 Mini Review
Mini Review

Affiliation:
1Department of Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL 33136, USA
2Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA
Email: nstrbo@med.miami.edu
ORCID: https://orcid.org/0000-0003-2152-2433
Affiliation:
1Department of Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL 33136, USA
ORCID: https://orcid.org/0009-0001-3866-6751
Affiliation:
1Department of Microbiology and Immunology, University of Miami Miller School of Medicine, Miami, FL 33136, USA
2Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA
Email: dfrasca@med.miami.edu
ORCID: https://orcid.org/0000-0002-9816-6679
Affiliation:
3IDI-IRCCS Istituto Dermopatico dell’Immacolata, 00167 Rome, Italy
ORCID: https://orcid.org/0000-0002-0916-7769
Explor Immunol. 2025;5:1003218 DOI: https://doi.org/10.37349/ei.2025.1003218
Received: April 21, 2025 Accepted: September 25, 2025 Published: September 29, 2025
Academic Editor: Francois Niyonsaba, Juntendo University, Japan
The article belongs to the special issue Immunosenescence: Mechanisms and Its Impact
The skin covers the entire surface of the body and therefore is the largest organ in humans. The skin has various functions, primarily defence from infections and trauma. With aging, profound changes occur that compromise its key functions, leading to impaired barrier protection and immune responses. This is in part due to the increased low-grade systemic inflammation known as inflammaging, driven by senescent cells, and release of pro-inflammatory cytokines, to which the skin also significantly contributes. As a consequence of inflammaging, the skin’s function is compromized. The cellular and molecular components involved are summarized in this review.
Skin is the largest organ within the human body and represents the first line of defense against pathogens, toxins and trauma, but also cancer and autoimmunity. In addition, it prevents water loss and regulates temperature and metabolism [1].
The immune system of the skin includes a large variety of immunocompetent, innate (dendritic cells, innate lymphoid cells, NK and Langerhans’ cells) and adaptive cells (B and T lymphocytes) that work together in a coordinated manner with skin resident cells (keratinocytes, fibroblasts, adipocytes and endothelial cells).
The skin is made of multiple layers: the epidermis (the superficial cell layer), the dermis (layer beneath the epidermis), the hypodermic or subcutaneous tissue (deepest layer), as well as several cutaneous appendages, such as hair follicles, sweat glands, and sebaceous glands [2]. Within each of these layers, immune cells communicate with other cells to maintain homeostasis within the skin microenvironment. Innate immune cells also provide the first protection against pathogens and toxins, rapidly detecting them and responding to them by secreting products with broad anti-microbial activities. In addition, antigen-presenting cells in the skin present antigens directly to B and T cells, leading to the secretion of antibodies and generation of memory responses to previously encountered microbes, and therefore providing long-term protection.
Keratinocytes, the predominant cells in the epidermis, act as sentinels and are crucial specialized components of the immune system of the skin [3]. Through their interactions with skin immune cells such as alpha-beta (αβ) and gamma-delta (γδ) skin-resident T cells [4], keratinocytes secrete cytokines, chemokines, and anti-microbial peptides that modulate the immune landscape of the skin [5]. Keratinocytes are also involved in the first step of vitamin D metabolic pathway, in which pro‐vitamin D3 (7‐dehydro‐cholesterol) is converted into vitamin D3, catalysed by ultraviolet (UV) light B, UVB. Vitamin D is a crucial modulator of immune responses and its metabolism contributes to barrier immunity [6].
The skin undergoes profound changes with age, summarized in Figure 1. Skin aging is a complex biological process where both intrinsic and extrinsic factors play a role, determining progressive structural and functional changes. While intrinsic aging is primarily driven by genetic and hormonal factors, extrinsic aging is mostly due to environmental exposure to UVs, pollution, and lifestyle factors, including diet, smoking habits and alcohol consumption [7, 8]. It has also been shown that metabolic processes regulate skin aging, as they influence cellular energy, nutrient availability and removal of cellular waste (dead cells, damaged proteins and lipids).
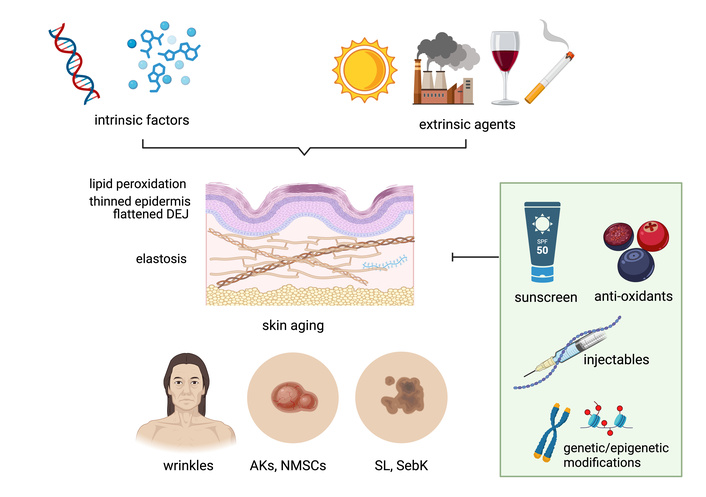
Overview of the key intrinsic and extrinsic factors driving skin aging, the underlying cellular and histological mechanisms, clinical manifestations, and potential preventive strategies. AKs: actinic keratoses. DEJ: dermoepidermal junction. NMSC: non-melanoma skin cancer. SebK: seborrheic keratosis. SL: solar lentigo. Created in BioRender. Paganelli, A. (2025) https://BioRender.com/8jgpgym.
The immune landscape of the skin is significantly remodeled during aging. One of the most relevant alterations that occur with age, as the skin becomes drier and thinner due to decreased production of collagen and elastin [9], is the decrease in the secretion of fat-soluble defensins, a family of small peptides which help maintain the skin’s defense against microbes by dysrupting bacterial membranes, blocking bacterial cell wall synthesis and inducing toxin neutralization [10]. Other reasons leading to age-related increase in trans-epidermal water loss and de-hydration are changes in the dermal-epidermal junction, which loses its structural integrity with age, decreased secretion of natural moisturizing factors, including sebum and decreased cellular turnover.
Exposure to sunlight and UVs over time damages the skin and accelerates skin aging. A recent analysis of photo-exposed skin biopsies from women 20–70 years of age revealed significant age-related biological changes. These included thinning of the epidermis, shortened rete ridge pathlength (projections of the epidermis that contact the dermis), an increase in the damage response biomarker p53-binding protein 1, and a dysregulation of gene expression, with a decrease in differentiation and senescent genes in older ages and an increase in genes associated with hypoxia and glycolysis [11]. Clinically, photoaged skin displays deep wrinkles, uneven pigmentation, telangiectasia, rough texture, and loss of firmness. Over time, photoaging predisposes to a range of benign, premalignant, and malignant cutaneous lesions. Seborrheic keratoses, for example, are benign epidermal tumors frequently observed in aging skin. These lesions typically present as waxy, pigmented plaques with a “stuck-on” appearance. Despite their occurrence not being closely related to chronic sun exposure, their prevalence typically increases with age [7]. Solar lentigines, commonly referred to as age spots, appear as well-demarcated, hyperpigmented macules on sun-exposed sites, particularly the face, forearms, and dorsum of the hands. These lesions result from localized melanocyte hyperactivity and increased melanin deposition, without cellular atypia. While benign, their presence indicates cumulative photodamage and an increased risk for other UV-induced lesions [12]. Similarly, actinic keratoses (AKs) are among the earliest clinical markers of photodamage. These erythematous, scaly macules arise on chronically sun-exposed areas. Histologically, AKs exhibit atypical keratinocytes confined to the lower layers of the epidermis, with potential to progress to squamous cell carcinoma (SCC) [7]. Importantly, chronic photodamage and cumulative DNA mutations in skin cells contribute to the development of non-melanoma skin cancers (NMSC), either SCC or basal cell carcinoma (BCC) [13, 14]. These manifestations reflect the cumulative effects of intrinsic and extrinsic aging processes and underscore the importance of therapeutic strategies to maintain skin health.
Skin remodeling with age compromises its key functions, leading to impaired barrier function and immune responses. This is in part due to a phenomenon known as inflammaging [15], the chronic, low-grade systemic inflammatory status driven by senescent cells and their continuous release of pro-inflammatory cytokines, associated with the decline of immune function over time [16]. These age-related changes disrupt the structural integrity of the cutaneous immune system, needed to maintain effective skin immune responses [17].
The skin itself is an initiation site of the inflammatory response and a contributor to inflammaging. Skin inflammation starts following barrier disruption and exposure to exogenous substances that then penetrate deeper, inducing a strong local inflammatory response mainly involving pro-inflammatory immune cell types. It has indeed been demonstrated that keratinocytes contribute to many inflammatory skin disorders, due to their capacity to secrete pro-inflammatory mediators, a reason for which they have been identified as “cytokinocytes” [18].
Although there are indications that inflammaging contributes to age-related immune decline, its exact mechanisms remain poorly understood, as many of the underlying pathways and interactions are still being uncovered. This uncertainty is reflected in age-associated skin conditions and diseases such as cutaneous SCC [19], atopic dermatis [20], and diabetic wound healing [21].
The skin of older individuals is enriched in cells characterized by the senescence-associated secretory phenotype (SASP). The SASP is responsible for the secretion of multiple soluble factors (inflammatory cytokines, chemokines, growth/survival/hemostatic factors, extracellular matrix macromolecules, proteases, ceramides, bradykinins, damage-associated molecular patterns) [22]. Many of these SASP factors disrupt the extracellular matrix and the normal function of the skin [23, 24] and can lead to chronic inflammatory conditions such as atopic dermatitis [25] and psoriasis [26]. External triggers often exacerbate these changes through the induction of oxidative stress and the activation of matrix metalloproteinases, further contributing to collagen degradation and cytokine release [27].
Skin cells undergoing immunosenescence experience significant changes in their phenotype and function. Cell senescence, initially described by Hayflick and Moorhead in 1961 [28], indicates the irreversible arrest of cell proliferation after exposure to different stressors [29, 30], like DNA damage, telomere shortening, X-rays, metabolic stressors [22, 31], chronic viral infections, industrial toxicants [32], and chemotherapic drugs [33, 34]. Senescent cells are characterized by the expression of p16INK4a (cyclin-dependent kinase inhibitors 2A, CDKN2A) and/or p21CIP1/WAF1 (cyclin-dependent kinase inhibitor 1A, CDKN1A) [35], and/or the senescence-associated β-galactosidase (SA-β-gal) [35], the enzyme that catalyzes the hydrolysis of β-galactosides into monosaccharides, and by the capacity to acquire the SASP [22, 36]. Although senescent cells are experiencing irreversible proliferation arrest, they are metabolically active, with intrinsic metabolic pathways supporting their secretory function. Cellular senescence plays a pivotal role in skin aging and is broadly driven by complex molecular mechanisms such as damage, telomere shortening, epigenetic remodeling, and dysfunctional immune cells. Not surprisingly, reduced cellular proliferation and activity are the hallmarks of cutaneous aging [24, 37]. Histologically, this manifests as a flattened dermoepidermal junction (DEJ), epidermal thinning, decreased collagen and elastin content [23]. Moreover, extracellular-matrix components undergo aging-related structural changes, determining solar elastosis, characterized by the accumulation of abnormal elastic fibers in the dermis, and increased collagen fragmentation [13]. Of note, altered lipid composition in the stratum corneum contributes to xerosis and barrier impairment typical of aged skin [7, 12].
Both stromal and immune cells in the skin display senescent phenotypes. Dermal senescent fibroblasts that accumulate in the skin with age express multiple SASP markers and are characterized by disruption of collagen homeostasis, delayed wound healing, reduced anti-microbial immunity and increased susceptibility to skin tumorigenesis [14]. Keratinocytes become shorter and fatter with age, and have reduced proliferation, differentiation and responses to exogenous stimuli [38]. Skin macrophages that are immunosenescent shift to a pro-inflammatory phenotype (M1) [39]. This pro-inflammatory status drives chronic inflammation and oxidative stress, disrupting immune regulation and tissue homeostasis. The decline in anti-inflammatory pathways, including reduced antigen presentation and phagocytosis, further exacerbates homeostatic imbalance [40].
Little is known about age-related changes in skin-resident αβ T cells, although a higher ratio CD4+/CD8+, with a decreased frequency of CD45RA+ naive T cells and an increased expression of PD1 on CD4+ T cells, has been reported in the skin of old subjects, suggesting a reduced generation of long-lived memory T cells [41]. However, it has also been reported that skin T-cell immunity is maintained more efficiently in advanced age as opposed to circulating T-cell memory [42]. Skin T cells maintain their density, diversity, and protective cytokine production despite the reduced T cell diversity and function in blood, with a shift towards more differentiated memory (effector and exhausted) T cells [43]. Interestingly, skin resident cytotoxic CD4+ T cells can efficiently eliminate senescent cells by targeting cytomegalovirus (CMV) antigen [44]. γδ T cells exhibit increased levels of expression of immune activation markers such as CD69, increased production of pro-inflammatory cytokines and a decreased proliferative response to exogenous stimuli [45]. Given the established role of γδ T cells in the anti-microbial response and in keratinocyte proliferation and migration upon wounding, our group recently described that human skin γδ T cells express novel intracellular antimicrobial protein Perforin 2 (P2), which expression is modulated by different microbes [46–49]. We found that P2 modulates morphology of DETC (dendritic epithelial T cells) and we have also shown that aged P2KO mice show significantly higher levels of systemic and intrinsic B cell inflammation, which are negatively associated with protective antibody responses to a vaccine [50]. Importantly, since aging is associated with susceptibility to numerous bacterial pathogens, including Staphylococcus aureus, Pseudomonas aeruginosa and γδ T cells are a crucial part of protection, we postulate that loss of γδ T cell cytotoxic function will most likely have a significant impact on the anti-bacterial host defence in the elderly, a topic that requires further investigation. With aging, Vδ2+ T cells, subset of more “innate-like” γδ T cells, are predominantly depleted, while “adaptive-like” Vδ1+ T cells are accumulating in the blood [51]. In addition, accumulation of terminally differentiated and senescent Vδ1+ T cells with age and with CMV seropositivity is similar to accumulation of CD8 αβ T cells [52, 53]. Frequencies and numbers of T regulatory cells were also shown to be increased in aged hosts, which may be linked to weakened antigen-specific T cell responses and also associated with reactivation of infectious conditions [54], which all potentially contribute to age-related inflammation.
As a result of immune dysfunction associated with immunosenescence, the skin’s ability to maintain homeostasis and respond to stressors is compromised, particularly evident in its impaired ability to respond to injury and initiate proper healing. Wound healing is a critical process for maintaining skin integrity, as skin is vulnerable to breaches that must be promptly sealed.
The normal wound healing process comprises four distinct phases: hemostasis, inflammation, proliferation, and maturation (tissue remodelling). Briefly, after hemostasis, clotting and vasoconstriction occur, the inflammatory immune response takes place, during which immune and non-specialized cells mobilize to clear bacteria and debris. Shortly after, the proliferation phase begins, initiating the re-epithelialization, angiogenesis and regeneration of damaged or missing tissue structures [55]. Lastly, the remodeling phase is the final stage of wound healing, where the wound matures and strengthens as temporary tissue is replaced with more permanent structures [56].
Aging increases the number of platelets that adhere to the injured epithelium. After adhesion, pro-inflammatory cytokines (PDGF/TGF-α/TGF-β) are secreted in large amounts and neutrophils and monocytes are recruited to the site of injury [57]. Recruitment of these cells is reduced, however, in the skin of aged versus younger controls, due to the reduced expression of adhesion molecules on the surface of both cell types [58].
In the skin of young individuals, neutrophils and macrophages secrete ROS, which are involved in microbe killing and prevention of infections in the wounds, as well as in vasoconstriction and reduction in blood flow [59]. Conversely, in the skin of old individuals, neutrophils and macrophages secrete significantly higher amounts of ROS, leading to sustained inflammation, and increased oxidative stress, protein breakdown and DNA damage [60]. These, altogether, will impair and delay wound healing through increased apoptosis and senescence. In the skin of old individuals, increased numbers of senescent cells that secrete higher amounts of IL-6, IL-1α and matrix metalloproteinases accumulate, and these contribute to the hostile and pro-inflammatory microenvironment that delays epithelization and leads to chronic inflammation and delay in wound closure [60]. In the early stages of normal wound healing, M1 macrophages upregulate pro-inflammatory markers (IL-12α/TNF-α/IL-6/IL-1β) and nitric oxide synthase-2. M2-macrophage anti-inflammatory markers (CD206/CD163/CCL17/Arginine/TGF-β1) are up-regulated at later stages of wound healing [61]. Predominance of M1 phenotype during immunosenescence significantly impacts wound healing, resulting in delays in the healing process and related complications.
Fibroblasts are one of the main contributors to the process of tissue remodeling. Fibroblasts facilitate several processes, such as helping to dissolve the fibrin clot, producing new extracellular matrix components, and synthesizing collagen to provide structural support for healing tissues [62]. In chronic inflammatory conditions, including aging, senescent fibroblasts exhibit pro-inflammatory and immunosuppressive characteristics [63]. These senescent fibroblasts express SA-β-gal, p16INK4A and SAHF (senescence-associated heterochromatin foci) [63], which are all key markers of cellular senescence in tissues. Single-cell transcriptomics has confirmed these phenotypic changes, as the expression of fibrous extracellular matrix components significantly declined in fibroblasts with aging, while inflammatory mediators are markedly increased [63]. Cultured human dermal fibroblasts isolated from aged individuals had an increase in senescent markers such as SA-β-gal and p16INK4A, while alpha-smooth muscle actin and collagens were decreased. These changes ultimately impact signaling pathways, such as an impairment in the activation of the TGF-β/SMAD3 signaling pathway [64], suggesting fibroblasts transition from a tissue-repairing phenotype to a more inflammatory state in aged tissues, although research still needs to better characterize this transition and how to target it.
γδ T cells play a key role in coordinating wound healing by releasing cytokines and growth factors (KGF-1/KGF-2/IL-13/IFN-γ/TNF-α/IGF-1/IL-2/IL-17), which help to orchestrate the inflammatory response during the initial days of the wound healing process [65, 66]. Studies in mice have shown that γδ T cells, and especially the Vg5+ T cell subset, exclusively expressed in the mouse epidermis, DETC that promotes wound by secreting KGF-1 and IGF-1, are present in comparable numbers in the skin of young and old mice, but after wounding, their numbers significantly decrease in old skin, leading to delayes wound re-epithelization [67]. In humans, Vδ1+ T cells isolated from chronic non-healing wounds have also been shown to be dysfunctional, unable to produce IL-2 and IGF1 upon stimulation [68].
Skin remodeling occurring with age leads to impaired barrier function and immune responses, including a decrease in cutaneous immune cell function [69], which is responsible for a variety of bacterial infections, including those with Staphylococcus aureus, β‐Haemolytic streptococcus, Pseudomonas spp. and Klebsiella spp. [70]. Skin infections by Proteus mirabilis and Pseudomonas aeruginosa [69], as well as by fungi (Candida) and viruses (shingles, herpes simplex virus‐1 and human papillomavirus) [70], have also been reported in people over 65 years of age. Major mechanisms for skin infections include a decrease in the autophagic capacity of immune cells, in the cytotoxic capacity of CD8+ T cells and in macrophage phagocytosis to clear microbes from the tissue. Decreased secretion of anti-microbial products is also significantly affecting the susceptibility of older adults to skin infections.
Skin cancer is among the most common types of cancer worldwide [71]. The age-associated changes summarized in Figure 1, together with inflammaging, have been linked to an increased susceptibility to cancer development. This occurs because of a gradual loss of the reparative capacity of the tissue that leads to the accumulation of cellular damage and cell dysfunction. It has recently been shown that the age-related changes leading the skin to stiffen and be less elastic may also contribute to higher rates of metastatic melanoma in older adults [72]. The proposed mechanism is the increased release of the protein ICAM1 by the skin of older individuals. ICAM1 not only stimulates the growth of blood vessels in the tumor, promoting its growth, but also makes the blood vessels leaky, allowing tumor cells to disseminate. Moreover, senescent cells in the tumor microenvironment may also support the pro-inflammatory milieu and tumor growth/progression [73].
Although the findings summarized in this review clearly demonstrate the effect of aging on the skin, more work is needed to better characterize the cellular and molecular mechanisms involved. Preventative measures, including sun protection and antioxidant use, alongside therapeutic strategies aimed at enhancing collagen synthesis and reducing oxidative damage, are essential in mitigating the effects of skin aging. Despite advancements in imaging technologies and molecular research—including telomere length modulation (with the specific goal in this case to reduce oncogenic risk)—there are currently no effective therapies capable of preventing or halting skin aging. However, because inflammaging and associated immune dysfunction significantly contribute to skin aging, immunomodulation may represent at least one key strategy for both the prevention and treatment of skin aging. The modulation of specific metabolic disorders in the skin may also represent an important anti-aging therapeutic strategy of intervention.
AKs: actinic keratoses
CMV: cytomegalovirus
DETC: dendritic epithelial T cells
P2: perforin 2
SASP: senescence-associated secretory phenotype
SCC: squamous cell carcinoma
UV: ultraviolet
αβ: alpha-beta
γδ: gamma-delta
The authors thank members of Drs. Strbo’s and Frasca’s labs for scientific reasoning and discussion.
NS and DF: Writing—original draft, Writing—review & editing, Funding acquisition. SY and AP: Writing—original draft. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Financial support of Dr. Strbo [NIH/NIDDK R01DK136241] and Dr. Frasca [NIH/NIA R01AG023717]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Fidaa Bouezzedine ... Georges Herbein
Pitu Wulandari
Valquiria Bueno
Rafael Moura Maurmann ... Brandt D. Pence
Roberto Paganelli, Angelo Di Iorio
