 Review
Review

Affiliation:
1Faculty of Medicine, Tbilisi State Medical University, Tbilisi 0160, Georgia
ORCID: https://orcid.org/0009-0009-7821-9292
Affiliation:
1Faculty of Medicine, Tbilisi State Medical University, Tbilisi 0160, Georgia
Email: islam1048@gmail.com
ORCID: https://orcid.org/0009-0007-4855-6226
Affiliation:
1Faculty of Medicine, Tbilisi State Medical University, Tbilisi 0160, Georgia
ORCID: https://orcid.org/0009-0000-0814-7937
Affiliation:
2Faculty of Medicine, David Tvildiani Medical University, Tbilisi 0159, Georgia
ORCID: https://orcid.org/0009-0005-0400-410X
Affiliation:
2Faculty of Medicine, David Tvildiani Medical University, Tbilisi 0159, Georgia
ORCID: https://orcid.org/0009-0005-3343-3632
Explor Neurosci. 2025;4:1006111 DOI: https://doi.org/10.37349/en.2025.1006111
Received: June 29, 2025 Accepted: August 21, 2025 Published: September 23, 2025
Academic Editor: Ryszard Pluta, Medical University of Lublin, Poland
The article belongs to the special issue Progress in Alzheimer's disease research: etiology, molecular mechanisms involved in disease progression, and advances in therapies aimed at slowing or reversing neurodegeneration
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder with declining memory and cognitive impairment, largely mediated by extracellular amyloid-beta (Aβ). Although the amyloid cascade and tau protein hypotheses have long served as established frameworks for AD pathology, recent evidence suggests that long-term infections, particularly with Chlamydia pneumoniae (C. pneumoniae), may contribute to disease progression. A systematic search strategy was used to identify relevant literature using PubMed, Scopus, Google Scholar, and Web of Science. Keywords and Boolean operators such as “Chlamydia pneumoniae and Alzheimer’s disease,” “neuroinflammation,” “amyloid-beta,” and “tau protein” were applied, with filters for peer-reviewed articles, human and experimental studies, and publications from the past 25 years. Epidemiological and background data were supplemented by official sources, including the Centers for Disease Control and Prevention (CDC) and the World Health Organization (WHO). This review examines the potential relationship of C. pneumoniae infection with AD pathogenesis. Studies have identified DNA and antigens of C. pneumoniae in AD-infected brain regions, often co-localized within Aβ plaques and neurofibrillary tangles (NFTs). Proposed mechanisms of CNS invasion include olfactory, hematogenous, and immune cell-mediated routes, leading to persistent glial activation, neuroinflammation, altered amyloid precursor protein processing, and tau protein hyperphosphorylation. Experimental models support these associations, with infected animals developing AD-like pathology. Diagnostic challenges persist due to the limitations of PCR and immunohistochemistry, though advanced approaches such as next-generation sequencing and TSPO-PET imaging are emerging. Potential therapeutic approaches include antimicrobial and immunomodulatory strategies, although human trials have shown mixed results. While current evidence suggests a possible link, causality remains unproven. Future research must prioritize large-scale, longitudinal, and mechanistic studies to clarify these relationships. Establishing a definitive role for C. pneumoniae in AD pathogenesis could reshape current understanding of disease etiology and inform the development of novel preventive and therapeutic strategies.
Alzheimer’s disease (AD) is the leading cause of dementia, marked by memory loss and cognitive decline. Its two main pathological features are extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) made of hyperphosphorylated tau protein [1–3]. The amyloid cascade hypothesis suggests that the misfolding of Aβ proteins triggers the development of AD, while tau protein pathology worsens neuronal damage. Despite decades of research, however, neither the amyloid nor tau protein hypothesis has fully explained the onset of AD, nor have therapeutic strategies targeting these pathways yielded significant clinical success [4]. This underscores the need to investigate alternative or complementary mechanisms underlying disease pathogenesis.
Recent perspectives have proposed the “ischemic theory of the brain,” in which chronic cerebral hypoperfusion or microvascular injury triggers oxidative stress, inflammation, and blood-brain barrier (BBB) disruption, thereby enhancing Aβ production, impairing its clearance, and promoting tau protein hyperphosphorylation [5]. These vascular mechanisms provide a compelling framework but remain insufficient to fully account for the complexity of AD. Similarly, tau protein pathology, though strongly correlated with cognitive decline, does not clarify the initiating factors driving its abnormal phosphorylation and aggregation [1, 3].
Growing evidence suggests that chronic infections may represent such triggers. Chlamydia pneumoniae (C. pneumoniae), a common respiratory pathogen, has been repeatedly identified in post-mortem AD brains, often co-localized with Aβ plaques and NFTs [2, 4]. Experimental data show that C. pneumoniae can invade the brain via the olfactory pathway, immune cell trafficking, or systemic inflammation, subsequently infecting astrocytes and microglia. This persistent infection promotes chronic neuroinflammation, impairs Aβ clearance, and accelerates neuronal damage [1, 2]. While these findings are suggestive, the literature is limited by inconsistent methodologies, small sample sizes, and the lack of longitudinal studies, leaving unanswered whether C. pneumoniae is a causal factor, a cofactor in predisposed individuals, or an incidental finding in vulnerable neural tissue.
Given these gaps, this review synthesizes pathological, mechanistic, and epidemiological evidence to evaluate C. pneumoniae’s potential role in AD progression. By addressing whether C. pneumoniae contributes meaningfully to AD pathogenesis, this work aims to refine the understanding of disease etiology and highlight implications for novel approaches to detection, prevention, and treatment.
C. pneumoniae, a respiratory pathogen, has been strongly associated with AD. It has been detected in the brains of AD patients and correlates with hallmark features such as amyloid plaques and NFTs. Elevated anti-C. pneumoniae antibody levels in affected individuals usually suggest chronic or prior infection. C. pneumoniae’s ability to persist inside immune cells allows it to spread to the brain, where it may contribute to the progression of the disease [6].
C. pneumoniae may enter the brain through different routes. Systemic inflammation caused by lung infection increases cytokine levels that affect the brain, with infected immune cells potentially facilitating the translocation of C. pneumoniae across the BBB, and alternatively direct invasion through the nasal cavity via the olfactory nerve may also occur. Once in the brain, C. pneumoniae infects glial cells, triggering neuroinflammation and neuronal damage, thereby promoting AD pathogenesis [4].
In addition to infectious agents, the microbiota-gut-brain axis is an important emerging factor in AD. This axis represents the bidirectional communication between the gut microbiota and the brain through neural, immune, and metabolic pathways. Disturbances in this system can lead to neuroinflammation and cognitive decline, highlighting the role of gut health in brain aging and neurodegeneration [4, 6].
The gut microbiota influences brain function and immune regulation. Dysbiosis can lead to systemic inflammation and alter neurotransmitter signaling, potentially exacerbating neurodegenerative changes. Modulating gut microbial composition may therefore offer promising avenues for preventing or slowing AD progression [6].
Emerging studies suggest that C. pneumoniae infection may contribute to the pathogenesis of AD through various mechanisms such as chronic neuroinflammation, amyloid-beta (Aβ) accumulation, and tau protein phosphorylation. Increasing evidence from histopathological and molecular investigations supports the presence of this pathogen in AD-affected brain tissue. In 1998, Balin et al. analyzed postmortem brain samples from individuals with and without AD. They found that 89% of brains from AD patients tested positive for C. pneumoniae in regions showing typical AD neuropathology, while 95% of control brains tested negative for the organism [7, 8]. Further studies, including those by Gerard and colleagues, supported these findings, which showed consistent presence of C. pneumoniae in AD-affected brains [8]. Their preliminary research using highly specific polymerase chain reaction (PCR) assays demonstrated the presence of C. pneumoniae DNA in 90% of postmortem brain samples from individuals with late-onset Alzheimer’s dementia [9, 10].
The brains of AD patients have been linked to three major bacterial species: C. pneumoniae, Porphyromonas gingivalis (P. gingivalis), and Borrelia burgdorferi [11]. Beyond its role in respiratory infections like community-acquired pneumonia, C. pneumoniae has been associated with several non-respiratory diseases such as atherosclerosis, inflammatory arthritis, and multiple sclerosis [10]. Immunohistochemical investigations have shown that astrocytes, microglia, and neurons in the AD brain can host C. pneumoniae. These infected cells are frequently found around neuritic plaques and NFTs, suggesting a possible role in AD pathophysiology. Activated microglia and astrocytes release pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), contributing to chronic neuroinflammation. This prolonged inflammatory state promotes neuronal damage, synaptic dysfunction, and eventual neurodegeneration [12].
Infected astrocytes show increased β and γ-secretase activity and reduced α-secretase activity. This enzymatic shift alters amyloid precursor protein (APP) processing toward increased Aβ production, promoting Aβ plaque formation [9]. Immuno-histological analysis demonstrated that C. pneumoniae antigens are co-localized with Aβ plaques in the frontal and temporal cortices of AD brains [9, 10]. Aβ oligomers generated during infection can activate neuronal surface receptors, leading to tau protein hyperphosphorylation and NFT formation. Inflammatory cytokines like IL-1β and TNF-α stimulate tau protein-related kinases such as glycogen synthase kinase 3 beta (GSK-3β) and cyclin-dependent kinase 5 (cdk5), further promoting tau protein pathology. Infection of human neuroblastoma cells by C. pneumoniae inhibits apoptosis, potentially prolonging tau protein modifications and promoting NFT development. Long-term inflammation and excess Aβ from C. pneumoniae infection contribute to the aggregation of hyperphosphorylated tau protein into NFTs [8].
The inflammatory pathway induced by C. pneumoniae has been strongly associated with altered APP processing, leading to the accumulation of Aβ, one of the pathological hallmarks of AD [12]. Chronic infection with C. pneumoniae triggers the activation of innate immune cells, particularly microglia and astrocytes, resulting in the release of a variety of pro-inflammatory cytokines, including IL-1β and TNF-α. These cytokines create a sustained inflammatory milieu within the central nervous system (CNS), which not only contributes to neuronal dysfunction but also promotes increased enzymatic cleavage of APP, ultimately resulting in elevated Aβ levels. Additionally, this inflammatory cascade supports conditions favorable to tau protein hyperphosphorylation and aggregation, another major feature of AD pathology [13]. See Figure 1 for the pathway from C. pneumoniae infection to the start of AD.
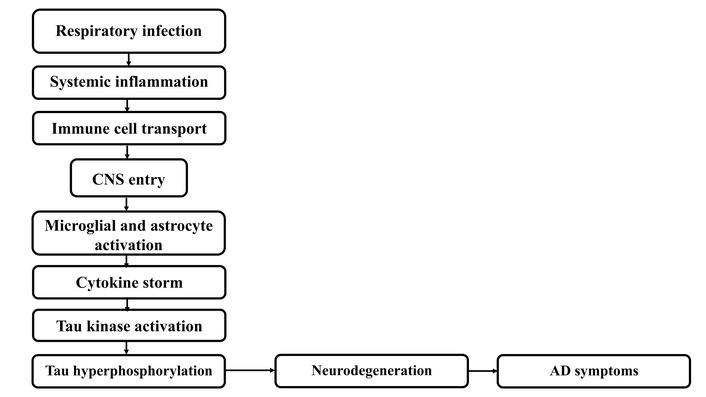
Pathogenesis and pathway of C. pneumoniae infection to AD. CNS: central nervous system; AD: Alzheimer’s disease.
Studies have shown that human monocytes infected with C. pneumoniae exhibit enhanced transmigration across an in vitro model of the BBB, which is composed of human brain microvascular endothelial cells. These infected monocytes upregulate pro-inflammatory mediators that increase vascular permeability, weakening the selective integrity of the BBB and facilitating the infiltration of pathogens and inflammatory cells into the brain parenchyma. Furthermore, if a similar inflammatory response occurs within CNS microglia, it may activate neighboring astrocytes, which play an essential role in regulating and maintaining the tight junctions and functional properties of brain endothelial cells that constitute the BBB [14, 15].
The brains of AD patients were also found to contain P. gingivalis DNA, with tau protein tangles and Aβ plaques co-localized with gingipains, the bacterium’s primary virulence factors. It was shown that gingipains were neurotoxic and could induce AD-like pathology in mice, including Aβ buildup and tau protein fragmentation [16]. Melvin Ball proposed in 1982 that herpes simplex virus type 1 (HSV-1) was the cause of AD. His notion is that latent HSV-1 in the trigeminal ganglia may reactivate and climb along established neural pathways into the limbic system and the areas of the brain most affected by AD [17].
Evidence from a variety of pathogens, including HSV-1, P. gingivalis, and C. pneumoniae, has come together to support the idea that infectious agents may play a role in the pathophysiology of AD. These pathogens may cause chronic neuroinflammation, damage the BBB, and encourage the development of tau protein hyperphosphorylation and Aβ accumulation, two hallmarks of AD. In addition to improving our knowledge of AD’s pathophysiology, more investigation into the role of microbes in the disease may lead to the development of new targeted antimicrobial and anti-inflammatory treatment approaches [16, 17]. Mechanisms of neurodegeneration have been summarized in Figure 2.
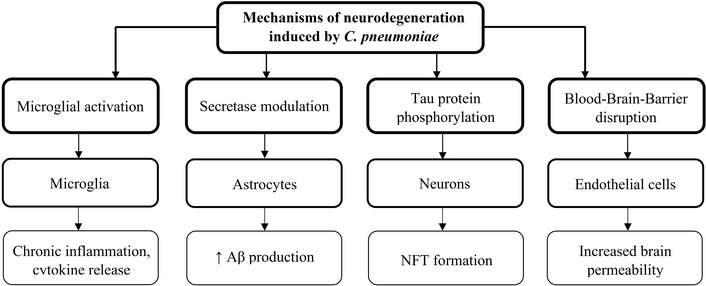
Mechanisms of neurodegeneration induced by C. pneumoniae. Aβ: amyloid-beta; NFT: neurofibrillary tangle. ↑: increased levels.
C. pneumoniae persists on surfaces and spreads via aerosols, particularly in cool, humid conditions. It is a slow transmission (30-day interval) resulting in long-lasting (5–8 months) epidemics with worldwide distribution. In developed nations, infection rates are low in preschoolers but rise significantly during school years, affecting over half of middle-aged adults, with a higher prevalence in males [18].
Historically, C. pneumoniae outbreaks have been noted in closed populations such as military recruits and nursing home residents. Transmission typically occurs through respiratory droplets [19]. Although primarily a respiratory pathogen, C. pneumoniae has been linked to rare neurological complications. Initially, the disease presents as a slow loss of short-term memory, which later evolves into major cognitive dysfunction characterized by behavioral issues, disorientation, language difficulties, and impaired daily functioning [20]. Two independent researchers found C. pneumoniae in the brains of AD patients with PCR and immunohistochemistry (IHC) [21, 22]. Symptoms may include impaired consciousness, neck rigidity, and altered cerebrospinal fluid (CSF) findings [23]. While uncommon, these cases suggest that C. pneumoniae should be considered in the differential diagnosis of meningoencephalitis, especially when respiratory symptoms are present. Studies have also shown a potential link between chronic C. pneumoniae infection and neurodegenerative conditions, like AD, suggesting the bacteria might contribute to neuroinflammation [24].
The prevalence of C. pneumoniae infection varies geographically. Studies across different countries show varying rates of infection, which can be influenced by factors such as the age group examined and the diagnostic methods used [25–30].
The most prevalent bacterial sexually transmitted infection in the US is C. trachomatis. It can lead to cervicitis and urethritis in women, nongonococcal urethritis and epididymitis in males, and reactive arthritis (Reiter’s syndrome) in both sexes. Furthermore, C. trachomatis can be transferred to the unborn child from an infected pregnant woman, resulting in pneumonia and conjunctivitis [25]. Approximately 10–20% of newborns exposed to C. trachomatis during birth may also develop respiratory issues, including pneumonia, within 2–12 weeks. Indeed, C. trachomatis is potentially the leading cause of pneumonia in infants [26]. An analysis of sub-Saharan African data revealed a significant burden of C. trachomatis in women, with higher prevalence observed in healthcare settings than in community-based research, underscoring the need for effective control techniques [27]. In China, a study on women with lower reproductive tract infections found that while Mycoplasma genitalium prevalence was lower than C. trachomatis, both were associated with genital dysbiosis [29]. An Internet-based testing service in Sweden identified that C. trachomatis infection in both men and women was associated with younger age, multiple sexual partners, and non-condom use, with women also showing increased risk if they believed themselves infected or were requested to test by a partner [30]. Although the highest incidence of lymphogranuloma venereum (LGV) has been reported from subtropical and tropical areas, Chlamydia species, including C. trachomatis, are known to cause respiratory infections, and occasional pulmonary involvement has been reported [31].
Several factors can influence the risk and prevalence of C. pneumoniae infection:
Environmental factors: Close contact in crowded environments, such as military barracks or schools, can facilitate transmission. Seasonal changes might also play a role, with some studies suggesting higher infection rates during winter months. Air pollution has also been suggested as a potential contributing factor in some studies, though its direct impact on C. pneumoniae specifically requires further research [26].
Comorbidities: Certain underlying health conditions can increase susceptibility to or the severity of C. pneumoniae infection. For example, one study noted a higher frequency of C. pneumoniae infection in patients undergoing hemodialysis for chronic renal disease [28]. Smoking is another significant comorbidity that increases the risk of respiratory infections in general, including C. pneumoniae-associated pneumonia, and can worsen their severity [26].
Genetic predisposition: While not as extensively studied as other factors, genetic susceptibility might play a role in how individuals respond to C. pneumoniae infection or their likelihood of developing associated complications. Genetic studies suggest that polymorphisms in host defense and immune-related genes influence susceptibility and immune responses to Chlamydia infections [32].
Host genetic variations can influence the risk of C. pneumoniae infection ascending to the upper respiratory tract, increasing the likelihood of infertility by modulating genes involved in immune responses, T-cell function, and fibrosis [32]. Vaccination is key to preventing respiratory outbreaks. Once an outbreak begins, it is recommended to use vaccination and infection measures. Antibiotics for exposed high-risk individuals might help, but resistance is a concern [18, 25, 26].
Despite the growing evidence, identifying and confirming the presence of this microorganism within neural tissue remains a formidable challenge. Traditional diagnostic approaches such as PCR and IHC have demonstrated limited sensitivity and specificity. PCR, though widely used, may fail to distinguish between active infection and residual DNA, especially in post-mortem samples where DNA degradation can further compromise accuracy. IHC, however, is limited by challenges such as antibody cross-reactivity and variability in antibody quality, which can result in inconsistent findings across studies [4]. Culturing C. pneumoniae from brain tissue is even more problematic due to the organism’s fastidious growth requirements and the sterile, immune-privileged environment of the CNS. These challenges necessitate the development and adoption of more advanced diagnostic tools to better elucidate the bacterium’s role in neurodegenerative processes.
Advancements in diagnostic methodologies are beginning to offer new hope for overcoming these limitations. Next-generation sequencing (NGS) has emerged as a powerful tool for unbiased microbial detection. Unlike targeted PCR, NGS allows for comprehensive metagenomic analysis, enabling the identification of low-abundance pathogens such as C. pneumoniae without prior assumptions. This technique has proven effective in detecting bacterial genetic material in CSF and brain biopsies with a higher degree of resolution. In parallel, positron emission tomography (PET) imaging is being explored for visualizing neuroinflammation associated with chronic infections. PET tracers target activated microglia, such as translocator protein (TSPO) ligands, offering insights into infection-induced neuroinflammatory responses, a hallmark feature of AD pathology that could be driven by chronic microbial presence [33]. Furthermore, research on peripheral biomarkers is gaining momentum, with efforts aimed at identifying specific signatures in blood and CSF that may reflect CNS infection or immune activation. For instance, elevated inflammatory cytokines, antibodies against C. pneumoniae, or even bacterial RNA fragments in CSF may provide indirect evidence of its involvement in AD progression [34]. All these methods have been summarized in Table 1.
Diagnostic techniques for detecting C. pneumoniae in AD.
| Method | Target | Sensitivity | Limitations |
|---|---|---|---|
| PCR | DNA | Medium | Cannot distinguish active infection |
| IHC | Antigens | Variable | Cross-reactivity, inconsistency |
| NGS | Metagenome | High | Expensive, requires expertise |
| PET imaging | Neuroinflammation | Emerging | Indirect marker |
AD: Alzheimer’s disease; PCR: polymerase chain reaction; IHC: immunohistochemistry; NGS: next-generation sequencing; PET: positron emission tomography.
Collectively, these emerging diagnostic strategies represent a crucial shift from hypothesis-driven to data-driven discovery, facilitating a more accurate understanding of microbial contributions to neurodegenerative diseases.
Despite decades of research, AD continues to lack effective disease-modifying treatments, with current interventions largely focused on symptomatic relief rather than halting or reversing neurodegeneration. A growing body of evidence implicates microbial pathogens, particularly C. pneumoniae, in the etiology of AD, potentially through chronic infection-induced neuroinflammation. However, targeting such microbial components presents significant challenges, including the BBB, intracellular persistence of the pathogen, and variability in infection across individuals [35]. These factors complicate the development of targeted antimicrobial therapies that can both eliminate pathogens and mitigate downstream inflammatory responses without causing off-target effects.
Among the most explored antimicrobial approaches are tetracycline-class antibiotics, particularly doxycycline and minocycline, which have shown promise due to their dual antimicrobial and anti-inflammatory properties. In preclinical models, doxycycline reduced amyloid burden and improved cognitive function, potentially by inhibiting matrix metalloproteinases and modulating microglial activation [35]. Similarly, minocycline demonstrated neuroprotective effects by suppressing proinflammatory cytokines such as TNF-α and IL-1β in models of CNS inflammation [36]. However, translating these findings into human trials has produced mixed outcomes, possibly due to poor BBB penetration, insufficient dosing, or late-stage intervention. To overcome these limitations, research has turned to antimicrobial peptides (AMPs), which can be engineered to cross the BBB and possess both direct bactericidal activity and immune-modulating functions. Synthetic AMPs like LL-37 and analogs have demonstrated efficacy in disrupting intracellular C. pneumoniae and modulating host immune responses, though clinical trials remain pending [37].
Beyond direct antimicrobial action, immunomodulatory therapies are being investigated to target the chronic neuroinflammation thought to result from persistent infection. Longstanding activation of microglia and astrocytes contributes to synaptic loss and neuronal damage in AD, and therapies targeting the reduction of this inflammation are currently under development. Agents targeting pathways such as NF-κB or the NLRP3 inflammasome may reduce the harmful neuroinflammatory milieu triggered by microbial presence [34]. Additionally, restoring balance in the gut-brain axis through probiotics and microbiome modulation has emerged as a potential adjunct strategy. Dysbiosis of the gut microbiota is known to influence systemic inflammation and BBB integrity, which may, in turn, affect susceptibility to brain infections and neurodegeneration [38]. Therapies used have been summarized in Table 2. Certain probiotic strains have been shown to lower systemic endotoxin levels and modulate CNS cytokine profiles, offering a non-invasive avenue to reduce infection-driven neuroinflammation.
Therapeutic approaches targeting infection-induced AD.
| Class of medication | Agent | Mechanism |
|---|---|---|
| Targeting microorganisms directly | ||
| Antibiotics | Doxycycline, azithromycin | Broad-spectrum antibacterial action targeting C. pneumoniae in CNS and systemic sites |
| AMPs | LL-37 analogs | BBB-penetrating, intracellular killing of persistent C. pneumoniae forms |
| Other therapeutic categories | ||
| Immunomodulators | NF-κB inhibitors, minocycline, NSAIDs | Suppression of microglial overactivation and chronic neuroinflammation |
| Cell biological treatment | Mitochondrial protectants, antioxidants | Stabilization of neuronal bioenergetics and reduction of oxidative stress |
| Supportive treatment | Cognitive enhancers, lifestyle interventions | Maintenance of cognitive function and delay of clinical progression |
AD: Alzheimer’s disease; CNS: central nervous system; AMPs: antimicrobial peptides; BBB: blood-brain barrier; NSAIDs: non-steroidal anti-inflammatory drugs.
Early analysis identifying C. pneumoniae in the brains of Alzheimer’s patients suggests a potential link between the bacterium and the neurodegenerative process [39]. The possible involvement of C. pneumoniae in AD has generated significant interest; however, the current findings are limited by small sample size and inconsistent research. To establish a more absolute relationship, large-scale clinical studies are required [40, 41].
Animal models have provided valuable insights into the pathogenic mechanisms of C. pneumoniae. For instance, mice intranasally infected with the bacterium developed amyloid plaques resembling those observed in AD, suggesting a potential contributory role [12]. Additionally, it was shown that C. pneumoniae infection in neural cells altered genes related to autophagy and apoptosis, which are mechanisms that are similar in the pathophysiology of AD [41]. Despite these discoveries, there hasn’t been enough research done on how these findings convert to human populations.
Extensive research is also required to determine the prevalence of C. pneumoniae in various AD populations through large-scale epidemiological investigations. A particularly important aspect of research involves the genetic susceptibility, especially associated with the APOE ε4 allele. This allele is a well-established risk factor for late-onset AD and is also implicated in modulating immune response and BBB integrity. However, further research is needed to establish whether APOE ε4 increases susceptibility to infection or if the infection exaggerates APOE ε4-mediated neurodegeneration [41, 42].
Furthermore, the risk of infection-driven neurodegeneration may be reduced by alterations in the environment and lifestyle, such as dietary habits, physical activity, and vascular health. Although adherence to an anti-inflammatory diet, such as the Mediterranean diet, has been associated with a reduced rate of cognitive decline, the effects of these dietary factors on the brain’s inflammatory response and susceptibility to microbial invasion remain unclear [43].
Strong multidisciplinary research is needed, even if early evidence points to a connection between C. pneumoniae, genetics, and lifestyle in AD. In order to clarify how infection interacts with host susceptibility in the setting of neurodegeneration, future research should combine genetics, microbiology, and epidemiology [41].
Accumulating evidence suggests a potential link between C. pneumoniae infection and AD, though the precise nature of this relationship remains under investigation. Studies have detected the presence of C. pneumoniae within the brains of AD patients, and in vitro and in vivo models have demonstrated the bacterium’s ability to induce AD-like pathological changes, including Aβ plaque formation and neuroinflammation. Furthermore, some epidemiological studies have indicated a higher prevalence or prior exposure to C. pneumoniae in individuals with AD compared to controls.
Critical knowledge gaps remain, specifically regarding whether C. pneumoniae is the primary initiator of neurodegeneration, an accelerating cofactor in genetically or environmentally predisposed people, or just an opportunistic colonizer of already impaired neuronal tissue. The application of recent results to therapeutic or preventive measures is constrained by this ambiguity. High-quality longitudinal studies with sizable, heterogeneous populations that integrate standardized neuropathological evaluation, neuroimaging, and precise molecular diagnostics are required for addressing these gaps. In order to understand mechanistic pathways, research should also look into the chronological sequence of infection and neurodegenerative changes, as well as host-pathogen interactions at the cellular and molecular levels.
In conclusion, while causality between C. pneumoniae and AD has not yet been established, the convergence of current evidence makes further research both justified and significant. Investigating this link may improve our understanding of AD pathophysiology and open up novel possibilities for prevention and treatment, perhaps changing the course of this dreadful illness.
AD: Alzheimer’s disease
AMPs: antimicrobial peptides
APP: amyloid precursor protein
Aβ: amyloid-beta
BBB: blood-brain barrier
C. pneumoniae: Chlamydia pneumoniae
CNS: central nervous system
CSF: cerebrospinal fluid
HSV-1: herpes simplex virus type 1
IHC: immunohistochemistry
IL-1β: interleukin-1 beta
NFTs: neurofibrillary tangles
NGS: next-generation sequencing
P. gingivalis: Porphyromonas gingivalis
PCR: polymerase chain reaction
PET: positron emission tomography
TNF-α: tumor necrosis factor alpha
TSPO: translocator protein
ASK: Conceptualization, Writing—original draft, Writing—review & editing. AWI: Conceptualization, Writing—original draft, Writing—review & editing, Supervision. SSA, SFZ, and DT: Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5257
Download: 40
Times Cited: 0
Octavio García ... Jesús Antonio Villegas-Piña
Graziella Martins Guimarães ... Márcia Maria de Souza
Zsolt G. Venkei, Masha G. Savelieff
