 Review
Review

Affiliation:
1Carol Davila University of Medicine and Pharmacy, 050047 Bucharest, Romania
ORCID: https://orcid.org/0000-0003-3584-9808
Affiliation:
2Victor Babes National Institute of Pathology, 050096 Bucharest, Romania
ORCID: https://orcid.org/0000-0001-8751-0043
Affiliation:
1Carol Davila University of Medicine and Pharmacy, 050047 Bucharest, Romania
2Victor Babes National Institute of Pathology, 050096 Bucharest, Romania
ORCID: https://orcid.org/0000-0002-5507-1020
Affiliation:
2Victor Babes National Institute of Pathology, 050096 Bucharest, Romania
ORCID: https://orcid.org/0000-0002-5071-9756
Affiliation:
1Carol Davila University of Medicine and Pharmacy, 050047 Bucharest, Romania
2Victor Babes National Institute of Pathology, 050096 Bucharest, Romania
Email: ana.enciu@ivb.ro; ana.enciu@umfcd.ro
ORCID: https://orcid.org/0000-0002-8122-1096
Affiliation:
1Carol Davila University of Medicine and Pharmacy, 050047 Bucharest, Romania
3Cajal Institute, Titu Maiorescu University, 040051 Bucharest, Romania
ORCID: https://orcid.org/0000-0002-2122-0235
Explor Neurosci. 2025;4:100688 DOI: https://doi.org/10.37349/en.2025.100688
Received: February 18, 2025 Accepted: April 16, 2025 Published: May 11, 2025
Academic Editor: Ryszard Pluta, Medical University of Lublin, Poland
The article belongs to the special issue Cerebral Ischemia, Genetics, Comorbidities, Risk Factors and New Therapeutic Options for Neurorestoration
CD36 is a transmembrane protein that plays a role in various biological processes, including oxidized low-density lipoprotein and fatty acid uptake as well as regulatory control for inflammation signaling. Its robust expression in monocytes and macrophages associated with its ability to translocate fatty acids linked this scavenger receptor to foam cell formation and atherosclerosis. In the context of ischemic stroke, CD36 has been shown to contribute to brain injury and inflammation. Preclinical studies have demonstrated that CD36 expression increases in the brain after stroke and that inhibiting CD36 can reduce infarction size and improve neurological outcomes in animal models. These findings suggest that CD36 may be a potential therapeutic target for ischemic stroke. However, no clinical trials addressing CD36 and acute ischemic stroke are registered in the American or European databases. This review will discuss the relationship between CD36 and ischemic stroke and present some clinical findings in patients with single nucleotide polymorphisms of the CD36 gene.
According to World Stroke Organization acute cerebral vascular events are currently the second leading cause of death worldwide and over 62% of all incident acute cerebrovascular events are ischemic [1]. Key risk factors for de novo ischemic stroke, recurrence of events, and unfavorable outcomes of reperfusion therapy include metabolic conditions—arterial hypertension, high body-mass index, high fasting plasma glucose/diabetes, dyslipidemia, sleep-related breathing disorders, and low glomerular filtration rate [2–4]. Focusing on tackling these factors is essential as they account for 71.0% of the global stroke burden [1]. Generally, ischemic stroke arises from a disruption in cerebral blood flow—but for better prognosis, prevention, and treatment purposes, ischemic strokes are divided into 5 subtypes according to TOAST classification: large-artery atherosclerosis, cardioembolic (cardiac source and aortic atherosclerosis), small-vessel occlusion (or amyloid angiopathy), stroke of other determined etiology (systemic hypoperfusion and coagulation disease), and stroke of undetermined etiology [5]. Stroke pathology involves a complex mechanism, including cerebral parenchymal necrosis and apoptosis, endothelial dysfunction, oxidative stress, neuroinflammation, and blood-brain barrier (BBB) dysfunction [6, 7], emphasizing the importance of targeting a key molecule that addresses multiple mechanisms in ischemic stroke.
CD36 is a transmembrane protein expressed on many cell types and also referred to as fatty acid translocase (FAT), glycoprotein 4 (GPIV), or scavenger receptor class B member 3 (SCARB3). It acts as a cell-to-matrix attachment protein, a receptor for thrombospondins (TSPs) and other matrix molecules responsible for apoptotic signaling in different cell types (such as endothelial cells [8] or macrophages [9]) as well as signaling for damage-associated molecular patterns (such as hydrophobic peptides, apoptotic cell fragments, advanced glycation end-products), and a transporter for long-chain free fatty acids and oxidized low-density lipoproteins (oxLDLs) [10].
Ligand binding-induced cellular effects of CD36 differ among various cell types. Many such effects were first described in bone marrow-derived cells (e.g., monocytes and platelets), including the production of pro-inflammatory cytokines and reactive oxygen species (ROS), chemotaxis, and substrate binding [11–13].
Cellular effects of CD36 activation (Figure 1) are related to activation of several signaling pathways. Following oxLDL binding, MAPK signaling pathway was reported to be activated in several cell types (adipocytes [14], macrophages [15]), leading to the activation of MAPKs c-Jun N-terminal kinase (JNK)-1, important in cell proliferation and apoptosis. Activation of nuclear factor-kappaB (NF-κB) and cytokine productions in macrophages was also demonstrated to be CD36-dependent [16], albeit it appears to be restricted to M1 macrophages [17].
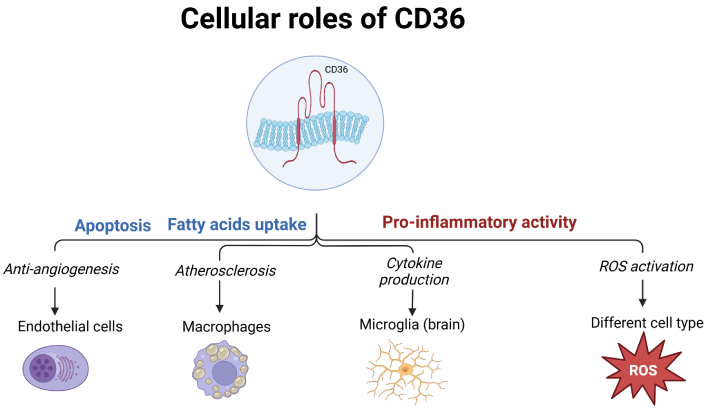
Involvement of CD36 in various cellular processes, in different cell types. ROS: reactive oxygen species. Created in BioRender. Enciu, A. (2025) https://BioRender.com/t73a673
In cell types relevant to vascular biology (endothelial and smooth muscle cells, but also platelets and macrophages), CD36 downstream signaling inhibits regulators of oxidative stress, such as nuclear factor erythroid 2-related factor 2 (Nrf2) [18]. In macrophages, binding of oxLDL and subsequent activation of JNK-1 results in CD36-dependent foam cell formation in vitro and in vivo mouse models [15]. In the vasculature, it impairs smooth muscle and endothelial function by increasing ROS and decreasing antioxidant gene expression. In blood cells, it promotes platelet dysfunction and arterial thrombosis, while in macrophages, it leads to foam cell formation and atherosclerosis. These effects are mediated by complex signaling pathways involving MAPK kinases [18] and several pharmacological approaches have been explored to modulate it.
Neuroinflammation detected locally in the post-ischemic brain was associated with activation of various transcription factors (reviewed in [19]), some located downstream of CD36 (such as NF-κB), while others, such as peroxisome-proliferator activated receptor (PPAR) alpha and gamma are known CD36 gene activators [20].
CD36 can also be detected in soluble form (sCD36) in serum and plasma, which facilitates its investigation in relationship to cardiovascular risk and metabolic syndrome [21–23].
In the context of ischemic stroke, CD36 has been shown to contribute to brain injury and inflammation and data from preclinical trials have extensively investigated the role of this protein in post-ischemic recovery, using CD36 null mice. This review will summarize the findings of CD36–/– mouse models in ischemia and stroke while extending the discussion to relevant clinical research.
There is twenty years’ worth of research on CD36–/– mice in cerebral ischemia, demonstrating free radical production and tissue injury in the absence of this protein. Most studies used transient middle cerebral artery occlusion [24–27] as the model for the ischemic injury and the reported effects usually refer to the early phase of the stroke.
CD36 expression was shown to increase in the ischemic brain, primarily in microglia/macrophages. Mice lacking CD36 had significantly less brain damage and better neurological function after ischemia, which was linked to reduced ROS production [24].
This outcome might be due to less neutrophil infiltration and glial reaction following ischemia, while, at the cellular level, it was correlated with reduced activation of NF-κB, a key regulator of post-ischemic gene expression [25].
The transcription factor NF-κB serves as a key regulator of inflammation pathways by inducing the expression of various pro-inflammatory genes [28], but there exist other receptors such as Toll-like receptors (TLRs) which are known to activate inflammation responses in immune cells. Abe et al. [26] investigated the crosstalk between CD36 and TLR2/4 in ischemic brain injury and found that CD36 is specifically required for TLR2/1 (but not TLR2/6 or TLR4) signaling in the brain, as CD36-deficient mice showed a reduced inflammatory response and less brain damage after TLR2/1 activation. Conversely, systemic inflammation triggered by TLR2/6 (but not TLR2/1) was reduced in CD36-null mice [26].
IL-1beta is also reduced after stroke in the brain of CD36–/– mice and further deletion of CD36 specifically in either microglia or endothelial cells also reduced brain injury (Figure 2) [29].
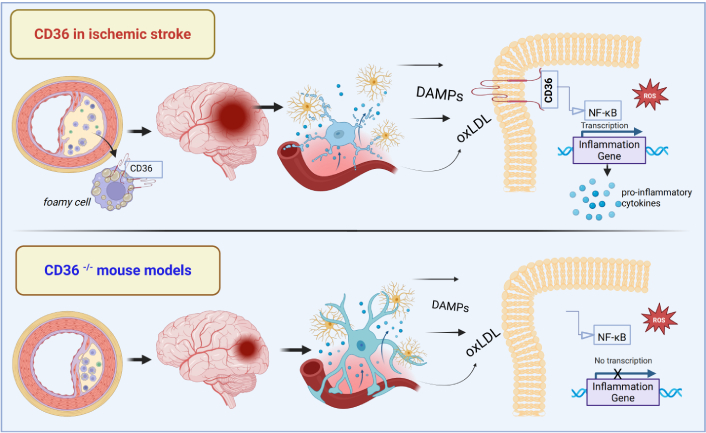
The role of CD36 in neuroinflammation after ischemic stroke: evidence from CD36–/– mouse models. DAMPs: danger associated molecular patterns; oxLDL: oxidized low-density lipoprotein; NF-κB: nuclear factor-kappaB; ROS: reactive oxygen species. Created in BioRender. Enciu, A. (2025) https://BioRender.com/t73a673
A CD36 gene knockout in endothelial cells was enough to improve the outcome of stroke in mice. These rodents also had reduced expression of monocyte chemoattractant protein-1 (MCP-1) and its monocyte receptor C-C motif chemokine receptor 2 (CCR2), leading to less monocyte infiltration into the brain and improved survival and motor function. This reduction in MCP-1 and CCR2 was observed even in severe stroke cases, suggesting a direct metabolic effect of endothelial CD36 deletion, independent of infarction size [30].
Another study showed, however, that this effect, although mimicked by pharmacological inhibition of CD36, is present only in a transient ischemic stroke model [31].
In the long term, CD36–/– mice displayed significantly attenuated BBB leakage and scar formation and improved long-term recovery across multiple behavioral tests, including motor activity, anxiety-related behaviors, depressive-like behaviors, and spatial learning/memory [32].
Protection against ischemia in CD36–/– mice is age-dependent, as demonstrated by Woo et al. [33] in a study using 9-day-old CD36–/– and wild-type (WT) mice. At 24 h post reperfusion, CD36–/– mice showed greater injury, increased caspase-3 activation, and impaired microglial clearance of dying neurons.
Another study performed on the same mouse model showed that transcriptomics changes induced by transient middle cerebral artery occlusion, related expression of key players in inflammation, leukocyte trafficking, and extracellular matrix pathways also affect other cell types in the brain, such as choroid plexus cells [34].
To highlight the impact of peripheral immune cells in the outcome of ischemic brain injury, Garcia-Bonilla et al. [35] transplanted WT bone marrow in CD36–/– mice, which decreased the infarction size. However, transplanting the CD36–/– bone marrow in WT mice did not impact the infarction size, elegantly demonstrating that local expression of CD36 (on microglia and endothelial cells) is the main responsible factor of ischemic stroke outcome [35].
Still, infiltrating monocytes play a part in the recovery phase of the stroke, where CD36 serves as a scavenger receptor for monocyte-derived macrophages which serve a reparative role [36].
One interesting finding was that of Qin et al. [27], which linked CD36 with brain derived neurotrophic factor (BDNF) (Val66Met) polymorphism and the motor outcome of stroke. They showed that lower BDNF levels were correlated with increased CD36 expression and reduced angiogenesis, which was mitigated with a CD36 gene knockout. This suggests that the poorer outcomes in BDNFMet/Met mice are partly due to an imbalance between the pro-angiogenic BDNF and the anti-angiogenic TSP-1/CD36 system [27].
Based on this data, it is demonstrated that the absence of CD36 positively influences the outcome of ischemic stroke in normal and hyperlipidemic models.
Pharmacological inhibition of CD36 was achieved with various peptides, mimicking known CD36 ligands. One cell-penetrating peptide, SS31, showed CD36-dependent protective effects in brain ischemia [37]. However, it was shown that the outcome of CD36 pharmacological inhibition, unlike the permanent loss of CD36 in null models, depends significantly on the moment of intervention. Post-stroke treatment with CD36 inhibitors did not improve and sometimes worsened outcomes, hinting that higher CD36 levels might be beneficial after stroke in these mice. However, pre-stroke (chronic) treatment with a CD36 inhibitor significantly reduced post-stroke brain swelling, with a trend towards smaller infarctions (though not statistically significant). This preventative benefit of CD36 inhibition aligns with previous findings of reduced infarction size and swelling in CD36–/– mice [38].
CD36’s role in cerebral ischemic events can be linked to a particular cause of microvascular impairment, such as amyloid beta clearance disturbance, BBB dysfunction, and oxidative stress [39].
With a smaller impact on the ischemic strokes causality, but noteworthy for its link with the CD36 receptor, is cerebral amyloid angiopathy (CAA; another etiology of small vessel ischemic disease), characterized by ischemic lesions such as white-matter hyperintensities, microinfarctions and cognitive impairment [40]. Studies showed that CD36 could be a potential therapeutic target for CAA as its absence reduces vascular amyloid deposition, preserves the Aβ clearance receptor LDL receptor related protein 1 (LRP-1), protects cerebral arterioles from Aβ-induced damage, and improves neurovascular function and cognitive performance [39, 41]. The lack of extensive human studies on CD36 in CAA specifically represents a significant knowledge gap, though the available evidence suggests CD36 could be a promising therapeutic target for CAA.
CD36–/– mouse model data convincingly demonstrated that CD36-mediated signaling is a detrimental factor for the outcome of acute ischemic stroke. In addition, the relationship between CD36 and fatty acid metabolism pinpoints it as an important player in atherosclerotic ischemic stroke. Without being a trigger for acute ischemic stroke, CD36 involvement in the neuroinflammation following the early phase justifies further scrutiny. Conditional knockout in relevant cell types, such as endothelial cells or bone-marrow-derived monocytes, highlighted the impact of CD36-mediated local response in acute stroke neuroinflammation, creating the premises for translational research in patients.
CD36 gene polymorphisms significantly influence several ischemic stroke risk factors such as the ones included in the metabolic syndrome [obesity, dyslipidemia, arterial hypertension, type 2 diabetes mellitus (T2DM)] through multiple mechanisms affecting lipid metabolism [42], vascular function, and inflammatory responses. Some recent studies have researched the role of CD36 also in cardiac pathology such as atrial fibrillation in overweight patients that correlated with increased cardioembolic risk and secondary cerebral ischemia [43].
In the past years, several genome-wide association studies have highlighted the role of CD36 polymorphisms in various diseases and phenotypes. Most studies focused their attention on linking CD36 single nucleotide polymorphisms (SNPs) to abnormal serum fatty acids [44], LDLs [45], coronary artery disease (CAD) [46], insulin resistance, and T2DM [47] as well as metabolic syndrome [48, 49] in different populations.
It is known that CD36 genetic variations (some listed in Table 1) significantly affect post-stroke inflammation and angiogenesis as they promote free radical production, pro-inflammatory cytokine expression, and control the recruitment of inflammatory cells. Understanding these genetic variations could help identify individuals at higher stroke risk, predict stroke outcomes, and develop personalized treatment strategies [50].
CD36 polymorphisms and cardiovascular diseases
| Pathological findings | CD36 variants | Population | Sample size | Effect size (OR/HR, 95% CI) | Key notes/observations | References |
|---|---|---|---|---|---|---|
| Abnormal serum fatty acids | rs3211938, rs1761667 | African Americans | HDL < 5.2 mg/dL (P = 0.00018); FA > 2.7 mg/dL/A-allele | rs3211938 reduces CD36 protein (41%), rs1761667 increases FA uptake | [47, 51] | |
| 30294G>C polymorphism | Caucasians | 585 | FFA levels (P = 0.02) | Men carrying the AGGIG haplotype had 31% higher FFA (P = 0.0002) and 20% higher triglycerides | [44] | |
| Abnormal LDLs | rs1761665, rs7755 | Caucasians | 1,117 | LDL particle numbers/20% (P < 0.01) | 3'UTR, rs7755 (A-allele), previously linked to risk of MetS and stroke was associated with higher LDL | [52] |
| SNP +30215/G-allele | Japanese | 494 | Difference in LDL-cholesterol concentrations was 10 mg/dL between GG- and AA-genotype carriers (P < 0.05) | Maker of the variation of LDL-cholesterol levels in male population | [45] | |
| Ischemic stroke risk | rs1761667, rs10499859 | Chinese | 1,387 | Increased risk of ischemic stroke with a 1.38 (95% CI 1.06–1.78, P = 0.020); rs10499859 risk was increased by 1.39 (95% CI 1.08–1.81, P = 0.012) | Dominant model risk independent of lipid levels | [53] |
| rs1761667 | Korean | 759 | Higher stroke risk of AG + AA genotypes (16.3% vs. 10.2%, respectively; P = 0.049) | No significant association with microvascular complication | [54] | |
| CAD | rs1761667 (AG genotype) | Egyptian | 147 | AG genotype rs1761667 association with increased risk of CAD (OR = 17.97, 95% CI 3.19–87.85, P = 0.001) | Plasma LDL levels in CAD patients with the AG genotype were significantly higher than those with the GG and AA genotypes (P = 0.046); AG genotype was significantly more prevalent among T2DM and metabolic syndrome patients (P < 0.05) | [55] |
| rs1761667 (G > A) | Iranian | 238 | Risk of CAD increased with rs1761667 (G > A) (OR = 5.677, 95% CI = 1.053–30.601, P = 0.043) | Dominant model of CD36 rs1761667 was found to have a protective role in hypertensive and CAD patients | [56] | |
| Insulin resistance & T2DM | rs3211938 (TG genotype) | Mexican Mestizos | 115 | HOMA-IR 3.2-fold (P = 0.001) | BMI 4.1 kg/m2 in diabetics vs. TT carriers | [47] |
| Stroke risk (LV mass & obesity) | rs1527479, rs1984112 | Australian | 275 | LV mass/2 g/m2 (P < 0.05) | Linked to exercise-induced lipid oxidation | [57] |
| rs1761663 | Finnish | 1,425 | rs1761663 effect on left ventricular mass measured either by echo (P = 0.017) or ECG (P = 0.007) | rs1761663 polymorphism has independent effects both on BMI and left-ventricular mass | [58] |
LDL: low-density lipoprotein; SNP: single nucleotide polymorphism; CAD: coronary artery disease; T2DM: type 2 diabetes mellitus; OR: odds ratio; HR: heart rate; HDL: high-density lipoprotein; FA: fatty acids; FFA: free fatty acids; 3'UTR: 3' untranslated region; HOMA-IR: homeostatic model assessment for insulin resistance; BMI: body mass index; ECG: electrocardiogram
There is evidence of CD36 as a potential biomarker for plaque vulnerability, outlined by Handberg et al. [59], how elevated soluble CD36 levels are associated with symptomatic carotid atherosclerosis and plaque instability in the absence of other inflammatory markers. In the context of assessing acute cerebral ischemic events, data highlighted that elevated soluble CD36 levels were observed in patients with recent symptoms, indicating a potential risk of ischemic events. This increase is likely due to the release of CD36 from foam cells in macrophage-rich regions of the atherosclerotic lesion [59].
More recently a genome-wide association study by Ikram et al. [60] identified 15 CD36 SNPs associated with stroke risk, as well as associations with left ventricular mass and obesity.
CD36 SNPs rs1761667 and rs10499859 are associated with an increased risk of ischemic stroke. The A-allele of rs1761667 increases stroke risk by 1.38-fold, correlates with abnormal lipid metabolism, altered HDL levels, and reduced CD36 protein expression. Similarly, the G-allele of rs10499859 raises stroke risk by 1.39-fold even after adjusting for multiple risk factors [53]. A study in Korean men with T2DM found that the rs1761667 SNP was significantly linked to a higher stroke prevalence in individuals with the AG + AA genotypes and a diabetes history of less than 10 years, adjusting for variables like age, body mass index (BMI), hypertension, HbA1c, and LDL-cholesterol. Regarding gender differences, there was no statistically significant association between rs1761667 and stroke risk in the female cohort [54].
Similar findings have been observed in other populations. Zhang et al. [53] identified a link between rs1761667 and rs10499859 variants and ischemic stroke risk in the Chinese Han population, this association was controlled by variables such as age, gender, BMI, smoking, hypertension, and diabetes. Though these adjustments strengthen the reliability of the findings, the precise mechanisms by which CD36 polymorphisms contribute to stroke risk remain unclear.
Further insights into the relationship between ethnicity, the CD36 rs1761667 polymorphism, and cardiometabolic risk factors for ischemic stroke are discussed in the review by Yazdanpanah et al. [50].
These findings underscore the role of CD36 genetic variations in stroke susceptibility [53].
As for environmental factors, dietary patterns can interact with CD36 variants to influence cardiometabolic risk factors and potentially stroke risk. For example, dietary patterns have been shown to interact with the rs1761667 polymorphism to affect body weight and metabolic syndrome risk [61].
CD36 genotyping may affect response to various treatments. For example, patients with CD36 variant rs3211938 (G/T) which reduces CD36 level by approximately 50%, had lower flow-mediated dilation, but a better response to sildenafil treatment than non-carriers [62].
To date, no active clinical trials involving the use of CD36 blockade in ischemic stroke can be found in USA and European databases, despite its well-documented role in infarction size determination and locally inflammatory response in ischemic stroke.
As the role of CD36 in the pathogenesis of atherosclerosis, inflammation, and lipid metabolism has been well-documented, as its contribution to the pathogenesis of cerebral ischemia, the importance of CD36 as a treatment target has emerged early but exhibited little or no efficacy in clinical trials. It was proposed that the failure of clinical trials may be due to the use of animal models of stroke that do not reflect traditional risk factors for stroke in humans [63]. Also, as previously mentioned, CD36 blockade was efficient in animal models as a preventive treatment, not as a curative one, which is difficult to address in patients, as no long-term studies with low dosages of CD36 inhibitors have been reported. Additionally, the wide distribution of this receptor on immune cells can be responsible for high toxicity and high-frequency adverse effects, which would limit chronic or high-dose treatments.
Furthermore, targeting a single pathway in experimental stroke models has proven insufficient to address the complex nature of stroke-related injuries in humans. CD36, activated by various ligands, triggers pathways that converge to promote inflammation and endothelial dysfunction, potentially contributing to both cardiovascular and cerebrovascular diseases [6].
Given the complexity of acute stroke physiopathology, a deeper understanding of its various subtypes could benefit from the identification of distinct protein biomarkers, as well as genetic and epigenetic changes (see [64] and citations therein). CD36 may serve as a reliable biomarker for predicting and prognosticating the outcomes of ischemic atherothrombotic acute stroke.
Although the role of CD36 signaling was convincingly demonstrated in the pathogenesis of ischemic stroke, intricate collaborations with other factors such as hyperlipidemia, metabolic syndrome, and chronic inflammation can be responsible for the lack of success of translation of preclinical data from bench to bedside. The wide cellular distribution of this receptor may contribute to the undesirable effects of its pharmacological blockade, thereby limiting its efficacy as a therapeutic target. However, CD36 could hold further potential as a prognostic biomarker in acute ischemic stroke associated with atherosclerotic vascular pathology, particularly in patients with specific genetic profiles. Additionally, further research is warranted in specific subtypes of stroke, such as CAA, which remains an area with significant knowledge gaps in the field.
BBB: blood-brain barrier
BDNF: brain derived neurotrophic factor
CAA: cerebral amyloid angiopathy
LDL: low-density lipoprotein
NF-κB: nuclear factor-kappaB
oxLDLs: oxidized low-density lipoproteins
ROS: reactive oxygen species
SNPs: single nucleotide polymorphisms
T2DM: type 2 diabetes mellitus
TLRs: Toll-like receptors
WT: wild-type
Figures were created in BioRender. Enciu, A. (2025) https://BioRender.com/t73a673. We thank Alexandru Diaconeasa for the English proofing of the manuscript.
AMDN: Writing—original draft, Writing—review & editing. IDP: Writing—original draft. EC: Writing—original draft, Funding acquisition. MD: Writing—original draft. AME: Writing—original draft, Writing—review & editing, Conceptualization. CT: Conceptualization, Writing—review & editing.
The authors declare no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This research was supported by the Core Program within the National Research, Development and Innovation Plan, 2022–2027, with the support of MCID, project NO. [10N/01.01.2023, PN 23.16.01.01, PN 23.16.02.01, PN 23.16.02.03]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2785
Download: 16
Times Cited: 0
Mihai Andrei Ruscu ... Aurel Popa-Wagner
Chaitanya Sanghadia ... Brandon Lucke-Wold
Vivian Molina Cuevas, Ambar Oyarzábal Yera
Divya Sharma, Rahul Kumar
Lorena Dellagnesi Depieri ... Lorena Souza Viana
