 Review
Review

Affiliation:
1Department of Pharmacology, Rungta College of Pharmaceutical Sciences and Research, Bhilai 490024, Chhattisgarh, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0006-7331-933X
Affiliation:
1Department of Pharmacology, Rungta College of Pharmaceutical Sciences and Research, Bhilai 490024, Chhattisgarh, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0006-1879-0989
Affiliation:
1Department of Pharmacology, Rungta College of Pharmaceutical Sciences and Research, Bhilai 490024, Chhattisgarh, India
2School of Pharmacy, Rungta International Skills University, Bhilai 490024, Chhattisgarh, India
Email: sanjay.gupta0311@gmail.com
ORCID: https://orcid.org/0000-0003-1084-3530
Affiliation:
3Department of Polymer Technology, Faculty of Chemistry, Gdansk University of Technology, 80-233 Gdansk, Poland
ORCID: https://orcid.org/0000-0002-7630-5834
Affiliation:
4Chitkara College of Pharmacy, Chitkara University, Rajpura 140401, Punjab, India
ORCID: https://orcid.org/0000-0002-3986-0337
Explor Immunol. 2025;5:1003219 DOI: https://doi.org/10.37349/ei.2025.1003219
Received: April 25, 2025 Accepted: September 02, 2025 Published: September 29, 2025
Academic Editor: Calogero Caruso, University of Palermo, Italy
Eosinophilic granulomatosis with polyangiitis (EGPA) is an uncommon form of necrotizing vasculitis that affects the respiratory system and other organs, characterized by asthma, eosinophilia, and multiple organ involvement that complicates the diagnosis and treatment. There are no definitive open-label clinical data on the diagnosis or treatment of EGPA to guide clinicians. The diverse presentation of the disease, distinct from other eosinophilic disorders; the presence of competing conditions such as allergy and asthma, which also have potential biomarkers; lack of a definitive test to confirm diagnosis; difficulties in obtaining endoscopic biopsies for histologic confirmation, etc., are significant barriers to early detection. Even with recent advances in imaging, immunological approaches, and molecular testing to determine the disease’s identity and characteristics, clinicians still misdiagnose or delay treatment, sometimes leading to life-threatening and irreversible complications. Though EGPA is pharmacotherapeutically controlled with glucocorticoids, it has typically included the use of cytotoxic agents such as cyclophosphamide for induction in cases of severity. Recently, several clinical trials have examined targeted biologic treatments (such as mepolizumab, benralizumab, and omalizumab) and demonstrated that these medications can reduce exacerbations, decrease the need for glucocorticoids, and improve asthma control. New drugs such as dupilumab and new anti-IL-5/IL-5R monoclonal antibodies are being studied in phase II and phase III trials, and these drugs may provide additional avenues for refractory disease. Treatment will be organized based on individualization of treatment strategy, depending on disease severity, organ involvement, and biomarker profile. Vertical investment in multicenter longitudinal studies is necessary to formulate therapeutic algorithms and evaluate new targets.
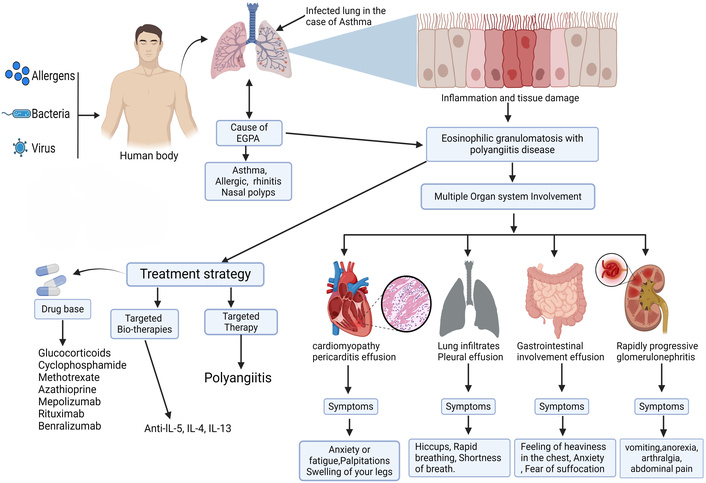
Eosinophilic granulomatosis with polyangiitis (EGPA) is an autoimmune connective tissue disease. Multiple organ systems are affected by vasculitis, which manifests as high eosinophil counts (HEC) in peripheral blood and tissues. Peripheral eosinophilia, defined as a HEC in the blood, and a history of presence of eosinophilic granulomatous inflammation in the lungs, are the two primary indications of EGPA [1]. Despite being classified, anti-neutrophil cytoplasmic antibody (ANCA) affects less than half of individuals. The disease is uncommon; it generally affects asthmatic people and spreads to the skin, lungs, and peripheral nervous system [2]. EGPA is also called Churg-Strauss syndrome, and it can cause people with asthma and other respiratory allergy symptoms to be sick. The exact cause of this condition is still uncertain. It is characterized by the presence of eosinophilic necrotizing vasculitis, extravascular granulomas, and slight to medium-sized arterial infiltration [3]. It was Churg and Strauss who first detailed the connection between allergy symptoms, extravascular granulomas, eosinophilic infiltration, and necrotizing vasculitis [4]. Afterward, Silver observed that many patients lacked some of the pathologic characteristics identified mainly with extravascular granulomas. They developed a clinical diagnosis predicated on the coexistence of extrapulmonary systemic vasculitis, eosinophilia (> 1.5 × 109/L or 1,500/mcL), and asthma in at least two locations [5, 6]. EGPA can present with a variety of symptoms and signs, and some people have more obvious vasculitis. Oral corticosteroids (OCSs) are a cornerstone for EGPA therapy, both in chronic daily and high (pulse) doses. An OCS-sparing effect, longer time to first remission, and a lower relapse rate in anti-IL-5 monoclonal antibody mepolizumab were shown over the phase III trial in patients with EGPA. As such, we do not yet know whether mepolizumab will have an effect on EGPA in people who predominantly suffer from vasculitis symptoms. People with and without a vasculitis EGPA phenotype were studied to determine mepolizumab’s sparing effects [6, 7].
This review aims to provide an outline of the pathophysiology, diagnostic methods, and treatment strategies for EGPA to help clinicians, healthcare agencies, and regulatory authorities in decision-making. Causes of endothelium-related pulmonary artery disease, such as EGPA, an inflammatory illness mostly affecting small and medium-sized arteries as well as the respiratory system, are also included in this study. Signs of it include elevated eosinophil counts in both blood and tissues. Antigens trigger the adaptive immune system, which in turn stimulates eosinophils, T-cells, and B-cells, damaging tissue and blood vessels [6–8].
ScienceDirect, Web of Science, PubMed, and Google Scholar were used as primary web resources in the search. This article mentioned all important available information about eosinophilic granulomatosis with polyangiitis from 1998 until 2025, including research and review publications. About 118 articles were examined thoroughly and found suitable for the required information and hence cited in this article. Articles that were deemed to lack traditional values were not included in the research. This study’s objective was to summarize the current understanding of EGPA, therapeutic approaches, biomarkers, mechanisms of action, routes of administration, pharmaceutical treatments, and diagnostic methods. This article was found using the following keywords: prognosis of EGPA, diagnostic techniques, biomarkers, ANCA mechanisms, mechanisms of action, and EGPA.
EGPA is an uncommon illness and a less frequent kind of vasculitis. Its incidence in the entire population varies from 10.7 to 13 cases/million person-year. The EGPA is more prevalent, with values that range from 34.6 to 64.4 cases/million patients. Unexpectedly, the prevalence of EGPA in asthmatics remains constant regardless of the prior treatments received [9]. EGPA may occur at any age; the typical diagnostic age is 48 years, and there is no significant sex difference in the majority [10]. It is thought that environmental and genetic factors both play a role in EGPA [11].
In patients diagnosed with vasculitis, research identified many risk factors for infection, including smoking, advanced age, renal failure, and low CD4 counts. Infection is a danger for people with vasculitis who also have dialysis, anemia, or hypoalbuminemia. An increased risk of infection is associated with factors that compromise cell-mediated immunity, such as low CD4 counts [12].
The clinical signs of EGPA are diverse and indicative of the multisystemic nature of the condition. EGPA typically advances via three consecutive stages [13]. An initial atopic or allergic phase, an eosinophilic phase came next, and culminated in a vasculitis phase. Patients often present first with allergic rhinitis or asthma, which may develop years before systemic symptoms appear. Allergic symptoms may antedate systemic symptoms by many years, with rhinitis and asthma being the first signs. As the disease progresses, eosinophilia may become more marked and is often associated with tissue infiltration by eosinophils into various organs, including the skin, digestive tract, peripheral nerves, or lungs. Respiratory symptoms, with cough, dyspnoea, and wheezing, are common findings that suggest pulmonary involvement, which may present as mild infiltrates to fulminant eosinophilic pneumonia [14]. Dermatological involvement may manifest as purpura, nodules, and livedo reticularis, whereas gastrointestinal involvement may be presented as abdominal pain, diarrhea, and, in severe cases, colonic perforation [15]. Vasculitic involvement of peripheral nerves may result in neurological findings such as mononeuritis multiplex or peripheral neuropathy. It is wise to understand that this type of inflammation can also cause myocarditis or pericarditis, although they are rare and carry unchanged high morbidity and mortality [16]. Renal involvement is rare, but it can manifest as rapidly progressive glomerulonephritis or it can be relatively benign [17]. Due to the differences in the clinical course, early recognition of the varied clinical manifestations of EGPA is very important for timely intervention and improved outcomes [18].
The diagnostic difficulty of EGPA is attributed to its diverse manifestations of symptoms that may impact different organ systems [19]. Wheezing, coughing, and shortness of breath are common asthmatic symptoms that patients first encounter and may even come on before systemic indications. Skin involvement can include ulcers, livedo reticularis, palpable purpura, and skin nodules. Sensory and motor compromise is a common feature of peripheral neuropathy, and mononeuritis multiplex may range from mild to severe. Respiratory involvement is common in EGPA and ranges from sinusitis and nasal polyps to severe manifestations such as pulmonary infiltrates and exacerbations of asthma. Furthermore, the likely vascular nature of the disease increases its multisystemic involvement, which may include cardiovascular complications such as myocarditis and pericarditis. Someone else may have diarrhea or other stomach symptoms, such as abdominal pain [20]. Considering the wide range of symptoms [21]. EGPA requires a multidisciplinary approach for efficient treatment to enhance patient outcomes and reduce the course of the disease [22].
EGPA has a polyorgan systemic involvement [13]. The most recognized system involved is the respiratory system, with the lungs being one of the most commonly involved organ systems. Respiratory symptoms can worsen if not treated quickly and may progress from mild asthma-like symptoms to severe eosinophilic pneumonia and diffuse alveolar hemorrhage, which can ultimately lead to respiratory failure [23]. In addition to the lungs, skin has a role in the majority of cases of EGPA with nodules, livedo reticularis, and palpable purpura, all signs consistent with small-vessel vasculitis. The cardiovascular involvement of EGPA may include coronary artery disease, myocarditis, or pericarditis, all of which have the potential to be serious complications [24]. Neurological features include peripheral neuropathy or mononeuritis multiplex, which represent peripheral nerve changes/damage due to vasculitis. Involvement can occur in other organ systems as well, with vasculitic changes in the gastrointestinal tract (GIT) and kidneys [25].
EGPA has an impact on several organs, such as the lungs, kidneys, heart, and GIT [22]. Before the vasculitic phase, asthma is frequently a respiratory problem in EGPA patients. Sinusitis and allergic rhinitis are frequent conditions that can develop either before or after asthma [26]. A frequent cause of morbidity and death in EGPA is myocarditis. Arrhythmias or heart failure might manifest as a result. Pericarditis: Chest discomfort and pericardial effusion can result from inflammation of the pericardium. One possible cause of myocardial ischemia or infarction is coronary vasculitis [27]. Peripheral neuropathy is a prevalent symptom that frequently appears as a broader polyneuropathy or as multiple sclerosis. Less frequent indications of CNS involvement are strokes, headaches, and encephalopathy [28]. Lower limbs are typically affected by palpable purpura. In the integumentary system, cutaneous vasculitis causes nodules or ulcers [29–31].
Cardiac function is not standard and can be fatal in EGPA patients, but it can also have serious outcomes. Endomyocardial biopsy showed eosinophilic aggregations in the sub-endocardium, suggesting injury to the ventricular endothelium, which can lead to thrombus formation [32–34]. Although rare, this multisystem vasculitis may cause important cardiac complications [35]. Ocular manifestations occurring in EGPA may include orbital myositis, conjunctival nodules, and blockage of the central retinal vein or artery, ptosis, ischemic optic neuropathy, and dacryocystitis [36]. Chronic or acute inflammation of the sinus and nasal mucosa, accompanied by abnormal mucus production, can be the result of many stressors that can influence the normal functioning of the immune system and/or the vascular tone, both locally and systemically. In certain situations, the inflammatory state of the lower respiratory tract can also be influenced by the upper respiratory tract [37]. The course of EGPA can progress through three distinguishing phases: prodromal, eosinophilic, and vasculitic phases. Each phase is characterized by a separate but closely linked group of manifestations and pathophysiology. The prodromal phase usually begins with sinusitis and asthma. The late phase of EGPA is marked by the development of heart failure, gastrointestinal involvement, and pulmonary infiltrates after 8 to 10 years [22, 38], representative of eosinophil-induced cardiomyopathy. The vasculitic phase of EGPA is characterized by patients with manifestations of glomerulonephritis, palpable purpura, and neuropathy [39].
Significant immunological dysregulation is a consequence of EGPA’s complicated pathogenesis. The eosinophilic and allergic reactions, as well as the granulomatous pulmonary inflammation characteristics, respectively, are caused by the participation of T helper (Th)-1 and Th-2 cells. In EGPA, lower levels of IL-10 show that regulatory T-cells are down, while higher levels of immunoglobulin show that humoral immunity is out of whack [11, 40].
Mucin may promote the proliferation of eosinophils. Mucus pathobiology is also expected to involve mast-cell activation, eosinophil-independent IL-13 production, and non-type 2 (T2) cytokines. Such as non-steroidal anti-inflammatory drug (NSAID)-exacerbated respiratory disorders, eosinophilic bronchiolitis, asthma with an eosinophilic phenotype, and eosinophilic chronic rhinosinusitis (ECRS) [41]. Though occasionally more than one illness develops concurrently or sequentially, most of the NSAIDs have an adult onset. When given systemic corticosteroids (CS) as an initial therapy, mast cells respond well to them [42] and generate precursors of eosinophils. As a chemotactic agent, IL-5 helps eosinophils migrate from bone marrow into the circulation and enhances their survival in tissue. The only cells that express the IL-5R are mast cells, basophils, and eosinophils [43].
ANCA vasculitis is associated with the most frequent systemic involvement of small vessel vasculitides. A predetermined list of cases was utilized to identify individuals with ANCA-associated vasculitis (Figure 1), including those with microscopic polyangiitis (MPA), EGPA [44]. The relationship between interstitial lung disease (ILD) and MPO-ANCA-associated vasculitis (AAV) is widely acknowledged. The diagnosis of AAV is associated with a poor prognosis and might happen simultaneously, post-, or even before pulmonary fibrosis [45]. AAV is an autoimmune-mediated inflammation of tiny blood arteries in several organs, including the kidneys [42]. In contrast EGPA is driven by an eosinophilic mechanism characterizsed by the excessive production of eosinophils, as well as elevated levels of IL-5. The resulting tissue damage is mainly caused by the accumulation of inflammation, chronic rhinosinusitis with eosinophilia and nasal polyps, toxic granules, and pro-inflammatory cytokines. This process leads to inflammation and the formation of granulomas (Figure 2).
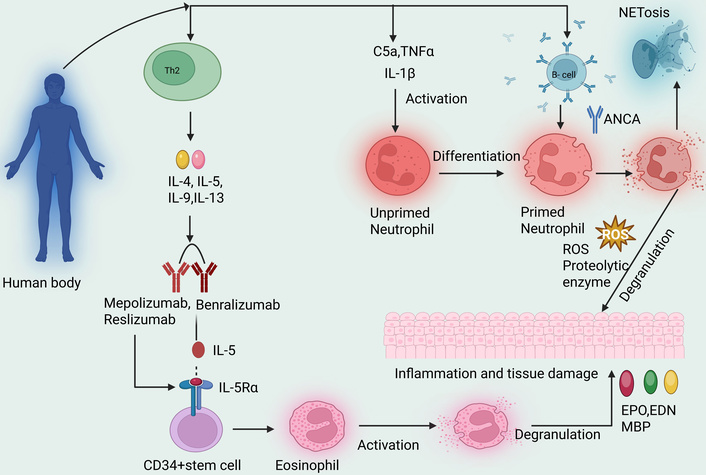
Diagrammatic view of eosinophilic-mediated and ANCA mechanisms of EGPA. ANCA: anti-neutrophil cytoplasmic antibody; EGPA: eosinophilic granulomatosis with polyangiitis; ROS: reactive oxygen species; IL-5Rα: interleukin-5 receptor alpha; CD34+ stem cell: cluster of differentiation 34 positive stem cell (hematopoietic stem cell marker); C5a: complement component 5a; TNFα: tumor necrosis factor alpha; IL-1β: interleukin-1 beta; EPO: eosinophil peroxidase: EDN: eosinophil-derived neurotoxin; MBP: major basic protein; NETosis: neutrophil extracellular trap-induced cell death. Created in BioRender. gupta, a. (2025) https://BioRender.com/1i4bmdi.
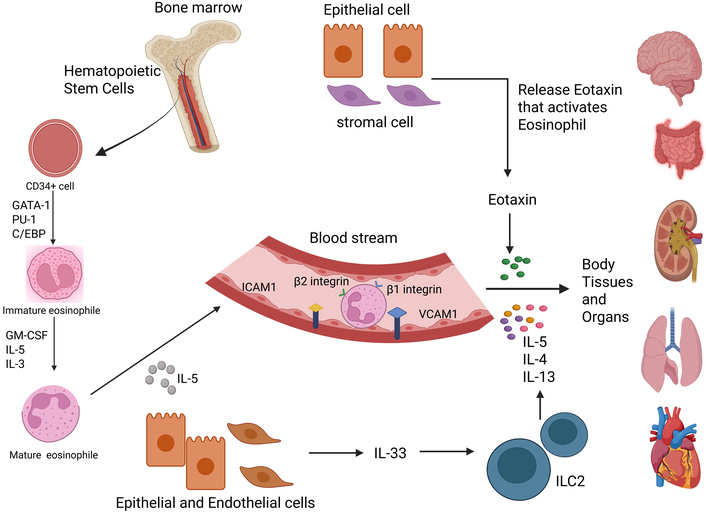
Eosinophilic production, maturation, and migration to the body tissues and organs. Eosinophils develop from cluster of differentiation 34 positive stem cells (CD34+), which are stem cells in the bone marrow. They differentiate through stages defined by the repression of the GATA-1 transcription factor, which regulates eosinophil differentiation, followed by the activation of PU.1, a transcription factor important for myeloid/lymphoid differentiation, and C/EBP, as well as cytokines such as GM-CSF, IL-5, and IL-3. They are released from bone marrow into the bloodstream by IL-5 and make it there with the help of intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) which allow them to bind to β1/β2 integrins. Subsequently, eotaxin, eosinophil chemotactic protein (chemokine that recruits eosinophils and cytokines, IL-4, IL-5, and IL-13, directs the selective recruitment of eosinophils to such inflammatory sites. ILC2: group 2 innate lymphoid cells. Created in BioRender. gupta, a. (2025) https://BioRender.com/tafg04w.
The pathophysiology of EGPA is hypothesized to have a genetic basis and environmental factors, which ultimately result in dysregulation of the immune system and a consequent inflammatory response. Genetically, some cases of EGPA are linked with certain variants of the HLA genes, indicating a genetic predisposition for this disorder. These genetic factors could mediate the regulation of the immune system, as well as promote an abnormal response to certain environmental factors [46].
In susceptible individuals, environmental factors are thought to act as triggers for EGPA. For instance, exposure to some medications has been linked to the change of EGPA in some individuals; these drugs include those used to treat asthma, such as leukotriene receptor antagonists. Infections have also been suggested as potential triggers, but this could be because they provoke an autoimmune response. Environmental allergens could also be involved in EGPA’s increased eosinophils, since such cells are frequently increased in allergic reactions [47]. The interaction between these genetic and ecological factors, therefore, induces the onset of the immune response, which then leads to eosinophilia production, ultimately leading to vasculitis affecting multiple organs. This complexity in pathophysiology highlights the possibility that genetic predisposition and environmental exposure converge to promote both initial eosinophilic proliferation as well as progression of EGPA [48].
Some pathological tests can diagnose the EGPA through the identification of biomarkers, and it can also be diagnosed through imaging or histopathological results, as discussed below in detail.
Currently, there are no accepted diagnostic criteria for EGPA. The proposal from 1984 stated that, in addition to eosinophilia and asthma, the patient should have systemic vasculitis of two or more organs. In 1990, the American College of Rheumatology (ACR) created six criteria classifications for EGPA. Criteria included asthma, eosinophilia greater than 10%, neuropathy, and paranasal sinus abnormalities, nonfixed biopsy-revealed lung infiltrates, and extravascular eosinophils. The categorization standards to distinguish the various vasculitides were also established by the ACR [12, 49]. The ACR criteria, however, may only be used to describe the kind of vasculitis in individuals who have been diagnosed with it, as they were developed for categorization (rather than diagnostic) purposes. In individuals who did not use prednisone, the association between laboratory tests was somewhat significant, but was dramatically decreased when prednisone was used [50]. Every patient at every study visit had the following tests performed at the Vector Control Research Center (VCRC) sites, and standard techniques were used to measure the eosinophil count, C-reactive protein (CRP), and erythrocyte sedimentation rate (ESR). As previously mentioned, ELISA was used to measure the serum levels of TARC (Thymus and Activation-Regulated Chemokine, also known as CCL17) and eotaxin-3, and total IgG and IgG4 content were examined using nephelometry [51].
Extravascular granulomas, eosinophilic infiltrates, and small and medium-sized vessel vasculitis are the most common histological findings in EGPA. An eosinophilic necrotic matrix, including interstitial and vascular granulomas, is encircled by large cells and palisading lymphocytes. The primary foci of the vasculitic process are tiny and medium-sized vessels, particularly small arteries. whether granulomas or eosinophils are present or not [45]. Fibrinoid necrosis of the arterial cell wall characterizes the systemic process, which may occur in small and medium vessels, particularly smaller veins, and is often related to or occurs apart from granulomas and eosinophilic infiltrates [13].
Recent studies have suggested that patients with seronegative EGPA might show ANCA activity in their sputum. This activity is associated with sputum eosinophilia and more severe respiratory symptoms. Sputum ANCA testing is being studied both as a way to reveal a separate subset of patients as well as a potential way to diagnose serum ANCA-negative EGPA [6]. It is essential to conduct a complete pulmonary and respiratory symptom evaluation, which will include pulmonary function testing and baseline imaging of the chest. At the same time, all patients should undergo chest X-rays; CT will allow for a more detailed assessment of pulmonary abnormalities. Confirmation of pulmonary eosinophilia can be made via bronchoscopy and evaluation of inflammatory cells in the bronchoalveolar lavage fluid (BALF). Assessing cardiac involvement, given the poor prognosis in sepsis with the cardiac participation at both diagnosis and relapse, is key; therefore, all patients should undergo a cardiological assessment at baseline. At a minimum, this would include an electrocardiogram (ECG), echocardiogram (ECHO), and recording of serum cardiac biomarkers, specifically BNP and troponin. A 24-hour symptom monitor can help detect arrhythmias that may not be seen with the resting ECG [52]. The differential diagnosis of EGPA involves mostly other eosinophilic disorders and small-vessel vasculitides. Microscopic polyangiitis GPA can generally be differentiated based on clinical presentation and histopathology. Still, at least some patients with EGPA may present with PR3-ANCA positivity and features of granulomatous vasculitis or eosinophilia. Peripheral or regional eosinophilia, although rare, can occur in EGPA [22, 53].
ANCA is an essential player in the pathogenesis of EGPA-associated vasculitis, with about one-third of patients testing positive for myeloperoxidase-ANCA. Conventional therapy for EGPA includes glucocorticoids (GCs), with cyclophosphamide (CYC) added for patients with adverse prognostic features. We identified a range of approaches to EGPA management in this review. The causes, mechanisms, symptoms, and treatment perspectives are given in Figure 3.
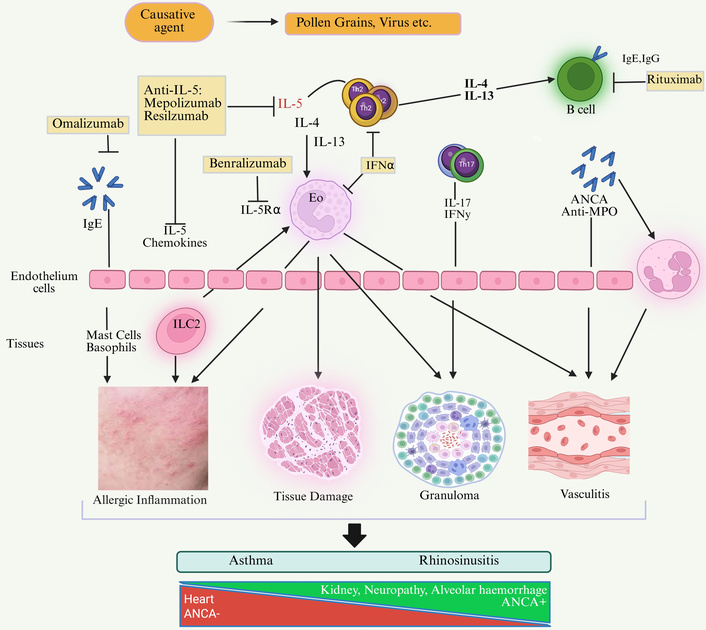
Etiology, symptoms, pathophysiology, and treatment perspectives of asthma and rhinosinusitis. IL-5Rα: interleukin-5 receptor alpha; IFNα: interferon alpha; IFNγ: interferon gamma; Th2: T helper cell type 2; Th17: T helper cell type 17; Eo: Eosinophil; ILC2: group 2 innate lymphoid cells; IgE: immunoglobulin E; IgG: immunoglobulin G; ANCA: anti-neutrophil cytoplasmic antibodies; Anti-MPO: anti-myeloperoxidase antibodies. Created in BioRender. gupta, a. (2025) https://BioRender.com/v8xt1oh.
A number of drugs have been developed for the treatment of EGPA with their unique mechanisms of action and indications (Table 1). EGPA management uses induction treatment with rituximab (RTX) or CYC. CYC is preferred more specifically when there is cardiac involvement. Maintenance treatment (with azathioprine, methotrexate, or mepolizumab, etc.) is initiated approximately two weeks after induction therapy and planned for 18 to 24 months to reduce relapse recurrences and glucocorticoid toxicity. Mepolizumab is currently a first-line option, while glucocorticoid monotherapy is used in mild disease. Interferon-α is rarely used since it has limited benefit compared to the risk of adverse effects, and also poor results in treating patients experiencing severe relapses.
Modern methods for the treatment of EGPA.
| Treatment strategy | Route | Dose/use case | Biomarkers | Mechanism of action | Indication | Results | References |
|---|---|---|---|---|---|---|---|
| GCs | Oral or IV; initial high doses followed by tapering | Prednisone: initial dose 0.5–1 mg/kg/day | Elevated eosinophil count, CRP, ESR | Anti-inflammatory; immunosuppressive, reduces eosinophils | First-line treatment; initial management of all patients | Rapid symptom improvement is essential for initial control | [54, 55] |
| CYC | Oral or IV | 2 mg/kg/day for 3–6 months | ANCA, eosinophil count, CRP | Alkylating agent; immunosuppressive, reduces B and T cells | Life-threatening condition | Effective in reducing remission in severe cases | [56, 57] |
| Methotrexate | Oral or SC | 15–25 mg weekly; folic acid supplementation recommended | CRP, ESR, liver enzymes | Antimetabolite; inhibits dihydrofolate reductase, reduces inflammation | For individuals on maintenance therapy and those with mild conditions | Useful in maintaining remission | [58, 59] |
| Azathioprine | Oral | 1–2 mg/kg/day | CRP, ESR, liver enzymes, TPMT enzyme levels | Purine synthesis inhibitor; immunosuppressive, reduces T and B cells | Maintenance treatment | Effective in maintaining remission; reduces steroid use | [60, 61] |
| Mepolizumab | SC | 300 mg every 4 weeks | Eosinophil count | Monoclonal antibody against IL-5; reduces eosinophil production and survival | EGPA is refractory or relapses, especially when there is eosinophilic tissue infiltration | Reduction in exacerbations and maintenance of remission | [62, 63] |
| Rituximab | IV | 375 mg/m2 each week for four weeks, or 1,000 mg on days | CD20, B-cell count, ANCA | The monoclonal antibody that suppresses CD20 and reduces B cells | Patients with cyclophosphamide contraindicated or who exhibit refractory instances | Effective in inducing remission in refractory cases | [64, 65] |
| Benralizumab | SC | 30 mg every 4 weeks for 3 doses, then every 8 weeks | Eosinophil count | Monoclonal antibody against IL-5 receptor alpha; depletes eosinophils | Considering refractory EGPA | Reduces exacerbations and maintains remission | [66, 67] |
TPMT: thiopurine methyltransferase; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; ANCA: anti-neutrophil cytoplasmic antibody; EGPA: eosinophilic granulomatosis with polyangiitis.
EGPA is a very uncommon condition; current treatment approaches are primarily based on research addressing other, more common AAVs. Asthma and ENT symptoms should be treated with topical medications first since they have a poor effect on the quality of life of the patient, may be effectively managed with general treatments, and often progress independently of systemic vasculitic manifestations. MPA, GPA, and EGPA treatment with systemic involvement must be phased in throughout the induction and maintenance phases [68–70]. It is reported that nearly 90% of EGPA patients attained remission, 18% relapsed, and 25.3% had flare-ups of either asthma or ENT, which supports continued GC usage [71, 72]. potential toxicity is a serious worry, particularly for young children who may require therapy for a long time, often their whole lives [73]. Before the availability of rituximab, CYC was used almost ubiquitously for initial induction therapy for severe vasculitis. There is a growing body of evidence across various patient groups that provides evidence for the use of the CYC induction regimen. The CYCLOPS study showed that patients treated with bolus IV of CYC experienced a safer induction than those treated orally daily and found fewer side effects because they had a reduced cumulative dose [74, 75]. The CYC regimen was suggested by the CORTAGE study, which included 14 EGPA patients. With regard to effectiveness, there were no important changes compared to the conventional regimen, but there were fewer adverse effects [76]. A total of six infusions, or three infusions every two weeks and three infusions every three weeks, make up the real dosage of 0.5–0.7 g/m2. In light of the growing experience with different AAVs, the recently released ACR recommendations recommend that patients with severe EGPA be administered RTX or CYC for the induction of remission. Because of its better clinical experience, individuals with a worse prognosis and active cardiac involvement should take CYC preferentially. In any case, it is still uncertain how well CYC and RTX work in comparison to EGPA induction regimens. To lessen dose-related CYC toxicity [71], RTX is also advised as an induction protocol for recurrent illnesses, regardless of the method chosen. Relapse rates in the absence of maintenance medication vary from 73.8% to 85.7%, depending on the length of CYC therapy time duration in 6 to 12 months [77]. It is suggested to begin maintenance therapy two weeks after the last CYC course. Again, there are no randomized controlled trials (RCTs) comparing the current maintenance therapy for EGPA or stating how long it should be continued. However, in RCTs, AAVs were found to be better than mycophenolate mofetil, and azathioprine and methotrexate are equal [71, 78, 79]. However, the recently published ACR recommendations urge this inclusion to reduce GC toxicity. Mepolizumab is now a part of first-line treatment with RCT evidence [30, 80, 81]. No RCT has evaluated methotrexate, azathioprine, or mycophenolate mofetil in EGPA patients. For recurrent disease, alternate immunosuppressants should be used. Glucocorticoid monotherapy can be used in select patients [67]. Interferon-α has demonstrated a potential to induce remission in EGPA patients and reduce eosinophil counts, but is limited in its clinical use because of significant side effects and poor efficacy in high-degree relapses [71, 82, 83].
Vasculitis, asthma-directed therapies, and pathways toward remission exacerbation and maintenance increasingly depend on new biologics, especially in relapsed or treatment-resistant EGPA, with some shared similarities in asthma, eosinophilic disorders, and systemic AAV [84, 85]. An improved understanding of the EGPA pathophysiology has revealed a target-rich landscape within the Th2 IL activation pathway and eosinophils. Agents have been developed targeting eosinophil biology, most importantly, mepolizumab, an anti-IL-5 monoclonal antibody. The role of the B-cell compartment in EGPA is unclear; however, rituximab has been studied in other AAVs and is being investigated for this purpose in EGPA [86].
A chimeric monoclonal antibody that targets CD20 is called RTX. All B-cell lineages, with the exception of pro-B-cells and plasma cells, have the B-cell membrane antigen CD20, which is essential for B-cell growth and interacts with T cells [83, 87]. Interest in using RTX for vasculitis exploded with a single case report in 2001 of a patient with GPA who had depletion of CD20-positive B-cells and a robust clinical response [88]. Because of the improved safety profile over CYC, the availability of biosimilars that lower costs and RCT indicating non-inferiority in general AAV as compared to CYC, RTX is currently authorized as both a protocol for induction and maintenance GPA and MPA. In any case, the investigations did not contain any EGPA patients [88–91]. Patients with relapsing EGPA typically have diminished CD20+ B-cells and also raised CD80+, CD27+, and CD95+ B-cells, and approximately 30% of patients are ANCA positive. Still, there is overlap, and there is often a persistent immune response towards IgG4. Serum IgG4 levels also reflect systemic disease activity, and affected organs are also invaded by IgG4-positive B-cells [92–95]. RTX specifically targets the CD20 on B-cells and induces cytotoxicity and apoptosis indirectly, while CYC is alkylating agent and exert its direct cytotoxic effect by cross-linking DNA. Open-label research and case report studies support RTX for relapsing EGPA or refractory EGPA [96–98].
In the recent prospective co-research, sixty-three individuals with refractory/relapsing EGPA who received RTX therapy were gathered from various sites across Europe. Approximately 87% of patients were prescribed RTX to alleviate a vasculitis flare-up, with symptoms primarily affecting the skin, kidneys, and PNS. Additionally, at the start of treatment, 83% of patients were diagnosed with active asthma [92, 96].
A crucial part of the immune system’s regulatory process is played by cytokines and chemokines. Eosinophil survival and activation are mostly mediated by IL-5, a cytokine produced by innate lymphoid and Th2 cells that controls innate immunity [99]. A humanized monoclonal antibody against IgG1 that is anti-IL-5, mepolizumab, prevents IL-5 from attaching to the α-subunit of its receptor, which is mostly expressed on eosinophils. When it was initially authorized, it was used to treat simple eosinophilic asthma. Mepolizumab has been studied in several allergy-related conditions, including hyper-eosinophilic syndromes, atopic dermatitis, and persistent rhinosinusitis, due to its role in the allergic pathway. The pathogenetic pathways linked to eosinophilic disorders are also present in EGPA. Because eosinophil products, such as eosinophil peroxidase and cationic protein, are present in large concentrations in serum, urine, and tissues, the proliferation of eosinophils in EGPA results in enormous tissue damage [100, 101]. Moreover, apoptotic genes CASP2, CARD4, and BLC2L13, and impaired EGPA eosinophils appear to display less apoptosis mediated by CD95 (Apo-1 Fas). In BAL fluid and EGPA sera, elevated levels of IL-5, IL-4, and IL-13 show that asthma is associated with an activated Th2 cell phenotype. EGPA patients had concentrations that were greater than normal of IL-5, which implies that this cytokine might be a target for treatment [102].
After favorable open-label pilot studies, mepolizumab was further assessed in 136 patients with uncontrolled predominantly non-severe EGPA in a double-blind study. Half of the patients were included in additional double-blind studies on other immunosuppressants, alongside GC [102]. Patients were administered 300 mg SC mepolizumab every four weeks for a full 48 weeks, which is three times the approved asthma dose. At 52 weeks, the mepolizumab group had fewer relapses, including a lower number of relapses with both asthma and vasculitic features. Patients reported improvement in asthma symptoms and no change in lung function test [100, 102, 103]. Recent funding-based evidence has shown that mepolizumab is also effective at lower individual doses (100 mg/month) than previously used (750 mg/month). In a European collaborative study, the 12-month remission rates were 76% in the 100 mg group and 82% in the 300 mg group, with similar GC sparing effects. Low-dose mepolizumab should be considered first-line therapy with escalation to 300 mg withheld for patients who show no response; 10% of patients studied noted benefit after switching to the higher dose. Additional work is required to show that low-dose mepolizumab is equivalent to the standard dose. Mepolizumab substantially improved asthma attacks and GC or immunosuppression-associated symptoms at modest doses [104–107].
Benralizumab, anti-IL-5α receptor, and reslizumab, anti-IL-5, are currently being studied in patients with EGPA, following successful phase 3 asthma trials. In phase-2 trials, reslizumab decreased oral GC usage significantly and was well-tolerated, while benralizumab had similar effects on GC usage and EGPA exacerbation. Both require larger controlled trials to show efficacy [107–110]. The pathology of EGPA involves several Th2 cytokines, including IL-4 and IL-13. Dupilumab, a fully human monoclonal antibody that blocks the IL-4 receptor α subunit and therefore IL-4 and IL-13 signalling, has already been approved for moderate-to-severe asthma, atopic dermatitis, and persistent rhinosinusitis accompanied by nasal polyposis in inadequately controlled patients, and represents a new treatment option for EGPA [108–110].
EGPA and IgE are both components of the same allergy pathway. The monoclonal IgG antibody, omalizumab, identifies free-circulating IgE and prevents it from binding to a particular high-affinity receptor, and acts by preventing basophil and mast cell degranulation [111]. Omalizumab is indicated for allergic rhinitis, Churg-Strauss syndrome, and severe asthma with elevated IgE, delivered via the SC route, and its role in EGPA has limited evidence and is sometimes conflicting. Omalizumab was initially begun in GC-refractory patients with severe EGPA-associated asthma with a good GC-sparing effect, although there is scant data on vasculitis control in EGPA. The studies that had been conducted on omalizumab in EGPA varied in their respective dosing schedules [112]. Omalizumab may be considered for maintenance treatment for people with EGPA who have severe, uncontrolled ENT or asthma symptoms. Still, it is less clear if this drug will lead to complete control of non-allergic manifestations. While rare, there are reports of asthma exacerbations occurring during therapy with omalizumab. In addition, there are isolated reports of EGPA onset at the same time that omalizumab was being used for severe asthma. These cases raise the possibility that omalizumab did not induce EGPA, but rather, steroid tapering for asthma treatment lost control for latent disease management as a result of the introduction of omalizumab [107, 111].
Treatments for EGPA that target IL-5, a cytokine that is intimately linked to eosinophils, have shown promise [113]. Anti-IL-5 medications, such as mepolizumab, can be used as a steroid-sparing therapy and have demonstrated effectiveness and safety in individuals with refractory or recurrent EGPA [114].
Compared to other forms of AAV, EGPA is a milder variant with lower fatality rates. Since survival rates have increased significantly as a consequence of effective treatment with CS and immunosuppressants, the illness is now considered chronic rather than fatal. The 10-year survival rate in a monocentric trial of 150 patients with EGPA was approximately 79%, which is comparable to the whole population. In another research, overall survival increased to around 90% after seven years [115, 116]. Based on whether Five Factor Score (FFS) are present or absent, patients with EGPA have varying prognoses (Table 2). Moreover, relapses, flare-ups of asthma or ENT, and disease-related organ degradation (sequelae) all have the potential to seriously harm a person’s quality of life with EGPA [52].
Modern diagnosis and treatment of EGPA.
| Category | Descriptions | References |
|---|---|---|
| Prognosis |
| [105, 107, 109, 115–118] |
| ||
| ||
| ||
| Treatment | Pharmacological interventions: | [115, 116] |
| ||
| ||
| ||
| Non-pharmacological interventions: | ||
| ||
| ||
| Outcomes |
| [116–118] |
| ||
| ||
| ||
| ||
|
EGPA: eosinophilic granulomatosis with polyangiitis.
Prior studies have shown that newly diagnosed patients with EGPA and highly active disease may have elevated levels of eotaxin-3, TARC/CCL17, and IgG4, but their significance for relapsing disease is unknown. A recent longitudinal cohort study measured serum levels of TARC/CCL17, eotaxin-3, IgG4, and IgG4/IgG ratios at every visit, along with epidemiological, clinical, and laboratory data. At the initial visit, 80% of patients were on GCs, and 68% were also taking immunosuppressants. Disease flares occurred at 18 visits, with median Birmingham Vasculitis Activity Score (BVAS) and BVAS/WG scores of 4 and 2 during the time of a relapse. At all sites, prednisone was the predominant treatment. None of the biomarkers consistently discriminated between active disease and remission. Similarly, various clinical studies have been conducted for the treatment of EGPA; some recent ongoing clinical trials are mentioned in Table 3.
There are several ongoing clinical trials and funding for the treatment of EGPA.
| S. No. | NCT numbers | Title | Status | Interventions | Phase | Populations 18 years to 50 years (adult, older adult) | Funding | Result |
|---|---|---|---|---|---|---|---|---|
| 1 | NCT06230354 | Explore the Efficacy and Mechanism of Action of Tezepelumab in Eosinophilic Granulomatosis with Polyangiitis (RACEMATE) | Enrolling by invitation | Tezepelumab, placebo | Phase 2 | 62 | Imperial College London | Publicly not available |
| 2 | NCT05979051 | A Study to Evaluate the Efficacy and Safety of SHR-1703 in Subjects With EGPA | Recruiting | SHR-1703, SHR-1703 Placebo | Phase 2–3 | 112 | Guangdong Hengrui Pharmaceutical Co., Ltd | The result is not publicly available |
| 3 | NCT05263934 | Efficacy and Safety of Depemokimab Compared with Mepolizumab in Adults with Relapsing or Refractory EGPA (OCEAN) | Recruiting | Biological: DepemokimabBiological: MepolizumabPlacebo matching mepolizumab, depemokimab | Phase 3 | 160 | GlaxoSmithKline | Publicly not available |
| 4 | NCT02807103 | Rituximab in Eosinophilic Granulomatosis with Polyangiitis (REOVAS) | Completed | RituximabPlacebo-rituximabCyclophosphamidePlacebo-cyclophosphamide | Phase 3 | 107 | Assistance Publique—Hôpitaux de Paris | Rituximab has shown a significant reduction in symptoms of EGPA compared to cyclophosphamide and glucocorticoids |
| 5 | NCT04551989 | Mepolizumab Long-term Study to Assess Real World Safety and Effectiveness of EGPA In Japan (MARS) | Completed | - | Observational | 120 | GlaxoSmithKline | Mepolizumab is effective in managing EGPA symptoms and reducing corticosteroid dependency |
EGPA: eosinophilic granulomatosis with polyangiitis.
EGPA is complicated to diagnose and manage, due to its heterogeneous clinical presentation and the absence of definitive biomarkers, leading to the challenge of differentiating it from other eosinophilia syndromes. In addition, even though many patients present with ANCA, the variation of ANCA presence adds an additional complexity and makes ANCA not a reliable diagnostic marker for all patients. Additionally, many patients need to develop a reliance on CSs throughout treatment, which can have significant long-term consequences. Some people do not receive the proper therapy options because biological therapies, mepolizumab included, have variable efficacy. Long-duration therapy is needed due to high rates of relapse, which puts patients at risk for long-term effects. Therefore, EGPA also presents multisystemically with various clinical manifestations, including some consequences that result from the delay in diagnosis and treatment, and have caused irreparable organ damage. There is also a wide variability in the course of patients’ disease states that makes it difficult to develop standard treatment protocols, and the absence of prognostic markers for determining disease progression and treatment response complicates the delivery of personalized treatment plans.
Future innovations within medicines and diagnostics promise to open up tremendous opportunities to improve the management of EGPA. Innovations in imaging may increase awareness and monitoring of organ involvement, while future exploration of biomarkers may improve our ability to detect EGPA early and correctly. The advances in newer therapeutic agents and new targeted biologics, such as anti-IL-5 and anti-IL-5R antibodies, should allow for more efficient and tailored therapy options for patients. Personalized medicine through the use of personalized genomic and molecular analyses will enable more personalized treatment. Better tracking of conditions supports new biomarkers, and advanced imaging may facilitate more exact dose alterations of drugs. We have the potential to implement a complete patient care model where quality of life and disease management are enhanced through incorporating management by a multidisciplinary team, along with providing patient education and patient support. For new potential opportunities to be realized in this area, the field must overcome its current limitations, and researchers, physicians, and patients must work together with these innovations (Table 4).
Rewinding the crucial facts for better understanding.
| Aspects | Facts |
|---|---|
| Definition | Vasculitis, a rare autoimmune disease, is characterized by eosinophilia and inflammation of blood vessels. |
| Etiology | Unknown; may be caused by a mix of environmental stimuli and genetic predisposition. |
| Epidemiology | Prevalence: 10–13 per million; more common in middle-aged adults, with slight male predominance. |
| Pathophysiology | Involves Th2-mediated immune response, eosinophilic infiltration, and ANCA (anti-neutrophil cytoplasmic antibodies) positivity in some cases. |
| Clinical features | Asthma, eosinophilia, vasculitis symptoms (e.g., purpura, neuropathy), and organ involvement (e.g., heart, lungs, kidneys). |
| Diagnostic criteria | 1. Asthma 2. Eosinophilia > 10% 3. Sinusitis 4. Pulmonary infiltrates 5. Vasculitis 6. Extravascular eosinophils. |
| Laboratory findings | Elevated eosinophils, positive ANCA (in 40–60% of cases), elevated IgE, increased inflammatory markers (ESR, CRP). |
| Imaging studies | Chest X-ray and CT scan showing pulmonary infiltrates, sinusitis, and sometimes cardiomegaly. |
| Biopsy findings | Vasculitis with eosinophil infiltration in tissue (such as the skin or lungs) occurs in. |
| Differential diagnosis | Churg-strauss syndrome, Wegener’s granulomatosis, hypereosinophilic syndrome, and allergic bronchopulmonary aspergillosis. |
| Treatment options | Corticosteroids, immunosuppressants such as cyclophosphamide or azathioprine, and biologics. |
| Therapeutic approaches | The treatment of eosinophilic vasculitis should be done with a stepwise approach beginning with corticosteroids and/or other medications such as bronchodilators and oxygen to help with symptoms. If the patient has severe or refractory symptoms, immunosuppressants or immunotherapy should be added. |
| Prognosis | Generally good outcomes with treatment, the 5-year survival rate is > 80%. Long-term complications are possible. Patients are also at risk of relapse. |
| Complications | Patients may experience cardiac involvement (e.g., myocarditis, cardiomyopathy), renal impairment, peripheral neuropathy, and pulmonary fibrosis. |
| Research & developments | Studies are ongoing regarding new biologics, targeted therapies, and personalized approaches to treatment. |
| Biomarkers | ANCA, eosinophil counts, IgE levels, and biomarkers are being evaluated to contribute to diagnosis and monitoring. |
| Prognostic factors | Involvement of the heart, renal impairment, and higher eosinophil counts are related to worse outcomes. |
ESR: erythrocyte sedimentation rate; CRP: C-reactive protein.
There are multiple compelling pathways of research into therapeutic targets for EGPA to ultimately improve treatment success and lessen the burden of this multifactorial disease. Eosinophils are integral to the pathophysiology of EGPA, and targeting eosinophils might be the ideal therapeutic direction. Mepolizumab and benralizumab, two biological therapies that specifically target IL-5 or IL-5R, have been shown to reduce eosinophil counts and improve clinical outcomes in some patients. IgE is another possible target because it is important for the eosinophils and mast cells. The anti-IgE monoclonal antibody omalizumab has shown promise in the treatment of severe asthma. EGPA: More data will be needed to confirm this. In addition to this, future research could explore targeting pathways that are involved in eosinophil recruitment, activation, and survival (e.g., CCR3 antagonists or inhibitors of eosinophil survival determinants such as IL-5 and GM-CSF). In addition, a fresh understanding of the pathophysiology of EGPA and mechanisms of T lymphocytes and their cytokines, especially involving Th2 and Th17 responses, may lead to new therapies that target these immunological pathways. It is hoped that as the complex mechanisms of EGPA, such as the processes involving T lymphocytes, are unravelled, these specific strategies may shift therapy paradigms and improve outcomes of patients living with this challenging vasculitis.
The clinical symptoms of EGPA are vital for diagnosis and management, in addition to following some sort of management strategy. The variability of this disease and the absence of clear biomarkers make it difficult to diagnose, treat, and manage. However, science is revealing possibilities of new types of therapies and increased knowledge of the disease to improve outcomes and quality of life. On top of these challenges, the disease also has multisystem involvement and variable ANCA positivity. This may lead to irreversible organ injury as identification and treatment are delayed. There is also a need for new interventions that are more effective and personalized, since we rely heavily on CSs, and biological treatments are variable. In contrast, there is a bright future for EGPA with the developments in diagnostics, especially the identification of specific biomarkers and better imaging capabilities. The emergence of targeted biological therapies and new pharmacological combinations offers hope for less toxic and more effective treatment options. Utilizing genetic and molecular profiling will allow for individualized therapy that can help enhance outcomes. In addition to improved outcomes and quality of life, quality whole-patient care strategies, such as a multidisciplinary approach and better patient education, are needed. Multi-disciplinary engagement among clinicians, researchers, and patients will help accomplish and sustain the promise of imminent developments in EGPA, while concurrently overcoming present challenges.
AAV: ANCA-associated vasculitis
ACR: American College of Rheumatology
ANCA: anti-neutrophil cytoplasmic antibody
BALF: bronchoalveolar lavage fluid
CYC: cyclophosphamide
ECG: electrocardiogram
EGPA: eosinophilic granulomatosis with polyangiitis
ENT: Ear, Nose, and Throat
ESR: erythrocyte sedimentation rate
GCs: glucocorticoids
GIT: gastrointestinal tract
HEC: high eosinophil counts
MPA: microscopic polyangiitis
OCSs: oral corticosteroids
RTX: rituximab
We extend our heartfelt gratitude to the management of Rungta International Skills University, Bhilai, for their unwavering support and encouragement throughout this work. Their dedication to fostering a research-conducive environment has been instrumental in completing this review.
PK: Writing—original draft, Conceptualization. AG: Methodology, Resources, Writing—original draft. SKG: Writing—review & editing, Project administration, Supervision, Validation. TMJ and DKM: Data curation. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 6606
Download: 41
Times Cited: 0
