 Review
Review

Affiliation:
1SciGency Science Communications, 1124 Budapest, Hungary
ORCID: https://orcid.org/0000-0003-1282-849X
Affiliation:
1SciGency Science Communications, 1124 Budapest, Hungary
2Department of Biomedical Sciences, University of North Dakota, Grand Forks, ND 58202, USA
Email: masha.savelieff@ndus.edu
ORCID: https://orcid.org/0000-0001-5575-2494
Explor Neurosci. 2026;5:1006139 DOI: https://doi.org/10.37349/en.2026.1006139
Received: February 22, 2026 Accepted: April 28, 2026 Published: July 01, 2026
Academic Editor: Ryszard Pluta, Medical University of Lublin, Poland
The article belongs to the special issue Progress in Alzheimer's disease research: etiology, molecular mechanisms involved in disease progression, and advances in therapies aimed at slowing or reversing neurodegeneration
Type 2 diabetes continues to grow in prevalence globally due to contemporary dietary patterns and physical inactivity. Among its multiple complications, neurological injury is a long-known consequence of diabetes, especially in the peripheral nerve as diabetic peripheral neuropathy. However, the adverse effects of diabetes on brain health are also increasingly appreciated, raising patients’ risk of developing cognitive impairment and eventual dementia along with brain structural changes. Thus, despite the highly heritable nature of Alzheimer’s disease, addressing modifiable risk factors, including type 2 diabetes, may help curb dementia development. This review covers epidemiological evidence for the link between diabetes and dementia as well as mechanistic evidence on similar underlying pathophysiological pathways, describing potential links between the two diseases. Given excess dementia risk from diabetes, this review also covers how optimal diabetes control and, ideally, diabetes prevention, may mitigate future dementia burden, concluding with some practical interventions.
Dementia is a decline in mental abilities and cognitive skills that impair an individual’s ability to function in daily living and poses substantial socioeconomic burdens [1]. An estimated 57 million people globally were living with dementia in 2019, projected to increase to 153 million by 2050 [2]. Alzheimer’s disease (AD) is the most common dementia [3], affecting 1 in 9 adults over the age of 65 years, per the Alzheimer’s Association [4]. AD exists within a family of neurodegenerative conditions that overlap clinically, pathologically, and epidemiologically, termed AD and related dementias, and also accounts for patients that exhibit a mixed pathology [5] (Table 1).
Alzheimer’s disease and related dementias.
| Key information | Key pathologies | Major risk factors |
|---|---|---|
| Alzheimer’s disease (AD) | ||
| Most common dementia, accounting for an estimated 60 to 80% of cases [4]. Most AD (> 95%) occurs as sporadic, late-onset AD (usually > 65 years of age) without autosomal dominant inheritance of highly penetrant APP, PSEN1, PSEN2 mutations, which instead give rise to familial or autosomal dominant, early-onset AD. | Extracellular amyloid-β plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau protein [202]. Chronic neuroinflammation, synaptic dysfunction, and neuronal loss. Often accompanied by vascular abnormalities and mitochondrial and metabolic dysfunction. | Age (strongest non-genetic factor); genetics (hallmark genes include APP, PSEN1, PSEN2, APOE4, TREM2, among others [21]); cardiometabolic risk factors (diabetes, obesity, hypertension); environmental and lifestyle risk factors. |
| Vascular dementia | ||
| The second most common dementia, accounting for approximately 15% of cases. | Dementia caused by cerebrovascular injury from large-vessel strokes, small-vessel disease (white matter lesions, lacunes), chronic cerebral hypoperfusion [203, 204]. | Age; cardiometabolic risk factors (hypertension, diabetes, obesity, hyperlipidemia, stroke); environmental and lifestyle risk factors; genetics (hallmark genes include APOE4, SPRY2, FOXA2, among others [21]) as well as rare genetic forms (e.g., NOTCH3 in the most common heritable cause of stroke and vascular dementia, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [205]). |
| Lewy body dementia | ||
| Includes dementia with Lewy bodies and Parkinson’s disease dementia [206]. | α-Synuclein aggregates of Lewy bodies and Lewy neurites accompanied by neuronal loss. | Age; genetics (hallmark genes include APOE4, SNCA, GBA, among others [21]). |
| Frontotemporal dementia | ||
| A leading type of early-onset dementia [207]. | Selective degeneration of frontal and temporal lobes. Abnormal accumulation of tau, TDP-43, or FUS proteins (depending on subtype). Neuronal loss leading to behavioral, language, or executive dysfunction. | Genetics is the primary known risk factor (hallmark genes include APOE4, GRN, C9orf72, MAPT, among others [21]). |
| Mixed pathology dementias | ||
| A substantial proportion, possibly the majority, of dementia cases develop mixed pathology. | AD neuropathology with 1, 2, and 3 other neuropathological features (microvascular brain injury, Lewy body dementia, hippocampal sclerosis, limbic predominant TDP-43 encephalopathy, cerebral amyloid angiopathy) [5, 109]. Dementia cases can also develop multiple neuropathological features without AD neuropathology. | Not applicable. |
APOE4: apolipoprotein E4; APP: amyloid precursor protein; PSEN1: presenilin-1; PSEN2: presenilin-2.
Although dementia prevalence is anticipated to rise in large part due to an aging population, modifiable risk factors are also expected to contribute. Type 2 diabetes (T2D) is prominent among them, classified by the Lancet Commission on Dementia prevention, intervention, and care as an acknowledged dementia risk factor [6]. Diabetes represents a major health crisis and affected approximately 536.6 million people in 2021 globally, predicted to rise to 783.2 million by 2045, if current trends persist [7]. Most diabetes (Table 2), about 90 to 95%, is T2D, which arises from insulin resistance with consequent hyperglycemia and is acquired through poor diets and physical inactivity [8, 9]. In this modern-day era of ultra-processed, calorie-dense, nutrient-poor foods [10, 11] and physical inactivity [12, 13], even children increasingly develop T2D [14], placing them already at a young age on a poor health trajectory. Diabetes is a long-known risk factor for neurological injury, established earlier for peripheral diabetic neuropathy in the peripheral nervous system [15] but now also increasingly recognized in the central nervous system [6, 16].
Diabetes and other select cardiometabolic conditions.
| Condition | Description |
|---|---|
| Type 2 diabetes | A type of diabetes that develops due to rising insulin resistance, an inadequate compensatory increase in insulin production by pancreatic beta-cells, and eventual beta-cell dysfunction as the disease progresses, resulting in hyperglycemia [8, 9]. |
| Type 1 diabetes | A type of diabetes that occurs due to autoimmune-mediated destruction of insulin-producing pancreatic β cells, resulting in hyperglycemia [9]. |
| Prediabetes | A state preceding frank type 2 diabetes, characterized by insulin resistance, glucose intolerance, and elevated fasting blood glucose not meeting the formal criteria for diabetes [9]. |
| Generalized obesity | Usually measured by body mass index (BMI; weight divided by the height squared; kg/m2), defining the classifications overweight (BMI 25.0–29.9 kg/m2), class I obesity (moderate; BMI 30.0–34.9 kg/m2), class II obesity (severe; BMI 35.0–39.9 kg/m2), and class III obesity (very severe/morbid; ≥ 40.0 kg/m2) although cutoffs are race/ethnicity-dependent [208]. |
| Central obesity | A measure of visceral adiposity, linked to cardiovascular risk, frequently measured by elevated waist circumference (≥ 88 cm in females, ≥ 102 cm in males, per the AHA/NHLBI Scientific Statement [209]) and waist-to-hip ratio [210]. Waist circumference cutoffs are sex- and race/ethnicity-dependent [211]. |
| Dyslipidemia | A state of blood lipids falling outside the normal range (elevated triglycerides ≥ 150 mg/dL; reduced high-density lipoprotein cholesterol < 50 mg/dL in females, < 40 mg/dL in males; per the AHA/NHLBI [209]). |
| Hypertension | A state of raised systolic (≥ 130 mmHg) or diastolic (≥ 85 mmHg) pressure, per the AHA/NHLBI [209]. |
| Metabolic syndrome | A state of metabolic dysfunction spanning three or more out of the five criteria: (i) elevated waist circumference, (ii) elevated fasting glucose (> 100 mg/dL; or on relevant medication), (iii) elevated triglycerides (or on relevant medication), (iv) reduced high-density lipoprotein cholesterol (or on relevant medication), (v) hypertension (or on relevant medication), per the AHA/NHLBI [209]. |
AHA/NHLBI: American Heart Association/National Heart, Lung, and Blood Institute.
As a modifiable risk factor, T2D represents a path towards minimizing dementia and AD risk and mitigating the future burden of disease. In this review, we describe epidemiological evidence of T2D on dementia and AD risk. We outline emerging mechanistic evidence on similar pathophysiological pathways in T2D and AD, and conclude with evidence that targeting T2D, either through appropriate disease control or prevention, may constitute an approach for dementia and AD prevention.
This was a scoping review. From December 1, 2025, to February 8, 2026, we conducted a search of PubMed for English language articles. In addition to “type 2 diabetes” and/or “cognitive impairment” and/or “dementia” and/or “Alzheimer’s disease”, we used one or more of the following terms: “amyloid-beta”, “bariatric surgery”, “biomarker”, “brain”, “comorbidity”, “diet”, “exercise”, “exposome”, “gene-environment”, “GLP-1R agonists”, “human”, “insulin resistance”, “longitudinal”, “meta-analysis”, “metformin”, “obesity”, “pathophysiology”, “preclinical”, “rodent”, “senolytic”, “sex”, “SGLT2 inhibitor”, “social determinants of health”, “sotagliflozin”, “tau”. From the search results, we focused on articles published from January 1, 2020, to February 8, 2026; however, older seminal papers were also considered along with articles from the authors’ personal reference list. Articles were selected based on relevance to this review. Clinical papers prioritized large, population-based and longitudinal studies that adjusted for confounding but also included meta-analyses and systematic reviews; however, smaller clinical studies were cited for certain topics when larger studies were sparse, e.g., studies in youths.
Most AD risk derives from non-modifiable risk factors. Aging is a major risk factor for sporadic AD, the focus of this review, and usually occurs over the age of 65 years. AD is also highly heritable, and genetic factors account for approximately 60 to 80% of risk based on twin studies [3, 17]. The common apolipoprotein E4 (APOE4) allele explains a substantial portion of AD heritability (about 9%) [18], increasing risk an estimated 3- to 4-fold. On the other hand, besides the highly penetrant APP (amyloid precursor protein), PSEN1 (presenilin-1), and PSEN2 (presenilin-2), most of the other over 80 identified risk loci are associated with odds ratios of only between 1.05 and 1.2 [19]. However, risk from these more minor contributing alleles is additive; a polygenic risk score can estimate an individual’s AD risk through a weighted sum of all risk-associated variants they harbor based on their effect sizes [20]. Dementias more broadly exhibit a complex genetic architecture (Table 1) with polygenic risk [21].
The balance in variation after accounting for 60 to 80% of AD heritability, i.e., the remaining 20 to 40%, is explained by individual environmental and lifestyle factors that differ between people (e.g., lifestyle, diet, etc.) [17], formalized by the concept of the exposome [22]. The AD exposome is the cumulative of environmental exposures, spanning toxicant (chemical, biological), lifestyle, social, and ecosystem factors [22], that a person experiences over their lifetime, which influences disease risk and progression. Components of the exposome can be either harmful or protective. The AD exposome is wide-ranging, but poor diet and impaired metabolic health, such as T2D and (central) obesity, are key detrimental components [23]. The Lancet Commission on Dementia prevention, intervention, and care was launched to generate authoritative, evidence-based guidance for reducing the global burden of dementia by identifying potentially modifiable risk factors [6]. In their latest 2024 report, they listed diabetes along with various additional cardiometabolic variables, as dementia risk factors.
Besides environmental and lifestyle risk factors, gene-environment interactions may influence a person’s individual AD risk [24]. For AD risk genes that are not highly penetrant, adverse and protective environmental and lifestyle factors may interact with genetic risk to raise and lower the odds of disease, respectively. Besides APOE4, gene-environment interactions are relatively understudied in the context of diabetes and dementia and AD. Nevertheless, individuals that harbor genetic risk from non-penetrant mutations can reduce their personal chances of developing dementia through appropriate protective measures.
A solid body of evidence has accrued from large, population-based and, increasingly often, longitudinal clinical studies showing diabetes raises the risk of cognitive decline and future dementia (Table S1) [25], now formally articulated by the Lancet Commission, which ascribes a relative risk of 1.7 with a weighted population attributable fraction of 2.3 [6]. In the community-based, prospective Atherosclerosis Risk in Communities study, conducted in the US, midlife diabetes was associated with 19% more cognitive decline over 20 years versus study participants without diabetes [26]. Even prediabetes, a state of impaired glucose tolerance not meeting formal diabetes criteria (Table 2), as well as poorly controlled diabetes and longer diabetes duration, was linked to more pronounced cognitive decline. Cross-sectional analysis of the Atherosclerosis Risk in Communities study participants by magnetic resonance imaging underscored brain structural changes and vascular pathology in diabetic participants with poorly controlled disease [glycated hemoglobin (HbA1c) ≥ 7.0%] [27].
In a similar vein, longitudinal analysis of the large, community-based UK Biobank study over a 12-year follow-up found that T2D, especially diagnosed before 50 years of age, was a risk factor for all-cause dementia, AD, and vascular dementia (VaD) [28]. In UK Biobank, an early type 1 diabetes diagnosis before 30 years of age was also a risk factor for all-cause dementia and VaD, as seen in other studies, for instance of the Swedish National Diabetes Register [29]. Since type 1 diabetes hyperglycemia results from autoimmune-mediated destruction of insulin-producing pancreatic β cells, it is not a modifiable risk factor, but the associated risk of dementia underscores the importance of optimal control and disease management. Another UK study, Whitehall II, with very long 32-year follow-up, also highlighted earlier T2D onset on greater dementia risk [30].
The China Health and Retirement Longitudinal Study observed diabetes significantly correlated to cognitive decline after adjusting for numerous covariates [31]; stratified by sex, diabetes was linked to cognitive decline in females but not males. These sex dimorphic results are echoed by other studies. Sex-stratified analysis of UK Biobank identified excess AD, but not all-cause or VaD, risk in females versus males [32], while a large meta-analysis of 14 studies found 19% excess VaD risk in T2D females versus T2D males [33]. Dementia prevalence is approximately 1.3 times higher in females versus males [34], attributed in large part to longer female life expectancy [2]; however, biological factors, such as menopause and hormones (e.g., estrogen, which exerts neuroprotective properties [35]) or social factors (e.g., education level) may contribute to real sex differences in dementia risk in T2D and remains an active research area. Besides sex, one meta-analysis examined dementia risk from diabetes by race but concluded cautious interpretation due to sample size [36].
Finally, and worryingly, T2D in youths may also predispose patients to poorer cognition and brain structural changes; although studies are presently sparse, they indicate an area of potentially grave concern [37]. A large nationwide, population-based study (n = 971,677 of which 3,570 developed T2D) found that adolescents in the lowest versus highest quintile of global cognitive Z-scores were at highest risk for T2D, with hazard ratios of 2.46 in males and 2.33 in females, adjusted for age, various metabolic factors, and socioeconomic status [38]. The relationship also held for cognitive subdomain scores. This finding is recapitulated by relatively small studies that show, versus obese youths without T2D [39] or lean controls [40] matched for various metrics, youths with T2D and obesity perform poorer in several cognitive domains, estimated intellectual functioning [39], working memory [40], verbal memory [39, 40], psychomotor efficiency [39], and processing speed [40]. Brain structural findings are also concerning, finding variably across diverse studies, lower global and regional grey and white matter volumes and microstructural integrity, with altered functional connectivity [37, 41]. Further study is needed in this critical area with emphasis on prevention of T2D and related cognitive impairment (CI).
As highlighted above, mounting evidence supports a link between diabetes and dementia and AD risk (Table S1). However, one prominent limitation has been lack of stratification by dementia subtype, restricting mechanistic interpretation of pathophysiology [25]. For instance, Zhou et al. [32], from examining UK Biobank, found that T2D was significantly linked to AD with a hazard ratio of 2.38 but to VaD with hazard ratio of 3.93, although they did not compute relative ratios. Thus, granular analysis is essential. Additionally, many observational studies relied on electronic health records and International Classification of Diseases codes rather than on clinical examination and disease biomarkers, e.g., amyloid-beta (Aβ), which may misclassify cases and dementia subtypes. Importantly, although several analyses adjusted for APOE4 status as a covariate [26–28, 32], many lacked the information. However, one investigation found that accounting for APOE4 status did not change their main findings on the T2D-dementia relationship [30].
Furthermore, several studies acknowledged that they lacked complete information on all potential covariates that could influence the T2D-dementia relationship, such as metabolic comorbidities, medication use and diabetes control, neuropsychological conditions (e.g., depression, sleep disorders), lifestyle habits, and socioeconomic factors. It is crucial to adjust for all covariates to derive meaningful insight. It is especially important to consider T2D is highly comorbid with many other cardiometabolic risk factors (e.g., obesity, dyslipidemia, hypertension) [15], which the Lancet Commission lists as dementia risk factors [6]. Thus, although the weighted population attributable fraction of diabetes to dementia is only 2.3, accounting for comorbid cardiometabolic risk factors increases the total weighted population attributable fraction, which is consequently more impactful on dementia and AD burden.
Insulin is a peptide hormone released by pancreatic β-cells in response to the post-prandial rise in blood glucose levels. Insulin acts on tissues throughout the body to promote cellular glucose uptake as well as fatty acid and amino acid uptake and energy storage. At the cellular level, insulin binding to insulin receptors activates several downstream signaling cascades; the insulin-IRS-AKT axis promotes translocation of glucose transporters (GLUTs) to the membrane for glucose uptake and regulates phosphorylation of various kinases, including GSK3, overall activating metabolic and anabolic processes, including DNA, protein, and lipid synthesis [42]. Insulin binding also engages the insulin-MAPK axis, which regulates various transcription factors and elements controlling transcription, translation, and post-translational modifications.
Although neuronal glucose uptake in the brain occurs primarily by insulin-independent GLUT3, the brain is nonetheless an insulin sensitive organ [42]. Insulin, entering via selective transport across blood-brain barrier (BBB) endothelial cells, binds to insulin receptors widely expressed across the brain, especially at pre- and postsynaptic compartments. Binding activates neurite outgrowth, modulates neurotransmitter release and uptake, and regulates expression, trafficking, and localization of excitatory and inhibitory neurotransmitter receptors. Insulin also mediates glucose uptake via GLUT4, also expressed on neurons, during periods of intense activity, acting in concert and supplementing uptake via insulin-independent GLUT3. Under homeostatic conditions, glia aid neurons in their activities; astrocytes recycle neurotransmitters, transfer energy substrates to neurons, and maintain BBB integrity [43]. Microglia, the resident macrophages of the brain, surveil their surroundings and conduct housekeeping activities, such as synaptic remodeling and clearing debris [44]. Astrocytes and microglia express insulin receptors and are insulin responsive, modulating their actions in supporting neuronal activity [42].
Insulin resistance in peripheral tissues is a defining and early characteristic during T2D progression, especially in muscle and adipose tissues, with failure of β-cells to compensate. A similar concept occurs centrally, defined as the development of brain insulin resistance in T2D and lack of a response of brain cells to insulin [42] (Figure 1). Consequently, neurons and their supportive glia fail to perform their usual activities prompting cognitive dysfunction in T2D. Drawing parallels to the periphery, it is tempting to speculate that brain insulin resistance is the initiating or an early process in the brain leading to CI. Indeed, subtle CI occurs in individuals with T2D early in the disease course [39, 45], suggesting this state of brain insulin resistance may underlie these cognitive deficits. Furthermore, brain-specific knockout of insulin signaling in animal models promotes CI along with disrupted hippocampal synaptic plasticity, mitochondrial dysfunction, reduced energy production, and oxidative stress [46, 47] (see sections below), but confirmatory investigation is needed. Additionally, further comorbid risk factors, e.g., central obesity, and pathophysiological processes also contribute to CI progression, including vascular pathology. The human T2D brain is associated with poor glucose metabolism [48, 49], also an early occurrence likely to coincide with the development of brain insulin resistance.
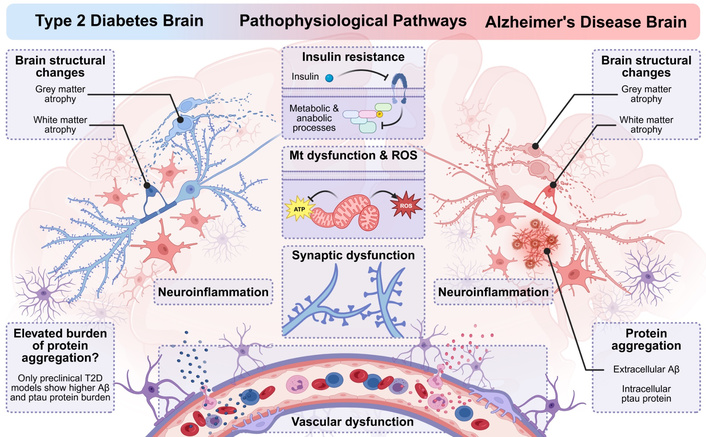
Similar underlying pathophysiological pathways in type 2 diabetes (T2D) and Alzheimer’s disease (AD). T2D and AD have several similar underlying pathophysiological pathways spanning brain structural changes, insulin resistance and altered (glucose) metabolism, neuroinflammation, mitochondrial (Mt) dysfunction and oxidative stress from reactive oxygen species (ROS) generation, synaptic dysfunction and loss, and vascular dysfunction. AD is also characterized by extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau (ptau) protein. In T2D, preclinical models show elevated burden of characteristic AD protein aggregates (Aβ, ptau protein); human clinical studies do not show elevated amyloid or tau burden in individuals with T2D versus controls. Created in BioRender. Savelieff, M. (2026) https://BioRender.com/6inzukd.
In parallel, insulin resistance [42, 50] is also a facet of the human AD brain, independent of diabetes, showing altered levels and phosphorylation of insulin signaling pathway proteins, potentially early in the disease course [51, 52]. Glucose hypometabolism is also a key feature in AD and other dementias thought to arise downstream of neurodegeneration and synaptic failure, and tracking with disease progression [53, 54].
In T2D, insulin resistance and consequent hyperglycemia trigger a series of inflammatory responses and mitochondrial dysfunction (Figure 1) [43]. In vitro models suggest insulin may modulate inflammatory cytokine secretion from astrocytes and microglia [55]; in vivo models of T2D [56] and high-fat diet (HFD)-induced prediabetes and obesity [57, 58] exhibit astrogliosis and microgliosis underscoring a neuroinflammatory state. Molecularly, hyperglycemia promotes the formation of advanced glycation end products (AGEs), which activate receptor for AGE (RAGE), promoting inflammation through NF-κB in diabetes [59]. Microglia express RAGE [60] and may potentially constitute inflammatory pathways in diabetes, although this remains understudied specifically in the context of T2D-related CI. T2D also compromises the BBB, prompting trafficking of immune cells and inflammatory factors from chronic peripheral inflammation [8] into the CNS [61].
Hyperglycemia raises mitochondrial fission [62] and depolarization [63] and lowers levels of mitochondrial complex components [64] with consequent reactive oxygen species (ROS) and aggravated insulin resistance [64] in vitro in hippocampal neurons [62–64] and in vivo [64]. Furthermore, hyperglycemia reduces mitochondrial motility in hippocampal neurons [65], which, in the brain, would impair their ability to direct mitochondria to areas of high energy demand to support cognitive function. Dyslipidemia, a frequent comorbidity in T2D, modeled by treating neurons with palmitate in vitro, also drives mitochondrial fission [66] and depolarization [66, 67], alters mitochondrial respiration [68], diminishes cellular ATP levels [69], and raises ROS generation [66, 67, 69].
In AD, neuroinflammation is a disease hallmark with both microglial and astrocytic dimensions (Figure 1) [44, 70]. Initially, during aging, acute microglial inflammatory responses via Toll-like receptor binding promote phagocytosis and clearance of amyloid aggregates, maintaining CNS homeostasis [71]; however, triggers, such as genetic predisposition, peripheral inflammation, or other risk factors, promote sustained microglial activation, which eventually fails to resolve and segues into chronic inflammation with ensuing synapse loss and neurodegeneration [44]. RAGE signaling via microglia [72, 73] may also contribute to neuroinflammation, Aβ aggregation, and CI in AD [74, 75]. Astrogliosis also occurs in AD, even prior to amyloid deposition; with progression, in tandem with activated microglia, reactive astrocytes also surround amyloid aggregates [76]. However, astrocytes more distant from amyloid may instead exhibit atrophy [77], with resulting deficits in neurotransmitter recycling and impaired neurotransmission [78].
Like in the T2D brain, mitochondrial dysfunction is also a potentially central AD characteristic [79, 80]. Mitochondria from AD patients show altered structure and function versus those in healthy neurons. AD mitochondria are smaller in size with swollen cristae, found associated with oxidatively damaged neurons [81]. Mitochondrial dynamics are impaired early in the AD disease course, with defects in biogenesis, fission:fusion balance, and mitophagy [82]. Functionally, based on human brain autopsy tissues, AD mitochondria are deficient in mitochondrial enzyme activities, such as pyruvate dehydrogenase complex [83], 2-ketoglutarate dehydrogenase [84], glutamate dehydrogenase, and ATP synthase [85]. Mitochondrial ATP production has been reported both as inhibited [86] and unaffected [82], requiring clarification. Nevertheless, these mitochondrial abnormalities may either contribute to or drive AD pathologies, which have birthed alternate theories on their involvement in disease development. From one predominant perspective, Aβ promotes mitochondrial dysfunction in AD, which is viewed as a neuropathology byproduct, a view aligned with the amyloid cascade hypothesis. Alternatively, a substantial body of evidence suggests mitochondrial dysfunction can occur independent of Aβ, potentially upstream, formulated as the “mitochondrial cascade hypothesis” [79], which has garnered interest [87, 88] but has not become mainstream; thus, questions persist on the precise contribution of mitochondrial dysfunction in AD. Oxidative stress is also present in AD, evidenced by elevated brain levels of oxidatively damaged proteins, lipids, DNA, and RNA [75].
Diabetes is long-known to cause vascular damage, giving rise to its diabetic complications, either as microvascular (small vessels) or macrovascular (large vessels) complications. Microvascular small vessel damage to peripheral nerves gives rise to diabetic peripheral neuropathy [15]. In the brain, diabetes injures the small vessels, promoting the development of white matter hyperintensities, lacunae, and microbleeds, and also damages the large vessels [25, 61]. Mechanisms underlying vascular dysfunction in diabetes are multifactorial; systemic hyperglycemia promotes AGE production [89], atherosclerosis development, buildup of proteinaceous deposits, and ROS formation along with oxidized lipotoxic species [90]. Brain endothelial-specific knockout of genes involved in removing proteinaceous deposits and inflammatory modulation compromises cognition and increases string vessel pathology, BBB disruption, and neuroinflammation, underscoring the contribution of vascular dysfunction to neurodegeneration [91, 92].
Vascular pathology in T2D manifests with slowed blood flow [93, 94], stiffening of the large vasculature that increases pressure and damage to the microvasculature, reduced vasoreactivity, basement membrane thickening, and increased capillary and BBB permeability [61]. Decreased perfusion leads to chronic ischemia and loss of neurovascular coupling, with consequent neuronal injury, CI, and eventual neurodegeneration. Indeed, the T2D brain exhibits both grey [95] and white matter atrophy in various regions, including hippocampus linked to memory and learning [96–98], although studies are not universally concordant [99]. T2D also increases vulnerability to stroke and post-stroke dementia [100, 101].
Likewise, the AD brain also exhibits vascular dysfunction [102, 103] and reduced cerebral blood flow [104], built from a body of epidemiological, clinical, and preclinical evidence. Aβ has vasoactive and vasculotoxic properties; Aβ interacts with blood vessel endothelial cells [105], BBB RAGE [106], and various other receptors [107], generating ROS and inflammatory responses that impair endothelial vasoactivity and cerebral blood flow. Additionally, cerebral amyloid angiopathy, the cerebrovascular deposition of Aβ, is a common age-related small vessel pathology associated with intracerebral hemorrhage and CI [108], that frequently occurs as an AD co-pathology [109]. Brain ischemia may also be a contributing factor to AD [110, 111].
Overall, studies demonstrate that similar multifactorial pathological processes occur in the T2D and AD brains, spanning insulin resistance, impaired glucose metabolism, mitochondrial dysfunction, inflammation, oxidative stress, and vascular dysfunction, although the interrelationship of these pathways, if any, in T2D to AD remains unclear. Moreover, the precise sequence of events and whether specific pathways are causal or associated with CI in T2D remain uncertain, and more mechanistic studies are needed. A deeper understanding of CI pathogenesis in T2D may yield therapeutic avenues warranting further investigation. The next sections discuss some points regarding the neuropathological hallmarks of AD, amyloid-β plaques and neurofibrillary tangles of tau protein, and their relationship to T2D.
Although T2D patients are at elevated risk of developing AD (see “Diabetes and comorbid risk factors raise dementia and AD risk” section), studies do not show a correlation between T2D measures, such as HbA1c [112], and amyloid burden, or a higher amyloid or neurofibrillary tangle burden in T2D patients [113, 114]. Rather, dementia in T2D patients is heterogeneous, encompassing vascular, AD, inflammatory, and metabolic processes with substantial inter-individual variability, which arises from a confluence of superimposing non-modifiable and modifiable risk factors spanning aging, genetics, extent of diabetes control, and lifestyle factors and other environmental exposures (Figure 2; see Biessels and Despa [25] for a review). Thus, T2D patients are at increased AD risk but not of AD neuropathology [25, 42]. In contrast (see section below), T2D in animal models and mechanistic studies does show a link to AD neuropathology. Herein, we discuss possible concepts to reconcile these differences.
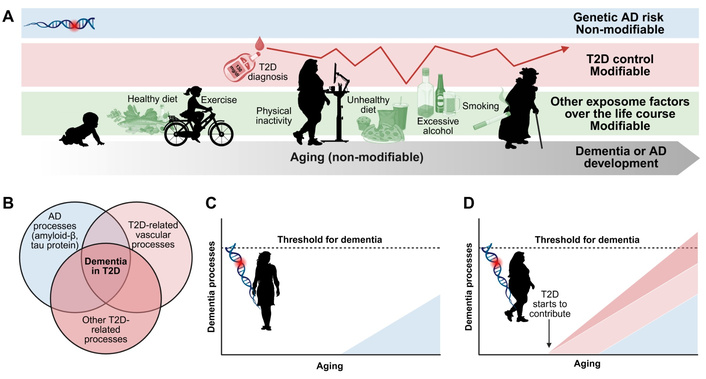
Dementia development in type 2 diabetes (T2D). (A) Dementia in T2D patients arises from a confluence of superimposing non-modifiable and modifiable risk factors spanning aging, genetics, extent of diabetes control [e.g., hyperglycemia, hypoglycemia events, glycated hemoglobin (HbA1c), etc.], and lifestyle factors and other environmental exposures. Each T2D individual will exhibit diverse risk factors that contribute to dementia or Alzheimer’s disease (AD) risk to varying extents. (B) Dementia in T2D patients is heterogeneous, encompassing AD processes (amyloid-β, tau protein), T2D-related vascular dysfunction, and other T2D-related processes (e.g., metabolic, mitochondrial, inflammatory, etc.) with substantial inter-individual variability. To illustrate how T2D might increase dementia risk, consider two individuals of equal genetic risk factors, (C) one lean and (D) one with T2D. AD genetic risk will contribute to AD-related processes in both the lean and T2D individuals. Vascular and other processes will superimpose on AD-related processes in the individual with T2D, who will be more likely to attain the threshold for dementia and AD development. The extent of T2D-related vascular and other processes will depend on the extent of diabetes severity and/or poor disease control. Color code in panels (C) and (D) same as in (B). Figure concept based on Biessels and Despa [25]. Created in BioRender. Savelieff, M. (2026) https://BioRender.com/gk6aikr.
T2D rodents develop CI, including genetic deficient leptin signaling db/db models [115, 116] and diet-based HFD mice with low-dose streptozotocin [116]. Even obese prediabetic HFD rodents already develop CI [117, 118] before T2D development. These animals also develop neuropathological AD hallmarks, with elevated Aβ [119, 120], tau, tau cleavage, and ptau protein levels [119, 121, 122] versus control counterparts. These rodent models are of metabolic dysfunction only, either T2D or obesity and prediabetes, and are not crossed to AD mouse models. Thus, T2D is linked to AD neuropathology in rodent models, contrary to human studies. Additionally, crossing T2D [123] or feeding HFD [124] to AD mice aggravates Aβ pathology, brain atrophy, and CI [125]. Molecularly, several pathways may link insulin resistance, hyperglycemia, and dyslipidemia in diabetes to Aβ and tau pathology [126, 127]. For instance, insulin resistance blunts insulin-IRS-AKT signaling leading to constitutive GSK3α and GSK3β activation [42]. GSK3α activation, as a kinase involved in APP processing, would promote amyloid pathology [128], while GSK3β activation, as the kinase phosphorylating tau protein, would lead to tau pathology [129]. Another potential pathway may be via hyperglycemia-induced increase in methylglyoxal levels and glycation [130], which, when modifying Aβ, slows fibril aggregation [131], maintaining them longer in their toxic oligomeric forms [132].
Although the T2D link to amyloid and tau burden differs in human and preclinical models, it is possible to reconcile findings. Epidemiological and clinical data show that T2D patients develop a mixed, heterogeneous vascular and AD-like dementia with inflammatory and metabolic aspects; thus, in humans, the added dimension of vascular pathology may contribute to higher AD risk rather than any impact of T2D directly on AD neuropathology. On the other hand, most transgenic AD animal models that are crossed to T2D models are genetically manipulated to express highly penetrant APP and PSEN mutations, a scenario distinct from most sporadic human AD; therefore, studies on the impact of T2D in AD animal models may bias findings towards pathways with a strong AD neuropathology component. In studies using metabolic T2D or obesity models, animals are usually not aged, and so may not fully mirror the features of vascular aging and other age-related pathologies in human T2D. In vitro studies in neuronal cultures mostly completely omit the vascular component, which is a crucial feature of dementia and AD in T2D patients. Thus, overall, preclinical models may not comprehensively recapitulate T2D-related AD [25], and research is needed to develop better models.
Since T2D does not enhance Aβ and tau neuropathology in humans, excess dementia risk instead arises from vascular and other diabetes pathways, e.g., insulin resistance, mitochondrial dysfunction. The amyloid cascade hypothesis [133] and related amyloid, tau, and neurodegeneration (ATN) [134] biological framework have dominated conversations in AD; the “pathology-first” mindset assumes that individuals with AD have the same underlying biology. However, although amyloid is integral to the definition of AD, burden does not necessarily correlate with cognitive performance [135]. Many cognitively normal individuals have substantial amyloid burden [136, 137]. Thus, it may be necessary to revise the amyloid cascade hypothesis [138] and centrally integrate additional multifactorial pathways in AD as processes upstream or concurrent with Aβ and tau protein. Furthermore, Aβ and tau protein have historically governed thinking, although AD is biologically heterogeneous with overlapping mechanisms. Thus, AD endotypes or molecular subtypes may exist, wherein multiple underlying mechanisms give rise to the same clinical phenotype [139], including a distinct T2D-related version [140]. This is a growing research direction.
These concepts have important implications for AD research, clinical trial design, participant stratification, and drug development. As many have noted, these similar pathways in T2D and AD, antidiabetic medications are increasingly considered for AD [141]. Although AD treatments are in dire need, the next section will focus on early interventions, in persons with T2D, and what approaches might help curb their risk of developing dementia or AD.
What are the implications of this relationship between T2D and dementia on patient clinical care? Compared to diabetic peripheral neuropathy, clinical guidelines for diabetes began to highlight the risk of cognitive decline and dementia in diabetes clinical care relatively more recently, only a decade and a half ago [25]. The American Diabetes Association guidelines and standards of care for older adults with diabetes recommend early screening for mild CI (MCI) or dementia in adults aged ≥ 65 years at the initial visit, and annually thereafter or as appropriate [142]. Routine screening for MCI in the clinic typically involves questionnaires [143]; an abnormal screen may prompt more detailed assessment using comprehensive neuropsychological testing and possibly, where indicated, magnetic resonance imaging. Emerging approaches involving blood biomarkers and retinal markers are also being investigated [144, 145] and may help detect T2D-related MCI earlier for earlier intervention.
Given that T2D is a dementia risk factor, can optimally controlling diabetes and its comorbid risk factors mitigate dementia risk and onset [146]? A study found that individuals with diabetes exhibited a stepwise decrease in excess dementia risk for each risk factor on target, e.g., guideline HbA1c, body mass index (BMI), blood pressure, albuminuria, physical activity, diet, and nonsmoking [147]. Individuals with diabetes with five to seven risk factors on target had no significant excess dementia risk versus controls, suggesting that good management of comorbidities can mitigate dementia risk. The study suggests, encouragingly, that controlling diabetes and its comorbidities could mitigate dementia risk. In this section, we highlight such approaches, spanning from delaying, or ideally preventing, T2D onset, to adopting a healthy lifestyle in the T2D context, social determinants, and antidiabetic pharmacological agents and interventions.
As outlined in the “Diabetes and comorbid risk factors raise dementia and AD risk” section, individuals with earlier T2D diagnosis are at greater risk of brain atrophy [28] and dementia [28, 30]. Analysis of UK Biobank at 12-year follow-up found excess all-cause, AD, and VaD risk in T2D overall [28]; however, in age stratified analysis, excess VaD risk was fully attributable to T2D onset prior to 50 years of age, while all-cause dementia and AD excess risk were most evident in T2D onset < 50 years of age although other onset age groups were also at risk. In analysis of Whitehall II at 32-year follow-up, compared to individuals without T2D aged 70 years, every 5-year increment in earlier T2D onset increased dementia risk [30]. Therefore, preventing or delaying T2D onset is imperative for better long-term brain health outcomes; T2D prevention and/or delaying approaches span lifestyle interventions through diet and exercise as well as pharmacotherapy [8]. Wearable technologies may track cardiometabolic metrics, e.g., weight, BMI, and lifestyle, e.g., dietary habits, exercise duration, helping individuals gauge their health status [8]. Technology through online coaching, social media, and gamification may also help. Methods that identify individuals most at risk of T2D can direct more intensive efforts or allocation of resources and public health policy can implement national and community programs [8].
Although the optimal approach would be prevention, some individuals nevertheless develop T2D, even despite efforts. This could be attributable to genetic predisposition to T2D [148], inability to adhere to recommended guidelines [8], or life factors [149] that thwart implementing ideal prevention approaches, e.g., socioeconomic status, lack of access to health resources, lack of access to green spaces or exercise. Nevertheless, some research shows that, even in individuals with T2D, adopting a healthy lifestyle can curb the risk of dementia development. Exercise, both resistance and endurance training, may exert salutary effects on cognition and brain structure and function in individuals with T2D [150]; however, further investigation, especially of exercise intensity and duration, is required to identify the optimal regimen or personalized approaches, and determine whether it can prevent dementia onset and development. The situation is similar regarding dietary habits; although there is evidence that diverse diets, both in composition [151] and time-restricted eating [152], may be beneficial to brain health, the mechanisms are incompletely understood, and the ideal approach or personalized strategies for preventing dementia and/or AD onset remain uncertain [153]. Nonetheless, T2D patients should be highly encouraged to adopt a healthy lifestyle; working from UK Biobank data, Zhou et al. [154] found that persons with diabetes who managed to adhere to more favorable healthy lifestyle habits were only at twice the risk while those not adhering were at three times the risk of all-cause dementia versus diabetes-free participants adhering to a healthy lifestyle. Adopted healthy lifestyles dose-dependently lowered VaD risk in diabetes cases but did not impact AD risk. Although the results are encouraging, reduced dementia risk from healthy lifestyle adoption was most pronounced in patients with shorter diabetes duration (< 10 years), suggesting the need to early implementation, or those without insulin use [154].
Just as a higher level of disadvantage can impact T2D [149] and dementia [155, 156] onset, similarly, these circumstances can also potentially raise dementia risk in individuals with T2D, although this is a less studied aspect. Social determinants of health (SDoH) are modifiable risk factors that constitute an avenue for mitigating future burden of dementia. Importantly, most individuals living with T2D [7] and/or dementia [2] reside in lower and lower-middle incomes countries, making SDoH a topic of global concern.
Adverse SDoH lower individuals’ access to high-quality care, which delays diabetes diagnosis, leads to suboptimal medication use and poor disease control, and lack of comorbidity management, e.g., obesity, hypertension [157]. Chronic stress from socioeconomic pressures increases allostatic load, promoting insulin resistance, elevated cortisol, and inflammation [158]. Socially disadvantaged people have limited access to healthy foods and green spaces for exercise and fewer opportunities for higher education and more cognitively stimulating jobs. All these conditions, as dementia risk factors, can predispose socioeconomically disadvantaged T2D patients to future AD and other dementia development.
Zhong et al. [159] analyzed T2D cases from UK Biobank (n = 17,321), extracting data on 17 SDoH to generate a composite [159]. Versus T2D participants in the highest SDoH tertile, those in the middle and lowest tertile had hazard ratios of 1.35 and 1.76, respectively, for incident dementia, adjusted for age, sex, diabetes duration, Hb1Ac, BMI, systolic blood pressure, comorbidities, and various lifestyle factors. This study represents emerging evidence that adverse SDoH may negatively impact brain health in T2D diabetes patients.
Antidiabetic medications and interventions can mitigate dementia and AD risk by controlling systemic cardiometabolic risk factors, for instance via weight loss and improved glucose handling. Moreover, antidiabetic medications, if BBB-permeable, and interventions can potentially exert neuroprotective mechanisms in the brain microenvironment, ameliorating cognition. Several investigations have examined whether antidiabetic medications and interventions exert salutary benefits to cognition and risk of dementia, including AD (Table 3).
Summary of select studies on the impact of antidiabetic interventions on cognitive impairment/decline or Alzheimer’s disease/dementia risk.
| Study | Region and timeline* | Study participants and design | Study tools and interventions | Study findings |
|---|---|---|---|---|
| Glucagon-like peptide-1 receptor agonists (GLP-1RAs) | ||||
| Lin et al. [162] | USA, 2017–2024 | Obese T2D participants on GLP-1RA (n = 30,430) propensity score-matched to other antidiabetic medication users (n = 30,430); real-world, longitudinal, retrospective cohort | TriNetX US collaborative network. GLP-1RA use included semaglutide, tirzepatide. Primary outcomes: incidence of neurodegenerative and cerebrovascular disease. Secondary outcome: all-cause mortality. Propensity score-matched for age, sex, ethnicity, race, HbA1c, BMI, estimated glomerular filtration rate, comorbidities, medications, lifestyle, health care use, socioeconomic status. | At least 1 year follow-up; GLP-1RA vs. other antidiabetic medication users at lower dementia (HR 0.63, 95% CI 0.50, 0.81) and all-cause mortality (HR 0.70, 95% CI 0.63, 0.78) risk but not MCI (HR 1.13, 95% CI 0.87, 1.46). By dementia subtype, GLP-1RA users not at lower AD or VaD risk. By GLP-1RA subgroup, semaglutide may be more protective against dementia, tirzepatide against stroke. |
| Nassar et al. [163] | Worldwide | T2D participants on GLP-1RA (n = 854,197) propensity score-matched to without GLP-1RA (n = 854,197); real-world, longitudinal, retrospective cohort | Global Collaborative Network via TriNetX analytics platform. Participants treated with or without GLP-1RAs. Diagnoses by ICD codes for incidence and risk differences in dementia, AD, and various other illnesses. Propensity score-matched for demographics and clinical characteristics. | At least a 5-year follow-up; GLP-1RA users at lower dementia (risk difference –0.010, 95% CI –0.010, –0.009, p < 0.001) and AD (risk difference –0.003, 95% CI –0.003, –0.003, p < 0.001) risk. |
| Siddeeque et al. [164] | Worldwide, 2010–2023 | Obese participants on GLP-1RA (n = 102,935) propensity score-matched to without GLP-1RA (n = 102,935); real-world, longitudinal, retrospective cohort | Global Collaborative Network via TriNetX analytics platform. GLP-1RA use included semaglutide, liraglutide, dulaglutide. Diagnoses by ICD codes for primary outcomes: incidence of AD, VaD, other. Secondary outcomes: all-cause mortality and incidence of other neurodegenerative diseases. Propensity score-matched for demographics and comorbidities. | Variable follow-up (shorter in GLP-1RA users); diabetes prevalence 54.1% after matching; GLP-1RA users at lower AD (RR 0.627, 95% CI 0.481, 0.817, p < 0.001) and VaD (RR 0.438, 95% CI 0.327, 0.588, p < 0.001) risk. By GLP-1RA subgroup, semaglutide but not liraglutide or dulaglutide users at lower AD and VaD risk. |
| Sodium-glucose cotransporter 2 inhibitors (SGLT2is) | ||||
| Youn et al. [168] | Worldwide, 2018–2023 | Systematic review, meta-analysis registered in PROSPERO (CRD42023461758) | Population: adults aged ≥ 18 years at enrollment; intervention: SGLT2i use; comparison: no SGLT2i and/or other antidiabetic medication use; outcomes: dementia incidence as adjusted HRs or ORs and/or cognitive function score changes; study design: longitudinal comparative studies. 11 included studies. | Vs. non-users, SGLT2i use linked to significantly lower dementia risk (HR 0.68, 95% CI 0.50, 0.92). SGLT2i use significantly linked to improved cognitive function (standardized mean difference 0.88, 95% CI 0.32, 1.44), particularly in MCI or dementia. |
| Zhang et al. [169] | Worldwide, 2005–2025 | T2D participants initiating SGLT2i propensity score-matched to without SGLT2i; real-world, longitudinal, retrospective cohort | Global Collaborative Network (> 150 health systems worldwide). Applied a 456-day washout to reduce protopathic and immortal time bias. Participants stratified by baseline HbA1c (< 7.0%, 7.0–8.9%, ≥ 9.0%). Primary outcome: incident all-cause dementia. Secondary outcomes: dementia subtypes, major adverse cardiovascular events, major adverse kidney events, osteoporotic fracture as a negative control. Propensity score-matched for demographics, comorbidities, and medication use. | Median 4.2 years follow-up; vs. Hb1Ac < 7.0%, higher baseline HbA1c (HR 1.32 for HbA1c 7.0–8.9%; HR 1.68 for HbA1c ≥ 9.0%) significantly linked to higher dementia risk. Associations consistent but attenuated in propensity score-matched models (HR 1.08 and 1.28). Associations strongest for VaD. |
| GLP-1RA, SGLT2is, versus other antidiabetic medications | ||||
| Li et al. [171] | Worldwide | Systematic review, network meta-analysis registered in PROSPERO (CRD42022379082) | Population: adults with T2D; intervention: antidiabetic vs. other antidiabetic agents or placebo; comparison: interchangeable with intervention; outcomes: dementia events; study design: observational studies (cohort, case-control, and cross-sectional) or RCTs. 41 observational studies (3,307,483 participants), 23 RCTs (155,443 participants) included. Follow-up period was not restricted. | In network meta-analysis of observational studies, vs. non-users, use of SGLT2i (OR 0.56, 95% CI 0.45, 0.69), GLP-1RA (OR 0.58, 95% CI 0.46, 0.73), TZD (OR 0.68, 95% CI 0.57, 0.81), metformin (OR 0.89, 95% CI 0.80, 0.99) significantly linked to lower dementia risk. Rank order SGLT2i > GLP-1RA > TZD > DPP-4i > metformin > AGI > GKA > sulfonylureas > glinides > insulin on cognitive benefits. Vs. non-users, use of SGLT2i (OR 0.43, 95% CI 0.30, 0.62), GLP-1RA (OR 0.54, 95% CI 0.30, 0.96), DPP-4i (OR 0.73, 95% CI 0.57, 0.93) significantly linked to lower AD risk, while SGLT2i (OR 0.42, 95% CI 0.22, 0.80), TZD (OR 0.52, 95% CI 0.36, 0.75) linked to lower VaD risk. In network meta-analysis of RCTs, dementia risks comparable among antidiabetic agents and placebo. |
| Schechter et al. [177] | Worldwide, 2010–2021 | T2D participants on GLP-1RA (n = 107,221) propensity score-matched to DPP-4i users (n = 107,221); real-world, longitudinal, retrospective cohort | TriNetX platform of EHRs of > 170 million people worldwide. GLP-1RA use included semaglutide, liraglutide, dulaglutide. Diagnoses by ICD codes for dementia, AD, VaD, and other. Propensity score-matching used 40 baseline characteristics of demographic variables, medical diagnoses, laboratory measurements, medication use. | Mean 4.0 years follow-up; GLP-1RAs vs. DPP-4i users at lower dementia (HR 0.76, 95% CI 0.72, 0.81), AD (HR 0.77, 95% CI 0.68, 0.87), VaD (HR 0.75, 95% CI 0.67, 0.85) risk, unadjusted for multiple comparisons. Results in the same direction GLP-1RAs vs. basal insulin users. |
| Seminer et al. [179] | Worldwide, 2015–2023 | Systematic review, meta-analysis registered in PROSPERO (CRD42024557562) | Population: adults aged ≥ 18 years; intervention: SGLT2is, GLP-1RAs, metformin, pioglitazone use; comparison: RCT control arm; outcomes: dementia, CI, and/or change in cognitive score; study design: RCTs with minimum 6-mo follow-up. Total 26 RCTs included 164,531 participants. 12 (SGLT2i), 10 (GLP-1RA) RCTs included in meta-analysis for all-cause dementia. | Overall, glucose-lowering therapy was not significantly linked to lower CI or dementia risk (OR 0.83, 95% CI 0.60, 1.14). By class, GLP-1RAs were significantly linked to lower dementia risk (OR 0.55, 95% CI 0.35, 0.86) but not SGLT2is (OR 1.20, 95% CI 0.67, 2.17). |
| Song et al. [175] | Worldwide, 2010–2022 | Systematic review, meta-analysis registered in PROSPERO (CRD420251037959) | Population: T2D adults aged ≥ 18 years; intervention: SGLT2is, GLP-1RAs, DPP-4i; comparison: interchangeable with intervention; primary outcome: incidence of all-cause dementia; secondary outcomes: AD, VaD, all-cause mortality. 9 included studies. | SGLT2i use significantly linked to lower all-cause dementia risk vs. DPP-4i (HR 0.67, 95% CI 0.59, 0.77) and GLP-1RA (HR 0.93, 95% CI 0.86, 1.00) use. SGLT2i use significantly linked to lower AD risk vs. DPP-4i (HR 0.49, 95% CI 0.35, 0.70) and GLP-1RA (HR 0.68, 95% CI 0.52, 0.88) use. Empagliflozin consistently most protective among SGLT2i agents. Subgroup analyses reveal more benefits for patients aged > 65 years. |
| Tang et al. [174] | Sweden, 2010–2020 | T2D participants (n = 88,381); incident dementia cases (n = 4,607) at follow-up; retrospective, emulated trial study | Various Swedish national registers. T2D by ICD codes or with any non-insulin antidiabetic medication prescription; medication initiation by Anatomical Therapeutic Chemical codes; dementia by ICD codes. Inverse-probability weighting by propensity scores balanced baseline characteristics between groups. | In intention-to-treat analysis, GLP-1RA initiation was significantly linked to lower dementia risk vs. sulfonylureas (HR 0.69, 95% CI 0.60, 0.79, p < 0.0001) and DPP-4i (HR 0.77, 95% CI 0.68, 0.88, p < 0.0001), adjusted for age, sex, enrollment year, health conditions, past medication use, socioeconomic status. Findings consistent in several sensitivity analyses, including a per-protocol analysis. |
| Wu et al. [176] | United Kingdom, 2007–2022 | T2D participants (n = 13,965 pairs of GLP1-RA vs. DPP-4i; n = 25,533 pairs SGLT2i vs. DPP-4i; n = 14,214 pairs GLP1-RA vs. SGLT2i); population-based, emulated trial study | UK Clinical Practice Research Datalink. Participants with HbA1c ≥ 6.5% or T2D diagnosis; GLP1-RA, SGLT2i, DPP-4i medication initiators; dementia by ICD and other codes. Propensity scores overlap weighting for various covariates. | In intention-to-treat analysis, dementia risk did not differ in GLP1-RA vs. DPP-4i initiators (HR 0.95, 95% CI 0.87, 1.04; 6.54 years mean follow-up); continuous GLP1-RA vs. DPP-4i use linked to 21% lower risk (HR 0.79, 95% CI 0.64, 0.97; 2.71 years mean follow-up). In intention-to-treat analysis, dementia risk lower by 14% in SGLT2i vs. DPP-4i initiators (HR 0.86, 95% CI 0.79, 0.94; 4.91 years mean follow-up); continuous SGLT2i vs. DPP-4i uses linked 30% lower risk (HR 0.70, 95% CI 0.60, 0.82; 2.43 years mean follow-up). Dementia risk similar in GLP1-RA vs. SGLT2i initiators and continuous users. |
| Zhang et al. [178] | USA, 2005–2023 | Participants initiating GLP-1RA, SGLT2i, DPP-4i; real-world, longitudinal, retrospective cohort | Optum Clinformatics data, Northwestern Medicine Enterprise Data Warehouse. GLP-1RA use included albiglutide, dulaglutide, exenatide, liraglutide, lixisenatide, semaglutide; SGLT2i use included canagliflozin, dapagliflozin, empagliflozin, ertugliflozin; DPP-4i use included alogliptin, linagliptin, saxagliptin, sitagliptin; use of insulin, metformin, sulfonylureas, TZD, other non-insulin blood glucose-lowering agents also collected. AD by ICD codes. | Vs. DPP-4i, GLP-1RA use (HR ≤ 0.69, p < 0.001) and SGLT2i use (HR ≤ 0.6, p < 0.001) linked to lower AD risk. |
| Bariatric surgery | ||||
| Alosco et al. [189] | USA | Obese participants with/without family history of AD (n = 94); prospective, multi-center | Longitudinal Assessment of Bariatric Surgery (LABS) study. Most participants underwent RYGB. Cognition assessed by WebNeuro in attention/executive function, memory, language. | 12-week follow-up; 26.6% had diabetes. In overall sample and participants without family AD history, memory and attention/executive function improved 12 weeks post-surgery. In participants with family AD history, only attention/executive function improved 12 weeks post-surgery. Family history of AD linked to higher post-surgery CI rates. |
| Chen et al. [188] | USA, 2000–2023 | Obese participants (n = 5,303) with bariatric surgery, propensity score-matched to obese participants (10,606) without bariatric surgery; retrospective, longitudinal, single-center | Participants underwent RYGB or SG. ADRD via EHR review with ICD-9/10 codes for AD, VaD, Lewy body dementia, frontotemporal dementia, primary progressive aphasia. MCI via EHR review with ICD-9/10 codes. | At baseline, 20.3% of participants overall had T2D. Bariatric surgery was significantly linked to reduced ADRD (SHR 0.37, 95% CI 0.15, 0.89, p = 0.03) and reduced MCI incidence (SHR 0.57, 95% CI 0.39, 0.85, p = 0.01), adjusted for demographics, medical history, and socioeconomic status. Bariatric surgery significantly delayed MCI development by 2.01 years (95% CI 0.70, 3.50, p = 0.004). |
| Custers et al. [185] | Netherlands, 2018–2020 | Obese participants (n = 133); longitudinal, single-center | See Custers et al. [184]. Additionally, MRI for brain volume, cortical thickness, white matter hyperintensities, cerebral blood flow (CBF), spatial coefficient of variation (sCOV). | 2 years post bariatric surgery, global cognition > 20% higher in 42.9% (n = 52) of participants; brain structure and perfusion lower in most brain regions; hippocampal and white matter volume stable; no change in CBF and sCOV in nucleus accumbens and parietal cortex; higher temporal cortex thickness and lower sCOV. |
| Custers et al. [184] | Netherlands, 2018–2020 | Obese participants (n = 107); longitudinal, single-center | BARICO study (BAriatric surgery Rijnstate and Radboudumc neuroImaging and Cognition in Obesity). Participants underwent RYGB. General cognition, working memory, episodic memory, shift attention, verbal fluency. Education level. | 3 years post bariatric surgery, global cognition > 20% higher in 38.6% (n = 39) of participants; inflammatory factors, leptin, matrix metalloproteinase-9, and apolipoprotein A1 levels remained lower, whereas adiponectin and angiopoietin-1 levels remained higher; depressive symptoms remained lower, whereas physical activity returned to baseline. |
| Ghanim et al. [190] | USA | Obese participants (n = 15) with T2D; longitudinal | Participants underwent RYGB. AD biomarkers and AD-related gene expression in peripheral blood mononuclear cells. | 6-month follow-up; post RYGB, significantly lower plasma glucose and insulin levels, lower homeostasis model of assessment for insulin resistance, lower APP mRNA and protein, lower presenilin-2, ADAM-9, GSK-3β, PICALM, SORL-1, and clusterin levels, lower C-reactive protein and monocyte chemoattractant protein-1 levels. |
| Reynolds et al. [182] | USA, 2015–2018 | Obese participants (n = 87); longitudinal, single-center | Most participants underwent SG. Cognition assessed by NIH toolbox cognitive battery (NIHTB-CB) and Rey Auditory Verbal Learning Test (AVLT). Primary outcome: Change in NIHTB-CB fluid composite score. | 2-year follow-up; 24 (27.6%) participants had diabetes, 25 (28.7%) on anti-hyperglycemic medications. Post bariatric surgery, no change in NIHTB-CB composite score [mean change (SD) –0.4 (13.9), p = 0.81, n = 66], significantly higher NIHTB-CB executive function [+6.5 (19.9), p = 0.01, n = 66], significantly lower Rey AVLT memory [−0.24 (0.83), p = 0.01, n = 87]. Improved cognition did not correlate with improved metabolic risk factors and diabetes complications. |
| Smith et al. [183] | USA, 2016–2019 | Obese participants (n = 35); longitudinal, single-center | Participants split between RYGB or SG. A cognitive battery spanning 4 cognitive domains: auditory attention, processing speed, auditory-verbal memory, executive function. Overall composite scores. | RYGB improved weight loss at 1 year follow-up to a greater extent vs. SG. Post SG and RYGB, significantly higher processing speed, auditory attention measures, executive function measures. Only post RYGB, significantly higher executive function. Baseline auditory attention and memory predicted 1 year weight loss post RYGB but not SG. Adjusted for baseline cognition, % total weight loss predicted 1 year auditory attention post RYGB but not SG. |
Entries arranged alphabetically by the first author’s name. *: incomplete years of study enrollment and duration rounded to the indicated year. 95% CI: 95% confidence interval; AD: Alzheimer’s disease; ADRD: Alzheimer’s disease-related dementia; AGI: α-glucosidase inhibitor; BMI: body mass index; CI: cognitive impairment; DPP-4i: dipeptidyl peptidase 4 inhibitors; EHRs: electronic health record; GKA: glucokinase activator; HbA1c: glycated hemoglobin; HR: hazard ratio; ICD: International Classification of Diseases; MCI: mild cognitive impairment; MRI: magnetic resonance imaging; OR: odds ratio; RCT: randomized clinical trial; RR: relative risk; RYGB: Roux-en-Y gastric bypass; SD: standard deviation; SG: sleeve gastrectomy; SHR: subdistribution hazard ratio; T2D: type 2 diabetes; TZD: thiazolidinedione; VaD: vascular dementia.
GLP-1RAs have become a significant facet of T2D management [160]. GLP-1RAs promote insulin secretion as well as satiety by acting on hypothalamic neurons, in turn potentiating weight loss. GLP-1RAs additionally exert effects in the brain milieu [161], augmenting central insulin signaling, promoting neurite outgrowth, enhancing mitochondrial biogenesis, and lowering inflammation and AD biomarkers [160]. A review of GLP-1RAs [160] of 17 studies, mostly of randomized clinical trials of T2D participants with or without MCI or AD concluded possible improvement in some brain markers (e.g., hippocampal connections, cerebral glucose metabolism) albeit without strong correlations to cognition. Several more recently published pharmacoepidemiological studies suggest GLP-1RAs in patients with T2D and/or obesity may delay progression to dementia [162, 163], AD [163, 164], and VaD [164] versus non-GLP-1RA users or users of other antidiabetic medications [162]. One study noted benefit from GLP-1RAs on dementia overall but not on either AD or VaD [162]. However, many studies to date have been retrospective, and some may have used overlapping populations [164], necessitating corroboration in prospective cohorts.
SGLT2 and more recently, dual SGLT1/2is are another increasingly important class of antidiabetic medications. SGLT2is block glucose resorption in the kidneys while SGLT1/2is additionally inhibit resorption in the gastrointestinal tract effectively managing glucose levels in T2D and, more broadly, improving weight and other cardiovascular comorbidities. SGLT2is may also potentially exert neuroprotective effects in the brain with anti-inflammatory, anti-oxidative stress, metabolism enhancing, and vascular health promoting properties [165, 166]. A recent scoping review of SGLT2is that spanned 14 mostly longitudinal studies found the drug class could exert neuroprotective effects in T2D patients, lowering incidence or progression of MCI and dementia but concluded further prospective studies were required [167]. Since, a meta-analysis and systematic review found SGLT2i use significantly lowered dementia risk and improved cognitive function on neuropsychological testing [168]. Retrospective analysis of real-world data from the large Global Collaborative Network found that T2D participants that had initiated SGLT2i with better-controlled diabetes at baseline (Hb1Ac < 7.0%) were at lower risk of dementia development by 4-year follow-up than those with poorly controlled diabetes at baseline (Hb1Ac ≥ 7.0%) [169]. The study highlights the need to implement any potentially effective pharmacotherapy earlier in the diabetes disease course, although validation is needed. We found no data on the sole SGLT1/2i in the market, sotagliflozin, on cognition or dementia in T2D.
Most T2D patients receive metformin as first-line treatment while GLP-1RAs and SGLT2is are relatively newer antidiabetic medications, albeit rising in prescriptions [170]. Metformin use in T2D is potentially linked to lower dementia risk [171–173] but may be less effective than GLP-1RAs and SGLT2is [171]. GLP-1RAs and/or SGLT2is may outperform other antidiabetic medications against dementia [171, 174–176], AD [175, 177, 178], or VaD [177], such as dipeptidyl peptidase 4 inhibitors [171, 174–178], thiazolidinediones, and sulfonylureas [171, 174] in meta-analyses [171, 175], real-world data [177, 178], and emulated trial study designs [174, 176]. Regarding GLP-1RAs versus SGLT2is, they are reported to be equally effective [176], GLP-1RAs more effective [179], or SGLT2is more effective [171, 175].
Although many studies have been conducted, they suffer from the usual issues of retrospective analysis, while meta-analyses are limited by whether the included studies fully adjusted for confounding [175] and the presence of heterogeneity. In pharmacoepidemiological studies, medication use does not necessarily translate to optimal T2D control; ideally, study controls need to be propensity score matched for HbA1c and other parameters, but it is not clear whether this is always performed. Additionally, in real-world settings, patients may be on multiple medications, which need to be accounted for. Large, prospective cohorts are required to more clearly define the optimal pharmacological approach for T2D management, including type, dose, and timing, but also, potentially, for mitigating complications, including future dementia or AD risk. Safety profiles are also important, especially for a chronic illness in the long-term. For instance, metformin has a long-established safety profile in real-world scenarios, whereas the newer classes of antidiabetic medications may have less established safety profiles, particularly in real-world settings. Finally, there are sex differences in the effectiveness of antidiabetic medications, e.g., GLP-1RAs [180], and this may impact their ability to prevent dementia by sex, as examined by some studies [162, 174, 176, 178]. Overall, although mounting evidence suggests pharmacological management of T2D may mitigate dementia and AD risk, further investigation is needed in prospective cohorts and the optimal approach remains uncertain.
Bariatric surgery is a management strategy for class II/III obesity [181], which is a frequent T2D comorbidity making many diabetes patients eligible. Guidelines are also expanding eligibility criteria to people with T2D more broadly [181]. Indeed, besides improving BMI and other obesity metrics, such as waist circumference [182–186], bariatric surgery can also ameliorate glucose metrics, e.g., HbA1c, fasting blood glucose [181, 182]. The most frequent bariatric procedure is sleeve gastrectomy, which removes 80% of the stomach, limiting food intake and lowering ghrelin release, an appetite-stimulating hormone [181]. Sleeve gastrectomy has surpassed Roux-en-Y gastric bypass, which reduces the stomach to a small pouch and connects it to the mid-jejunum. Roux-en-Y gastric bypass limits food intake but also decreases macronutrient absorption, which can similarly prevent micronutrient absorption with consequent deficiencies. A 2021 review emphasized emerging evidence that bariatric surgery may ameliorate cognition in obese patients, concluding that further investigation was necessary [187]. More recent studies built upon this early evidence [182–185], potentially with long-term benefits by reducing the risk of AD development [188] though possibly only in patients without family history [189]. A very small study found bariatric may reduce circulating AD biomarkers in participants with T2D and obesity [190]. The efficacy of bariatric surgery for effectively preventing or delaying dementia or AD onset in patients with T2D and whether it outperforms pharmacological or lifestyle approaches presently remains an area of research.
Finally, there is interest in combatting brain senescence to prevent dementia onset. With normal aging, the cells of the brain develop genomic instability, epigenetic changes, and impaired proteostasis, with consequent senescence and loss-of-function in the essential activities needed for cognition [191, 192], manifesting as normal age-related cognitive decline. T2D accelerates senescence and aging [193], which may contribute to the development of cognitive decline and dementia. Brain rejuvenation by reversing senescence through diet and exercise is being explored [194–197], as are senolytics, a class of compounds designed to selectively eliminate senescent cells to slow or counteract aging. However, to date, most work on senolytics against CI development has been conducted in preclinical models [192]. A phase 1 feasibility trial of dasatinib and quercetin in mild AD just concluded (NCT04063124) [198], and preliminary exploratory biomarker results have been published [199]. A phase 2 trial of dasatinib and quercetin for MCI and early AD is planned, anticipated to end by 2029 (NCT04685590).
It is now well-established that diabetes adversely affects brain health, predisposing patients to dementia and AD development. However, questions remain about the precise mechanistic and molecular links between T2D and dementia, and should be an avenue of future research to inform possible therapeutic avenues. As communicable diseases wane as a public health issue, non-communicable diseases are gaining in prominence, in substantial part attributable to metabolic dysfunction from T2D and obesity, which promote neurological illness, including AD and dementia [34, 200].
First and foremost, prevention strategies must guide public health policy as well as community and individual efforts, prioritizing availability of healthy foods and diets, green spaces for exercise, healthcare access, and health literacy. Health awareness campaigns, for instance, modeled after “World Diabetes Day” launched by the International Diabetes Federation or “World Brain Day” promoted by the World Federation of Neurology can disseminate knowledge. Additionally, discussions with primary care providers during preventative healthcare visits and grants and public resource earmarking for parks and other recreational spaces could serve as potential avenues to facilitate prevention efforts. It is especially crucial as much as possible to urge T2D prevention in youths, who would otherwise have to grapple with earlier onset of associated complications with poorer health over their life course. This could be addressed by healthy meal options at school, after school sports activities, regular preventative health care checks, and targeted efforts for high-risk groups, such as funding for healthy foods for youths from lower-income backgrounds. For patients who do develop T2D, efforts should emphasize early steps to manage hyperglycemia and comorbidities by adopting healthy lifestyle habits and appropriate pharmacological control. Again, public health policy and community and individual choice can prioritize mitigating the modifiable risk factors that aggravate T2D and risk for dementia.
As the command center for most bodily functions, thought, and engagement, “there is no health without brain health” [201]; safeguarding it should be of utmost importance.
95% CI: 95% confidence interval
AD: Alzheimer’s disease
AGEs: glycation end products
APOE4: apolipoprotein E4
APP: amyloid precursor protein
Aβ: amyloid-beta
BBB: blood-brain barrier
BMI: body mass index
CI: cognitive impairment
GLP-1RAs: glucagon-like peptide-1 receptor agonists
GLUTs: glucose transporters
HbA1c: glycated hemoglobin
HFD: high-fat diet
MCI: mild cognitive impairment
PSEN1: presenilin-1
PSEN2: presenilin-2
RAGE: receptor for glycation end product
ROS: reactive oxygen species
SDoH: social determinants of health
SGLT2is: sodium-glucose cotransporter 2 inhibitors
T2D: type 2 diabetes
VaD: vascular dementia
The supplementary table for this article is available at: https://www.explorationpub.com/uploads/Article/file/1006139_sup_1.pdf.
The authors thank Emily J. Koubek, PhD, for general assistance.
ZGV: Writing—original draft, Writing—review & editing, Visualization. MGS: Conceptualization, Writing—original draft, Writing—review & editing, Visualization. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 552
Download: 21
Times Cited: 0
Atif Salim Khatib ... Daniya Tasnim
Octavio García ... Jesús Antonio Villegas-Piña
Graziella Martins Guimarães ... Márcia Maria de Souza
