis, as the blockade of IL-4/IL-13 signaling by dupilumab is effective in patients with severe AD [30]. The IL-24 gene has been identified as a dominant Th2 lineage-specific gene [31].
IL-4 increases IL-24 expression in normal human keratinocytes [9, 15], human monocytes [27], and T lymphocytes [28]. IL-24 expression is increased in the inflammatory skin lesions of IL-4-transgenic mice compared with that in wild-type mice [8]. IL-4 functions synergistically with IL-2 or lipopolysaccharide to induce IL-24 expression in natural killer (NK) cells and macrophages [32].
IL-13 upregulates periostin production via signal transducer and activator of transcription 6 (STAT6) activation, and periostin enhances IL-24 production in human keratinocytes [9]. STAT6 is critical for IL-4/ IL-13-induced IL-24 expression [9, 31]. In Th2 cells, the transcription factors STAT6, GATA3, and the activator protein-1 (AP-1) family member c-Jun were revealed to participate in the regulation of IL-24 production [31, 33, 34].
IL-31 stimulates keratinocytes to produce IL-24. The inhibition of Janus kinase (JAK), p38, and extracellular signal-regulated kinases (ERKs), but not c-Jun N-terminal kinases or phosphoinositide 3-kinases, interfere with the upregulation of IL-24 induced by IL-31 [35].
IL-17A upregulated IL-24 expression in normal human keratinocytes [15, 36]. Chong et al. [37] found that IL-17A induced IL-24 production in Th17 cells. Considering that IL-17A signals through the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway [38] and two potential NF-κB binding sites are present in the IL-24 gene, the researchers proposed that the binding of IL-17A to its receptor activates NF-κB signaling, leading to the transcription of IL-24 in Th17 cells [37]. IL-22 induced the production of IL-24 in normal human keratinocytes [15, 39, 40]. IL-17 synergistically induced the expression of IL-24 in keratinocytes with IL-22 or tumor necrosis factor alpha (TNFα) [36, 39, 40]. The injection of IL-23 into mouse skin elevated IL-24 expression [41].
IL-1β stimulation induced IL-24 protein secretion in human keratinocytes [14, 15, 42]. Inhibition of the p38 mitogen-activated protein kinase (MAPK) pathway was reported to significantly decrease IL-1β–induced IL-24 expression by reducing IL-24 mRNA stabilization, suggesting that p38 MAPK regulates IL-24 gene expression at the post-transcriptional level [43–45].
IL-6 induced the production of IL-24 in human keratinocytes [15, 46]. Suppressor of cytokine signaling-3 (SOCS3) inhibited JAK1, JAK2, and tyrosine protein kinase 2 but not JAK3, and it downregulated STAT3 activation [47]. The dysregulation of SOCS3 resulted in excess STAT3 activation in response to IL-6, leading to the upregulation of IL-24 expression by keratinocytes [48]. In an experimental model, physical stimulation (i.e. shaving) induced IL-24 expression at the site of stimulation in SOCS3 deficient mice, but not in control mice [48].
TNFα also upregulated IL-24 expression in keratinocytes [15, 49, 50–52]. Anti-TNFα antibody treatment almost completely inhibited IL-24 expression in psoriatic skin, indicating that IL-24 production is mainly driven by TNFα in psoriasis. Reactive oxygen species-activated ERK signaling was illustrated to mediate the upregulation of IL-24 in response to TNFα stimulation in keratinocytes [52].
The human IL-24 gene is located on chromosome 1q32-33. IL-24, together with IL-19, IL-20, IL-22, and IL-26, belongs to the IL-20 cytokine subfamily and IL-10 family, which further includes IL-10, IL-28, and IL-29. IL-24 signals through two heterodimeric receptors: IL-20R1/IL-20R2 and IL-22R1/IL-20R2. Notably, IL-24 shares the same two receptors with IL-20 and shares IL-20R1/IL-20R2 with IL-19 receptor, indicating that IL-24 may have partially overlapping biological activities with IL-19 and IL-20 [53].
In most immune cells, only IL-20R2 is expressed, whereas IL-20R1 and IL-22R1 are undetectable [14]. By contrast, these three receptor subunits are all expressed in several non-hematopoietic cells, such as keratinocytes and bronchial epithelial cells [14, 54–56]. Interestingly, IL-24–induced STAT3 activation was detected in keratinocytes but not in peripheral blood mononuclear cells, probably because of the lack of IL-24 receptor expression in immune cells [14]. These findings suggest a preferential effect of IL-24 on non-immune cells, including keratinocytes.
IL-24 binds to its receptor, leading to specific activation of JAK1/tyrosine protein kinase 2-STAT3 and ERK1/2 MAPK signaling pathways in keratinocytes [14, 15, 53, 54, 56]. In addition, STAT3 activation in keratinocytes is elevated in the skin of patients with AD [9].
As mentioned previously, the cardinal features of AD include skin barrier disruption, immune dysregulation, chronic pruritus, and abnormal skin colonization by pathogens. The following sections will discuss the role of IL-24 in AD concerning these features (Figure 2).
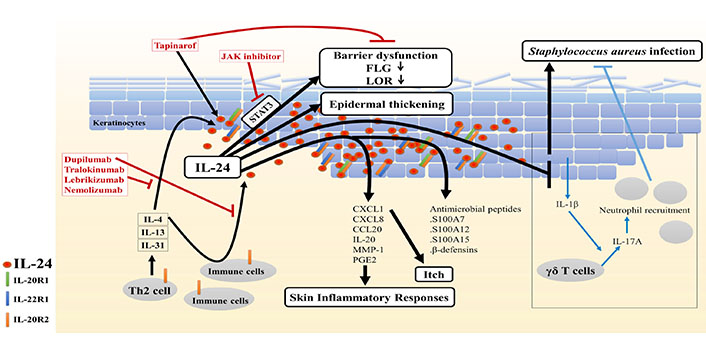
Potential role of IL-24 in AD. Type 2 cytokines stimulate immune cells and keratinocytes to secrete IL-24, leading to the upregulation of IL-24 expression in the epidermis of AD skin. Furthermore, IL-24 contributes to skin barrier disruption and hyperplasia in AD, and it may exacerbate skin inflammatory responses, itch, and S. aureus infection. Therefore, therapeutics that suppress IL-24 signaling, including the IL-4/IL-13 antagonist dupilumab, IL-13 antagonists tralokinumab and lebrikizumab, IL-31Rα antagonist nemolizumab, and JAK-STAT inhibitors, might be promising for managing AD, including combination use with tapinarof
Skin barrier dysfunction is a critical factor in the pathogenesis of AD. The disrupted barrier permits allergen penetration, accelerating immune response, pruritus, and trans-epidermal water loss [3]. In the epidermal barrier, FLG and LOR, markers of late differentiation of keratinocytes [3], are essential barrier-related proteins. They aggregate and align keratin bundles, contributing to skin barrier strength and integrity. In addition, FLG degradation products form a natural moisturizing factor, which has a role in skin hydration and barrier function [57]. In AD, overexpressed IL-4/IL-13 repressed FLG and LOR expression, leading to the impaired terminal differentiation and barrier function of the epidermis [2, 7]. IL-4/IL-13–mediated barrier dysfunction may be, at least in part, attributable to IL-24 because type 2 cytokine-induced IL-24 downregulates FLG and LOR expression via STAT3 [7, 9]. Depletion of IL-24 significantly restored the IL-13–induced downregulation of FLG [9]. Calmodulin-like 5 (CALML5) was revealed to be highly expressed in the differentiating epidermis. CALML5-knockout keratinocytes exhibit deficient FLG and LOR expression [58]. It was reported that IL-4 and IL-13 pathways converged on p63 to diminish CALML5 and FLG expression [59]. Interestingly, because IL-24 was also illustrated to inhibit the gene expression of CALML5 [39], the possible involvement of p63 in IL-24 signaling needs to be further studied. Collectively, these results indicate that IL-24 is a pivotal mediator of abnormal epidermal differentiation downstream of type 2 cytokine signals in AD.
Common inflammatory skin diseases, including AD, feature keratinocyte hyperproliferation [60, 61]. IL-24 was revealed to induce normal human epidermal keratinocyte proliferation in an epidermal growth factor receptor-independent manner, as blockade of this receptor in the monolayer culture system did not inhibit IL-24–mediated keratinocyte proliferation. However, keratinocyte proliferation might be further augmented through the upregulation of the EGF family ligands amphiregulin and heparin-binding epidermal growth factor induced by IL-24. IL-24 also increased the thickness of the reconstituted human epidermis [54]. Histological analysis revealed that IL-24 transgenic mice had a compact stratum corneum and markedly thickened epidermis compared with the findings in wild-type mice [42]. Additionally, IL-24 was reported to contribute to epidermal hyperplasia induced by IL-23, a major cytokine implicated in psoriasis, in mice [41]. Furthermore, expression of the epidermal proliferation-associated proteins (keratin 16 and S100As, particularly S100A7) was increased in IL-24–treated reconstituted human epidermis [54].
In summary, these data suggest that IL-24 induces abnormal epidermal differentiation and hyperplasia, contributing to skin barrier dysfunction in AD.
IL-24 might participate in a complex cascade of cytokines involved in skin inflammatory responses, as IL-24 can induce the expression of several cytokines and chemokines. When added to cultured human keratinocytes and/or reconstituted human epidermis, IL-24 upregulated the expression of many inflammatory mediators, including chemokine (C-X-C motif) ligand (CXCL)1 and IL-20 gene expression and CXCL8/IL-8, chemokine (C-C motif) ligand (CCL)20/macrophage inflammatory protein (MIP)-3α, matrix metalloproteinase (MMP)-1, and prostaglandin E2 (PGE2) protein secretion [15, 40, 52, 54]. Notably, IL-24 transgenic epidermis exhibited elevated induction of monocyte chemoattractant protein-1, a critical chemokine that mediates recruitment of monocytes/macrophages [42]. Furthermore, it was reported that IL-24-deficient mice were partially protected against allergic contact dermatitis and epidermal inflammatory infiltrate, particularly neutrophils, after exposure to paraphenylenediamine [16]. Altogether, these results indicate that IL-24 can promote skin inflammation, thereby contributing to the maintenance and exacerbation of AD.
Intense itch induces scratching and skin lesion exacerbation that disturbs the quality of life of patients with AD [62]. The role of IL-24 in pruritus remains poorly explored. IL-31, a major pruritogenic cytokine in AD [62], induced IL-24 gene expression in keratinocytes [35]. IL-24 acts as an activator of STAT3, which is also activated by IL-31 to promote elongation of the nerve fibers, followed by enhanced itching [63]. Additionally, IL-24 upregulated the expression of CXCL1 [54], a chemokine that can evoke itch through multiple pathways [64, 65]. These data suggest the involvement of IL-24 in the mechanism of pruritus in AD.
The skin of patients with AD is prone to microbial infections [66]. Atopic skin is preferentially colonized by S. aureus, and its colonization further contributes to disease exacerbation [67, 68]. Myles et al. [25] reported that S. aureus-induced IL-24 inhibits the induction of IL-1β in keratinocytes, followed by a decrease in IL-17A expression in γδ T cells, leading to reduced neutrophil recruitment and eventually resulting in more severe infection. They also suggested that IL-24 can promote cutaneous infection by S. aureus and that IL-20R2 blockade may have therapeutic potential for patients with S. aureus infection [25]. Conversely, in the human epidermis system, IL-24 was illustrated to induce several antimicrobial peptides that have a protective role against microbial infection, including S100A7, S100A12, S100A15, and β-defensins [52, 54]. These controversial findings imply that the anti-infection effects of IL-24 may vary depending on the infection conditions and timing.
The aforementioned evidence suggests that IL-24 plays a significant role in the pathogenesis of AD and supports the potential efficacy of therapeutics that block the upstream and downstream pathways of IL-24 signaling. Dupilumab, an anti–IL-4Rα monoclonal antibody that inhibits IL-4 and IL-13 signaling, is efficacious in the treatment of moderate-to-severe AD. The drug significantly improves the clinical severity of AD. In parallel, the upregulation of IL-24 is suppressed in the lesional and non-lesional skin of patients with AD after 4 and 16 weeks of treatment with dupilumab [69]. It remains unclear whether other emerging drugs such as the IL-13 antibodies tralokinumab and lebrikizumab [70–72] and anti-IL-31Rα antibody nemolizumab [70, 73] affect IL-24 expression in AD.
JAK inhibitors, such as topical delgocitinib and oral baricitinib, have demonstrated efficacy against AD, mainly owing to their inhibitory action on JAK/STAT signaling, and they also possess the potential to inhibit the IL-24 signaling [74–76].
Tapinarof has been illustrated to attenuate the disease activity of AD in clinical studies [4–6]. Our recent study demonstrated that tapinarof upregulates FLG and LOR expression in an AHR-dependent manner in human keratinocytes, thereby improving barrier dysfunction in AD. Paradoxically, tapinarof induced the secretion of IL-24, and IL-24 sequentially activates JAK–STAT3 signaling, which negatively regulates FLG and LOR expression, partially via AHR activation. Therefore, inhibition of the IL-24–STAT3 axis during AHR activation using JAK inhibitors may further increase FLG and LOR expression induced by tapinarof treatment.
Therefore, co-treatment of tapinarof with JAK a inhibitor may be a promising strategy for managing AD [7].
IL-24 is an integral part of the pathogenic pivotal cytokine network in AD. Type 2 cytokines stimulate immune cells and keratinocytes to secrete IL-24. IL-24 contributes to epidermal barrier disruption and hyperplasia. By inducing several inflammatory mediators, IL-24 may orchestrate and stimulate skin inflammation and itch. Additionally, IL-24 may promote cutaneous infection by S. aureus. These findings underpin the fundamental role of IL-24 in the development of AD. Targeting the upstream and downstream pathways of IL-24 signaling represents a potential strategy for treating AD.
AD: atopic dermatitis
AHR: aryl hydrocarbon receptor
CALML5: calmodulin-like 5
CXCL: chemokine (C-X-C motif) ligand
ERK: extracellular signal-regulated kinase
FLG: filaggrin
IL-24: interleukin-24
JAK: Janus kinase
LOR: loricrin
MAPK: mitogen-activated protein kinase
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
SOCS3: suppressor of cytokine signaling-3
STAT: signal transducer and activator of transcription
Th2: T helper 2
TNF: tumor necrosis factor
YHV wrote the first draft of the manuscript. GT and MF reviewed the manuscript. All authors contributed to manuscript revision, and all authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2021.
Copyright: © The Author(s) 2021. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 14037
Download: 286
Times Cited: 0
Masutaka Furue, Mihoko Furue
Mariko Seishima ... Kuniaki Saito
Ichiro Katayama ... Mari Wataya-Kaneda
Kanami Orihara
Xinhui Ni, Yuping Lai
