 Review
Review

Affiliation:
1Facultad de Psicología, Unidad de Investigación en Psicobiología y Neurociencias, Red Interdisciplinaria en Neurodesarrollo (RINDe), Universidad Nacional Autónoma de México, Coyoacán, Ciudad de México 04510, México
†These authors share the first authorship.
Email: ogarciag@unam.mx
ORCID: https://orcid.org/0000-0002-5792-0209
Affiliation:
2Departamento de Genética y Biología Molecular, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Gustavo A. Madero, Ciudad de México 07360, México
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0002-5445-6711
Affiliation:
1Facultad de Psicología, Unidad de Investigación en Psicobiología y Neurociencias, Red Interdisciplinaria en Neurodesarrollo (RINDe), Universidad Nacional Autónoma de México, Coyoacán, Ciudad de México 04510, México
Affiliation:
1Facultad de Psicología, Unidad de Investigación en Psicobiología y Neurociencias, Red Interdisciplinaria en Neurodesarrollo (RINDe), Universidad Nacional Autónoma de México, Coyoacán, Ciudad de México 04510, México
Affiliation:
1Facultad de Psicología, Unidad de Investigación en Psicobiología y Neurociencias, Red Interdisciplinaria en Neurodesarrollo (RINDe), Universidad Nacional Autónoma de México, Coyoacán, Ciudad de México 04510, México
Explor Neurosci. 2025;4:1006104 DOI: https://doi.org/10.37349/en.2025.1006104
Received: February 16, 2025 Accepted: July 10, 2025 Published: August 05, 2025
Academic Editor: Ryszard Pluta, Medical University of Lublin, Poland
The article belongs to the special issue Progress in Alzheimer's disease research: etiology, molecular mechanisms involved in disease progression, and advances in therapies aimed at slowing or reversing neurodegeneration
Down syndrome (DS), caused by trisomy 21, is strongly associated with an increased risk of early-onset Alzheimer’s disease (AD). This work explores the cellular, genetic, epigenetic, and neuropsychological mechanisms that underlie the accelerated development of AD in individuals with DS. We review key contributors such as amyloid-β accumulation, mitochondrial dysfunction, oxidative stress, tau pathology, neuroinflammation, and chromosomal and epigenetic instability in the neuropathology of AD in DS. Particular attention is given to genes, microRNAs, and chromatin remodeling factors encoded by human chromosome 21 (Hsa21) that regulate these pathological processes. We also highlight the roles of non-coding RNAs and altered DNA methylation patterns in modulating gene expression and neuronal vulnerability. Additionally, the writing evaluates current pharmacological and non-pharmacological interventions and addresses the critical need for inclusive, person-centered health services. Integrating molecular biology with clinical perspectives, the review emphasizes the importance of early diagnosis and coordinated care strategies for individuals with DS at risk for AD.
Down syndrome (DS) is the most common genetic cause of intellectual disability, affecting approximately six million individuals worldwide [1]. It occurs in an estimated one out of every 700 live births, with incidence rates ranging from one out of 400 to one out of 1,500 newborns [2, 3]. Karyotype analysis shows that approximately 95% of DS cases result from an extra, complete copy of chromosome 21, a condition known as trisomy 21 [4]. Around 4–5% of cases result from a translocation involving chromosomes 21 and 14 or 22, causing a partial trisomy (the duplication of only a segment of chromosome 21). A smaller proportion (1–2%) of cases result from mosaicism, wherein only a subset of cells contains an extra copy of chromosome 21 [4]. In addition to karyotype studies, the development of noninvasive methods, such as cell-free fetal DNA analysis in maternal blood, has enabled the timely identification of DS [3].
More than 250 distinctive features associated with DS have been identified phenotypically [4, 5]. The phenotypic variability observed in individuals with DS is attributed to the combined effects of multiple genes located on chromosome 21 and throughout the genome [5]. Although the exact causes of DS development remain unclear, advanced maternal age is considered a significant risk factor for trisomy 21 [6]. Environmental factors such as smokeless chewing tobacco use and oral contraceptive use also increase the risk of DS, particularly among younger and older mothers, respectively [7].
The typical physical features associated with DS include hypotonia, a short neck with excess skin at the nape, a flat facial profile, a small head, ears, and mouth, and upslanting eyes with epicanthic folds. Other characteristics include Brushfield spots (white spots on the iris), short and broad hands with short fingers, midfacial and mandibular recession, congenital heart defects, cerebellar hypoplasia, obstructive sleep apnea, recurrent respiratory infections, thyroid dysfunction, alopecia, diabetes, psoriasis, hematologic disorders (including leukemia), immune deficiencies, gastrointestinal anomalies, and infertility [4, 5, 8].
DS is also characterized by a distinct behavioral phenotype, including intellectual disability, cognitive delays, adaptive functioning difficulties, and early deficits in speech, language production, and auditory short-term memory. Additional common features include developmental delays, hearing and vision impairments, accelerated aging, as well as neurological or psychiatric conditions such as anxiety, depression, conduct disorder, epilepsy, and early-onset Alzheimer’s disease (AD) [5, 9, 10].
The relationship between DS and AD was first recognized in the mid-19th century when John Fraser and Arthur Mitchell studied 62 individuals with DS who experienced functional decline in adulthood. They observed that deaths in this population were often attributed to “general decay” or “premature senility”, marking the earliest recognition of early-onset senility in individuals with DS [11]. In 1929, Friedrich Struwe furthered this understanding by describing the neuropathological features of AD in individuals with DS. Struwe’s work identified isolated and perivascular plaques in the brains of individuals with DS, a hallmark of AD. His work significantly contributed to the understanding of the relationship between the two diseases [12]. In 1948, George A. Jervis published an article in the American Journal of Psychiatry that further elucidated this link, providing critical insights into the pathology of DS and AD [13]. Subsequent studies have confirmed the high risk of Alzheimer’s-like neuropathology and dementia in individuals with DS, with symptoms often appearing in the third or fourth decade of life [14]. In 1984, George G. Glenner and Cai-Wai Wong isolated and purified amyloid protein from the brain of an adult with DS and demonstrated its homology to amyloid-β (Aβ), the protein characteristic of AD. This discovery provided the first chemical evidence of the relationship between DS and AD, suggesting that DS could serve as a predictive model for AD and highlighting that the genetic defect responsible for AD is located on chromosome 21 [11]. Currently, DS is recognized as a genetically determined form of AD, similar to autosomal dominant AD [14, 15].
In this review, we explore the primary neuropathological, neuropsychological, and genetic features underlying the development of AD in individuals with DS, aiming to better understand the mechanisms of neurodegeneration in this high-risk population.
DS has been found to be strongly associated with AD, with virtually all individuals with DS developing AD-related neuropathological changes, such as amyloid plaques and neurofibrillary tangles (NFTs), by the age of 30 to 40 [16, 17]. This phenomenon is primarily attributed to the triplication of chromosome 21, which leads to the overexpression of amyloid precursor protein (APP) and subsequent Aβ plaque formation [18]. Despite the early onset of neuropathology, clinical symptoms of dementia typically do not appear until around age 50 or later. This indicates a separation between neuropathological changes and the beginning of cognitive decline [19, 20].
The progression of AD in individuals with DS is influenced by various biological and genetic factors, such as oxidative stress, neuroinflammation, the apolipoprotein E (APOE) genotype, and aging-related genes on chromosome 21 [14, 18]. The prevalence of dementia in adults with DS between the ages of 40 and 59 varies significantly, ranging from 4% to 55%. The prevalence increases considerably after age 50, reaching 75% and 90% of people over 60 years of age [14, 17]. The mean age of AD onset in this population is 53.8 years, with an estimated age of death of 58.4 years [21]. Thus, AD is classified as one of the primary causes of mortality among adults with DS.
Although cognitive deterioration in aging individuals with DS is often attributed to AD, it is important to consider other age-related conditions that may lead to functional decline. These include musculoskeletal and sensory impairments [22]. Clinically, cognitive decline is often characterized by impairments in memory, attention, language, and social functioning, which can progress to dementia [23]. Neuropathologically, AD in individuals with DS shares many features with sporadic AD, including synaptic loss, neuroinflammation, and selective neuronal degeneration in the entorhinal cortex, hippocampus, and neocortex [14, 18, 24] although they have notable differences in their origin and manifestation (Table 1). However, AD associated with DS exhibits distinctive characteristics, including earlier and more substantial amyloid accumulation, increased tangle density in the hippocampus, and a distinct M2b neuroinflammatory profile in older individuals [22, 24].
Comparison between Alzheimer’s disease (AD) in Down syndrome (DS) vs. sporadic AD (sAD)
| Feature | AD in DS | sAD |
|---|---|---|
| Age of onset | Early onset (commonly 40–50 years) | Late onset (typically > 65 years) |
| Genetic factors | Trisomy 21→Overexpression of APP gene (chromosome 21) | APOEε4 allele is a major risk factor; no APP gene overexpression |
| Amyloid-β deposition | Begins in teens–twenties; nearly universal by age 40 | Later onset; variable presence |
| Neurofibrillary tangles (tau pathology) | Develop later than amyloid but at younger ages than in sAD | Correlates with disease severity; gradual accumulation |
| Brain atrophy pattern | Early hippocampal and cortical atrophy; cerebellar involvement may be less pronounced | Prominent hippocampal, temporal, and parietal lobe atrophy |
| Cognitive profile | Baseline intellectual disability complicates diagnosis; early signs: behavior/personality changes, decline in adaptive skills | Prominent memory loss; later language, visuospatial, and executive deficits |
| Neuroimaging (MRI/PET) | Early amyloid PET positivity; structural MRI shows earlier atrophy | Amyloid PET positivity later; progressive cortical atrophy |
| Cholinergic system involvement | Significant degeneration of basal forebrain cholinergic neurons | Similar degeneration observed |
| Seizure prevalence | Higher incidence of seizures (up to 50% late in disease) | Lower seizure incidence (~10–22%) |
| Rate of progression | May be faster after onset | Variable, but generally more gradual |
| Neurological comorbidities | Congenital brain differences, hypothyroidism, epilepsy | Vascular risk factors (hypertension, diabetes, etc.) are more common |
| Diagnosis challenges | Harder to detect due to pre-existing cognitive impairment | Easier to detect due to prior cognitive baseline |
| Neuropathological hallmarks | Classic AD pathology is universally present by age 40 | Pathology variable and age-dependent |
APP: amyloid precursor protein; APOEε4: apolipoprotein E epsilon4; MRI: magnetic resonance imaging; PET: positron emission tomography
The manifestation of AD in individuals with DS is influenced by intellectual disability and reduced cognitive reserve. This often results in non-amnestic symptom patterns and a complicated trajectory of decline due to comorbid health conditions [25]. The early and frequent emergence of AD in this population intersects with broader challenges, including accelerated aging, increased dependence on care, and a growing burden on aging family caregivers [26]. These factors give rise to substantial clinical, familial, and public health concerns. Consequently, regular health screenings, timely diagnoses, and multidisciplinary interventions are crucial for enhancing quality of life and postponing functional deterioration in individuals with DS at risk for or living with AD [27].
Conventionally, DS has been diagnosed primarily through karyotype analysis and prenatal screening using biochemical and ultrasound markers [28]. Recently, advanced genetic and molecular techniques, such as next-generation sequencing and whole-exome sequencing, have significantly improved the accuracy and efficiency of diagnoses, with sensitivities exceeding 99% [29]. Additionally, non-invasive prenatal testing, which analyzes cell-free fetal DNA in maternal blood, has achieved detection rates above 98%. Proteomic and metabolomic studies have identified additional promising DS biomarkers in maternal serum, placental tissue, and amniotic fluid [30]. These studies have revealed numerous differentially expressed proteins and metabolic alterations.
In contrast, diagnosing AD in individuals with DS requires a multifaceted approach. This approach includes physical and neurological examinations, cognitive assessment batteries, and caregiver and informant questionnaires. It also involves neuroimaging techniques such as magnetic resonance imaging (MRI) and positron emission tomography (PET). PET scans targeting amyloid, tau, and fluorodeoxyglucose (FDG) detect changes in cognitive, behavioral, and functional domains [31]. These scans facilitate the identification of different stages of cognitive decline and dementia.
Biomarker studies in DS show progression similar to that of sporadic AD: early reductions in cerebrospinal fluid (CSF) Aβ42 and elevated plasma neurofilament light chain (NfL) levels are followed by amyloid PET changes, tau accumulation, brain atrophy, and hypometabolism [15]. Cognitive decline in DS correlates closely with regional glucose metabolism, as assessed by FDG-PET [31]. The most extensively studied biomarkers are amyloid and tau. DS individuals exhibit elevated baseline plasma Aβ levels due to APP overexpression [32].
In addition to imaging and clinical evaluations, established AD biomarkers, such as Aβ40, Aβ42, total tau, and phosphorylated tau 181 (pTau181), can now be measured in the blood of individuals with DS [33]. Emerging biomarkers, including pTau217, NT1-Tau, NfL, and glial fibrillary acidic protein (GFAP), have also demonstrated the potential to enhance diagnostic accuracy [34]. Furthermore, metabolomic biomarkers reflecting alterations in glucose metabolism, oxidative stress, cholesterol pathways, inflammation, and amino acid metabolism are being investigated for their potential to facilitate early AD detection in DS [34, 35].
Early diagnosis is critical for improving quality of life; however, it is often complicated by comorbidities, pre-existing intellectual disability, and functional variability [36–38]. Studies suggest that artificial intelligence and machine learning methods could improve the early detection of AD in individuals with DS. Algorithms such as k-nearest neighbors, Naive Bayes, decision trees, and ensemble methods have shown potential in enhancing diagnostic precision when applied to MRI and PET imaging data [30, 39–41].
Research into anti-Aβ antibodies in DS continues to reveal the complexity of disease mechanisms. Individuals with DS exhibit elevated plasma levels of Aβ1-42 and corresponding antibodies with higher affinity and titers than observed in the general population. These findings suggest a potential state of autoimmunization against Aβ [42, 43]. Higher plasma Aβ40/42 ratios and increased anti-fibril antibody titers have been associated with a greater risk of dementia [44]. Postmortem brain analyses confirm the presence of soluble Aβ protofibrils and insoluble Aβ40 and Aβ42, resembling the pathology seen in sporadic AD [45]. These findings reinforce the therapeutic potential of targeting Aβ pathology in individuals with DS and AD [46].
Diagnosing cognitive impairment and AD in individuals with DS presents unique clinical challenges due to the baseline presence of intellectual disability and atypical symptoms [47, 48]. Early indicators of AD in this population include visual memory decline, diminished learning capacity, and reduced social functioning [49]. Reliable diagnostic tools such as the Cambridge Examination for Mental Disorders of Older People with Down’s Syndrome and Others with Intellectual Disabilities (CAMDEX-DS) have demonstrated high accuracy in detecting the prodromal and dementia stages of AD [50]. Other effective instruments include the Modified Cued Recall Test and the Dementia Questionnaire for People with Learning Disabilities [51]. An accurate diagnosis requires a multidimensional approach that integrates observer-rated assessments, direct neuropsychological testing, and evaluations of functional abilities in daily life [48]. Longitudinal assessment and exclusion of alternative causes of cognitive decline are important, even among individuals with severe baseline impairments [49].
The cognitive and behavioral phenotype of individuals with DS evolves throughout life, showing greater decline with age [47, 52]. Neuropsychologically, individuals with DS who develop AD often follow a similar trajectory to individuals with sporadic or autosomal dominant AD. The initial clinical hallmark is typically a decline in episodic memory, especially among individuals who are amyloid-positive at diagnosis or become so over time [53]. Early impairments in attention and executive functioning are also common, followed by deficits in visuospatial skills, verbal fluency, motor coordination, and planning abilities (Figure 1) [54]. Nonverbal skills tend to remain relatively stable in adulthood, while verbal skills typically decline more steeply [54–56].
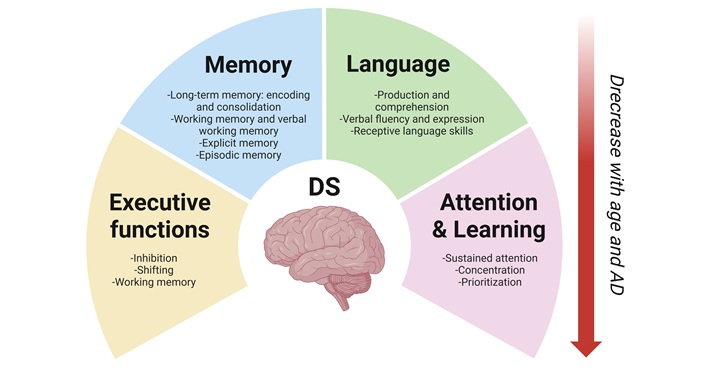
Cognitive domains affected by aging in individuals with Down syndrome. The neuropsychological profile of individuals with Down syndrome (DS) changes throughout life and declines further with age, potentially due to neurological disorders such as Alzheimer’s disease (AD). The primary affected domains are language, attention, learning, and memory. Created in BioRender. Dominguez, E. (2025) https://BioRender.com/vc7pgmf
Language is one of the cognitive domains most severely affected in DS, with expressive abilities typically being more impaired than receptive ones [23, 57]. In adulthood, language impairments become a central component of the cognitive decline associated with AD-like dementia (Figure 1). These deficits have been shown to negatively impact behavior, social relationships, and overall quality of life. Although relatively stable during earlier stages of life, receptive language abilities tend to decline with age, particularly in older adults with DS. Figueroa and Darbra [57] found that older individuals with DS performed significantly worse on the Token and Peabody tests, suggesting that receptive language may be more vulnerable to age-related decline than expressive abilities. However, declines in verbal fluency and expressive language have also been documented in individuals without AD, while comprehension often remains intact [9, 23, 58].
Deficits in working memory, particularly in the verbal domain, are well-documented in individuals with DS and tend to persist across the lifespan (Figure 1) [59, 60]. Long-term memory is also compromised, with difficulties in encoding and retrieval processes becoming evident in adolescence and early adulthood [16, 61, 62]. These memory issues are linked to temporal lobe and hippocampus dysfunctions [63] and progressively worsen with age [64]. Episodic memory remains relatively intact in early adulthood but deteriorates significantly with the onset and progression of dementia. Some studies suggest that episodic memory is the first cognitive domain to deteriorate during aging in individuals with DS [65]. As cognitive decline progresses, explicit memory, including verbal and visual encoding, also deteriorates [66].
Executive functions, including inhibitory control, cognitive flexibility, and working memory, are essential for self-regulation, planning, and goal-directed behavior [67]. Adults with DS exhibit significant impairments in these domains and score lower than their typically developing peers on standardized executive function measures such as the Global Executive Composite, Plan/Organize, Working Memory, Task Monitor, and Metacognition Index scales [68, 69]. However, it remains unclear whether these neuropsychological changes follow a distinct progression, occur independently, or converge over time to culminate in AD.
Neuroanatomical changes, particularly in the hippocampus and corpus callosum, are also linked to cognitive deterioration. Additionally, psychosocial factors may influence cognitive function and AD progression in DS [49, 70]. Therefore, a comprehensive diagnostic approach that incorporates clinical, imaging, and biomarker data is crucial for the early detection of AD in this population [34, 35].
Despite the clinical heterogeneity observed in older individuals with DS, age-related AD neuropathology is a consistent and defining feature. Postmortem studies of DS brains have identified structural abnormalities such as reduced total brain weight, hypoplasia of the frontal and temporal lobes, a simplified gyral pattern with fewer sulci, generalized cortical atrophy, ventricular enlargement, and cerebellar and brainstem atrophy, particularly in the middle cerebellar lobe [71]. Interestingly, while subcortical regions such as the lenticular nuclei and posterior parietal and occipital cortices often retain relatively normal volumes, the hippocampus shows disproportionately severe volume reduction, and the amygdala exhibits shrinkage consistent with overall brain size reduction [72].
Neuroimaging studies have further elucidated these structural changes. For instance, MRI findings reported by Pujol et al. [73] revealed significant volume reductions in the hippocampus, basal forebrain (specifically the substantia innominata), and the temporal and parietal lobes in adults with DS and dementia. Similarly, Mullins et al. [74] observed an increased lateral ventricular volume in individuals with DS, even in those without dementia. Fortea et al. [15] corroborated these findings, describing a distinctive DS brain morphology characterized by diminished frontal lobe, hippocampus, cerebellum, and brainstem volumes. These patterns closely mirror the neurodegeneration seen in sporadic AD.
Crucially, evidence suggests that some neuropathological differences in DS originate developmentally rather than degeneratively. Pinter et al. [64] found that children with DS have smaller hippocampal volumes than their typically developing peers, indicating early neurodevelopmental alterations. Neuropsychological evaluations of school-aged individuals with DS reveal hippocampal-specific dysfunctions alongside broader cognitive impairments [75]. In adults with DS without clinical dementia, age-related atrophy affects the hippocampus and corpus callosum, suggesting changes in allocortical and neocortical structures [72].
Advancements in pediatric medical care, particularly surgical interventions for congenital heart defects, have significantly increased the life expectancy of individuals with DS. Life expectancy rose from an average of four years in 1950 to approximately 60 years by 2010 [4, 76–78]. However, this extended lifespan has not been matched by proportional improvements in overall health outcomes. By age 30, many individuals with DS exhibit clear signs of accelerated aging. This is characterized by the early onset of age-related conditions and premature physical decline compared to the general population [79–81].
This is reflected by the high prevalence of comorbidities, including early menopause, osteopenia, osteoporosis, hypothyroidism, abnormal fat distribution, obesity, atherosclerosis, hypertension, and immune system dysfunction [16, 19, 82–84]. Furthermore, there is a possibility of the development of neurological or psychiatric disorders, including anxiety, depression, conduct disorder, epilepsy, cognitive impairment, and early-onset AD [16, 65].
Notably, the accelerated aging process in DS begins prenatally, as evidenced by epigenetic age acceleration observed in newborns with the condition [85]. Studies using epigenetic clocks have shown that individuals with DS have a biological age approximately 6.6 years older than their chronological age, based on blood and brain tissue [86].
Premature aging in DS is associated with multiple established hallmarks of aging, including Aβ accumulation, oxidative stress, and chronic neuroinflammation [87]. Additional mechanisms, such as cellular senescence and disruptions in metal homeostasis (e.g., copper imbalance), are believed to contribute to accelerated aging, age-related diseases, and cognitive decline in this population [80, 81].
AD is an incurable and progressive neurological disorder that ranks as the sixth leading cause of death worldwide and is the most common form of dementia, accounting for approximately 80% of all diagnoses [88]. The disease typically progresses over a period of 8 to 10 years during its clinical stages, following prodromal and preclinical phases that may span up to two decades [89]. The annual incidence of the condition ranges from 1% to 3%, with a prevalence that varies from 10% to 30% among individuals over the age of 65 [90]. Globally, the number of individuals affected is expected to reach approximately 152 million by 2050, with the most significant increase occurring in developing countries [88].
The main relationship between DS and AD is due to the overexpression of the APP gene located on chromosome 21 [91]. This genetic alteration leads to the excessive production and accumulation of Aβ peptides, particularly Aβ42. Intraneuronal Aβ deposits can be detected at an early age in individuals with DS and precede the formation of extracellular amyloid plaques [92, 93]. This early accumulation triggers a cascade of pathological events, including oxidative stress, mitochondrial dysfunction, chronic neuroinflammation, glial alterations, vascular abnormalities, and the formation of NFTs, hallmark features of AD pathology (Figure 2) [20, 93, 94].
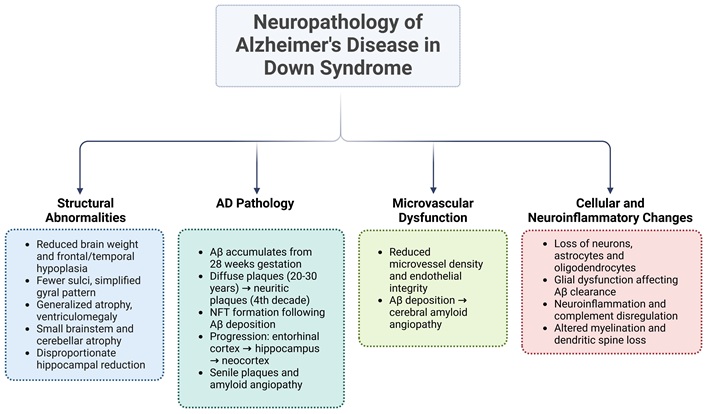
Neuropathology of Alzheimer’s disease in Down syndrome. During aging, trisomy 21 may trigger a series of processes, including structural brain alterations, early amyloid-β deposition, microvascular changes, neuroinflammation, and cell loss. These processes may be interrelated, contributing to different stages of cognitive decline and the development of Alzheimer’s disease. AD: Alzheimer’s disease; Aβ: amyloid-β; NFT: neurofibrillary tangle. Created in BioRender. Dominguez, E. (2025) https://BioRender.com/ccmz1u4
The pathological cascade of AD in DS begins early, with intracellular Aβ accumulation detectable in endosomes and lysosomes as early as 28 weeks of gestation [18]. By age eight, extracellular Aβ deposits are present, with a dramatic increase in deposition occurring between the ages of 35 and 45 [53, 92]. Diffuse Aβ plaques begin forming between ages 20 and 30 and evolve into neuritic plaques during the fourth decade of life. NFTs appear after Aβ deposition, reflecting the progressive nature of AD pathology [15]. Individuals with DS over the age of 40 exhibit AD-associated neuropathological hallmarks, including extracellular senile plaques, perivascular Aβ deposits (amyloid angiopathy), intraneuronal NFTs, and neuritic plaques, regardless of dementia symptoms [15, 20, 53, 95]. This pathology typically begins in the entorhinal cortex, then progresses to the hippocampus, and eventually spreads to the neocortical association areas [72]. Together, these interconnected mechanisms contribute to the accelerated onset and progression of AD in individuals with DS.
Studies have linked the overexpression of superoxide dismutase 1 (SOD1) to the accumulation of reactive oxygen species (ROS) and the oligomerization of Aβ in individuals with DS [96]. Trisomy 21, a characteristic of DS, leads to the overexpression of SOD1 and APP, contributing to increased oxidative stress and neural dysfunction [97]. An imbalance between SOD1 and glutathione peroxidase activity has been associated with increased free radical production [96]. High levels of oxidative damage, particularly the accumulation of 4-hydroxy-2-nonenal (HNE)-bound proteins, have been observed in the frontal cortex of individuals with DS and correlate with Aβ levels [98]. Additionally, increased SOD and catalase activity have been reported in plasma samples from adults with DS and cognitive impairment [99, 100]. SOD1 deficiency in an AD mouse model has been shown to accelerate Aβ oligomerization and worsen memory impairment, suggesting that cytoplasmic superoxide plays a critical role in Alzheimer’s pathogenesis [101]. Similarly, neurons obtained from the brains of individuals with DS have been shown to be highly sensitive to oxidative stress [102]. Taken together, these findings underscore the intricate relationship between SOD1 overexpression, oxidative stress, and Aβ aggregation in DS and AD, highlighting the potential of antioxidant-based therapeutic strategies.
Mitochondrial abnormalities have been linked to various clinical manifestations in DS, including intellectual disability, premature aging, and AD-like dementia [103–105]. Studies have demonstrated that mitochondrial DNA (mtDNA) mutations accumulate with age and are notably higher in the brains of individuals with DS and AD [104, 106]. This leads to a reduction in mtDNA copy number and impaired mitochondrial function [107, 108]. This dysfunction contributes to increased oxidative stress, elevated ROS production, and neuronal cell death in DS [102, 106, 109]. Overproduction of Aβ is also associated with mitochondrial dysfunction, suggesting that energy deficits in DS may contribute to altered processing of Aβ [110, 111]. Furthermore, the relationship between mitochondrial dysfunction and autophagy has been investigated in DS, revealing that autophagy can lead to inflammation and the development of a redox imbalance, as well as changes in protein homeostasis and signal transduction [112]. Consequently, therapeutic strategies that correct mitochondrial defects and enhance autophagy are being investigated as potential interventions to improve cognitive function and quality of life in individuals with DS and AD [106, 112].
Microvascular dysfunction is a critical contributor to the development and progression of AD, particularly in individuals with DS. Postmortem studies of brains from individuals with both conditions reveal significant vascular abnormalities, such as reduced microvessel density and compromised endothelial integrity [113]. These findings closely resemble those observed in sporadic AD. They also contribute to decreased cerebral blood flow, impaired nutrient delivery, and reduced metabolic waste clearance. These factors exacerbate neurodegeneration [113].
Additionally, the excessive accumulation of Aβ in individuals with DS leads to cerebral amyloid angiopathy (CAA). In CAA, Aβ deposits within cerebral vessel walls promote further vascular injury and accelerate AD pathology [114, 115]. Despite the absence of typical systemic vascular risk factors, such as hypertension or atherosclerosis, individuals with DS frequently exhibit cerebrovascular lesions, including white matter hyperintensities and microbleeds. These lesions increase with age and disease progression [116, 117]. These vascular injuries may intensify astrocytosis, promoting tau hyperphosphorylation and amplifying neurodegeneration [118].
The interplay among vascular pathology, Aβ accumulation, and neuroinflammation creates a reinforcing cycle that drives cognitive decline in DS. The mechanisms of vascular amyloidosis, endothelial dysfunction, and chronic inflammation are shared by sporadic and familial forms of AD. This underscores the value of DS as a model for studying the early and aggressive pathogenesis of AD [119, 120].
Understanding this complex network is essential for developing targeted preventive and therapeutic strategies that address vascular health, inflammation, and Aβ pathology for individuals with DS and the broader population affected by AD [118, 120].
Postmortem studies reveal significant reductions in neurons, astrocytes, and oligodendrocytes in the cortex and basal ganglia of adults with DS, indicating widespread cellular degeneration [121, 122]. Glial dysfunction disrupts key metabolic and homeostatic processes, including Aβ clearance, oxidative stress regulation, and inflammatory control, thereby amplifying neurodegeneration [123, 124].
In both DS and AD, astrocytes display pathological reactivity, characterized by elevated secretion of inflammatory mediators such as interleukin-1 and S100β. These mediators contribute to sustained neuroinflammation [125]. DS astrocytes also exhibit reduced expression of thrombospondin-1 (TSP-1), a critical molecule in synaptogenesis and spinogenesis [126–128]. This deficiency impairs dendritic spine formation and the production of synaptogenic proteins in DS and AD [126, 128, 129]. TSP-1 secretion is further diminished in astrocytes exposed to Aβ, which reinforces the impact of Aβ pathology on astrocytic function [129, 130], suggesting a potential association between TSP-1 and the aging process as well as the development of age-related diseases [131].
Single-nucleus RNA sequencing has identified disease-associated astrocyte subtypes in AD mouse models and aging human brains. This indicates a convergence of genetic predisposition and age-related changes in astrocyte biology [132]. Together, these findings suggest that dysfunctional astrocytic support plays a critical role in Aβ accumulation, synaptic failure, and cognitive decline.
In individuals with DS, the deposition of Aβ triggers early microglial activation and a sustained neuroinflammatory response marked by elevated levels of proinflammatory cytokines and immune-related proteins [125]. These changes are detectable from early developmental stages and are associated with synaptic dysfunction, impaired neurogenesis, and neuronal loss [125]. The complement component C1q, which binds to thioflavin-S-positive Aβ plaques, increases with age in the brains of individuals with DS, further linking Aβ accumulation to immune activation [133]. Veteleanu et al. [134] further underscore immune dysfunction in DS and AD in their study, reporting elevated levels of complement activation products (TCC and iC3b), core complement components (C1q, C3, and C9), and abnormalities in regulatory proteins, such as factor H and clusterin. Furthermore, mitochondrial dysfunction in cerebral endothelial and glial cells exacerbates Aβ-induced inflammation, contributing to vascular and neuronal damage [135].
In both human tissue and mouse models [e.g., Ts65Dn and Dp(16)], microglia shift from a homeostatic to a reactive phenotype. This phenotype is characterized by excessive synaptic pruning, heightened cytokine production, and tau-induced senescence [136–138]. Interferon signaling contributes to this dysfunction and inhibiting it has shown potential to restore spine density, normalize neuronal activity, and improve cognition.
However, recent evidence challenges the traditional view that activated microglia are the primary drivers of pathology. Instead, it suggests that dystrophic (senescent) microglia may play a more significant role in tau-related degeneration in sporadic AD [138–140].
Oligodendrocyte dysfunction and myelination deficits contribute to AD pathology in DS. Similar to changes observed in sporadic AD, hypomyelination and reduced oligodendrocyte populations have been identified in DS brains [141]. These alterations are accompanied by dysregulated gene expression involved in oligodendrocyte differentiation and myelin production [142]. This dysregulation can potentially impair neural conductivity and contribute to slowed information processing and cognitive decline [142]. Structural abnormalities in white matter may underlie the age-related cognitive deficits commonly observed in DS [143].
Significant neuronal alterations contribute to the pathological landscape of AD in DS. Ferrer and Gullotta [144] reported pronounced dendritic spine loss and morphological abnormalities in the CA1 and CA2–3 regions of the hippocampus in adults with both conditions, likely due to the accumulation of neuritic plaques and NFTs. Reduced spine density may be associated with decreased levels of TSP-1 [126–128]. In addition, reduced TSP-1 levels have been reported in sporadic AD brains [145] as well as in AD-related synaptic pathology [129, 130]. Another hallmark of AD pathology, present in individuals with and without DS, is the degeneration of the basal forebrain cholinergic neurons [146], which are crucial for cognitive function and are especially vulnerable in individuals with DS and AD. This contributes to impairments in memory and language [146]. A study of 33 adults with DS revealed developmental neuronal deficits and AD-related neuronal loss. The degree of neuronal reduction correlated strongly with age and dementia stage [147].
Together, these glial and neuronal alterations form a multifaceted, self-reinforcing pathology that accelerates cognitive decline in individuals with DS. The convergence of Aβ overproduction, mitochondrial alteration, chronic neuroinflammation, glial dysfunction, synaptic damage, and vascular abnormalities resembles the mechanisms seen in sporadic AD, highlighting the importance of DS as a model for studying the early stages of AD pathogenesis.
Human chromosome 21 (Hsa21) is one of the smallest autosomes in the human genome. Comprehensive genomic analyses have identified 233 protein-coding genes, 423 non-coding genes (including 69 small and 300 long non-coding RNAs, as well as 29 Hsa21-specific non-coding RNAs), and 188 pseudogenes on this chromosome [148]. Despite these findings, the molecular mechanisms underlying the phenotypic variability of DS and its strong association with AD are not fully understood [149]. Two predominant hypotheses have been proposed to explain the genetic basis of DS and its contribution to AD pathogenesis.
The first, known as the “gene dosage hypothesis” or the “chromosome 21 overdose effect”, suggests that the clinical features of DS result from the trisomic state of Hsa21. This state leads to an approximately 1.5-fold increase in gene expression. This overexpression includes key genes such as APP, dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), and SOD1 [148, 149]. These genes have been associated with AD-like dementia and premature aging in individuals with DS (Figure 3). Triplication of the APP gene, in particular, has been shown to be both necessary and sufficient to induce early-onset AD [91].
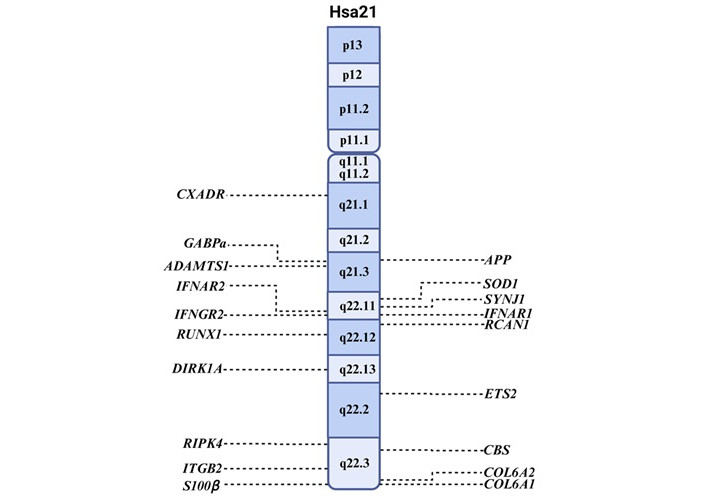
Location of genes on chromosome 21 associated with AD. The locations on the long (q), and petite (p) arms are shown within the chromosome, and the genes are shown on the sides. Hsa21: human chromosome 21; APP: amyloid precursor protein; SOD1: superoxide dismutase 1; SYNJ1: synaptojanin 1; RCAN1: regulator of calcineurin1. Created in BioRender. Garcia, O. (2025) https://BioRender.com/yaqw41n
The second hypothesis, referred to as “chromosomal instability”, posits that trisomy 21 disrupts transcriptional homeostasis, affecting not only Hsa21-encoded genes but also the expression of genes located on other chromosomes [148, 150], resulting in diverse phenotypic outcomes such as facial dysmorphology. Increased fluctuating asymmetry in facial features, particularly those derived from mandibular prominence, supports this view [151]. This global transcriptional dysregulation may play a critical role in the onset and progression of dementia. For example, the APOE epsilon4 (APOEε4) allele on chromosome 19 has been associated with increased amyloid deposition and a higher risk of AD in the general population, including individuals with DS [152].
Chromosomal instability is a well-documented phenomenon in DS. Individuals with trisomy 21 exhibit increased genomic instability, including higher frequencies of chromosomal aberrations, micronuclei, and nuclear anomalies compared with controls [153, 154]. This instability intensifies with age and contributes to mosaicism through additional chromosomal segregation errors [155]. Taken together, these findings suggest that chromosomal instability is a defining feature of DS that affects cellular and developmental processes.
The triplication of Hsa21 genes in individuals with DS alters gene dosage and regulation. This leads to widespread disruptions in cellular homeostasis. These changes affect multiple biological processes, including protein aggregation, mitochondrial function, inflammation, and synaptic signaling. Collectively, these changes accelerate the onset and progression of AD. Understanding the molecular mechanisms linking DS to early-onset AD requires knowledge of the specific contributions of Hsa21 genes and their interactions with genes on other chromosomes [4, 148].
Amyloid plaque formation is a defining feature of AD pathology, driven by a complex interplay of genetic factors (Figure 4). The most extensively studied gene in this context is the APP gene, which encodes APP. In DS, APP overexpression due to trisomy 21 increases the production of Aβ, particularly the aggregation-prone Aβ42 isoform [156]. This leads to neuritic plaque formation. DYRK1A, a kinase overexpressed in DS, amplifies this process by phosphorylating APP and promoting its cleavage via the amyloidogenic pathway [157]. Furthermore, Aβ peptides can upregulate DYRK1A, forming a pathogenic feed-forward loop [158].
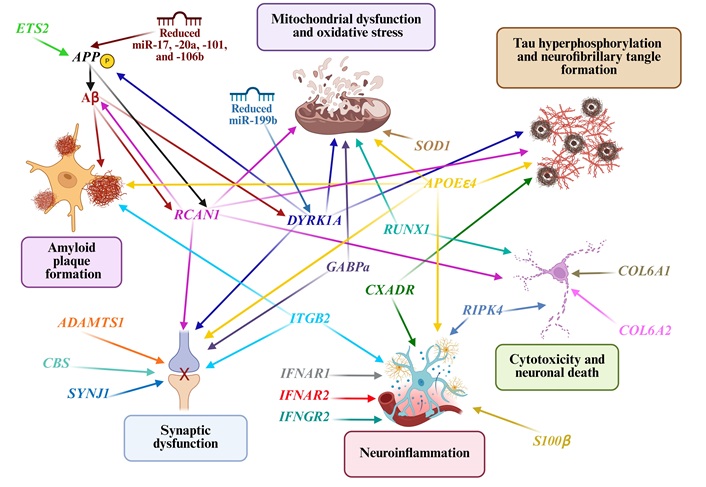
Overexpression of genes located on chromosome 21 that contribute to the neuropathology associated with Alzheimer’s disease in individuals with Down syndrome. The arrows illustrate the various connections between amyloid plaque formation, neurofibrillary tangles (NFTs), mitochondrial dysfunction, oxidative stress, neuroinflammation, synaptic dysfunction, and neuronal death. APP: amyloid precursor protein; Aβ: amyloid-β; SOD1: superoxide dismutase 1; APOEε4: apolipoprotein E epsilon4; RCAN1: regulator of calcineurin1; DYRK1A: dual-specificity tyrosine-phosphorylation-regulated kinase 1A; SYNJ1: synaptojanin 1. Created in BioRender. Dominguez, E. (2025) https://BioRender.com/66zeti5
NFTs, which are composed of hyperphosphorylated tau, are another hallmark of AD. DYRK1A phosphorylates tau at multiple residues, promoting its aggregation. Elevated DYRK1A expression correlates with cognitive impairment and NFTs burden in DS and AD [157]. These tangles disrupt axonal transport, impair synaptic function, and drive neurodegeneration [158].
Regulator of calcineurin1 (RCAN1) promotes tau pathology by inducing oxidative stress and disrupting calcium signaling, thereby enhancing tau phosphorylation and NFTs accumulation [159]. These effects contribute to cognitive deficits, circadian rhythm disturbances, and other symptoms associated with AD and DS [160]. CXADR, a gene involved in hippocampal plasticity, is downregulated during inflammation and is linked to increased tau phosphorylation via MAPK-p38 signaling [161]. Furthermore, APOEε4 promotes tau phosphorylation by activating the calpain-CDK5 pathway, highlighting its multifaceted role in AD progression [162].
Mitochondrial dysfunction and oxidative stress are early events in AD pathophysiology [105–108]. In DS, the overexpression of RCAN1 and DYRK1A impairs mitochondrial dynamics by disrupting calcium signaling and phosphorylation cascades (Figure 4). RCAN1 contributes to oxidative damage by dysregulating the redox pathway [160] and ETS2 induces mitochondrial-regulated apoptotic death [163, 164].
GABPa, which is triplicated in DS, encodes a transcription factor involved in mitochondrial biogenesis and neuronal homeostasis. While it is typically neuroprotective, its overproduction in DS can lead to epigenetic modifications that impair neuronal function [165]. Meanwhile, SOD1, which encodes the antioxidant enzyme SOD1, exhibits paradoxical behavior; it is overexpressed in DS but decreased in AD [96]. Misfolded SOD1 aggregates can exacerbate mitochondrial dysfunction and oxidative damage, accelerating memory decline and Aβ aggregation [99, 100, 166]. Additionally, RUNX1 overexpression impairs mitochondrial function, contributing to neurodegeneration in AD [167, 168].
Neuroinflammation is markedly elevated in DS-associated AD due to the overexpression of several immune-related genes [125]. S100β, which encodes a calcium-binding protein secreted by astrocytes and microglia, acts as a proinflammatory cytokine that amplifies glial activation [124]. Its upregulation in DS promotes neuroinflammation.
ITGB2 is highly expressed in microglia and the hippocampus, disrupting synaptic function and enhancing immune-mediated neurotoxicity in AD. The overexpression of IFNAR1, IFNAR2, and IFNGR2 leads to aberrant interferon signaling, triggering excessive synaptic pruning and impairing microglial plasticity [138, 169]. Conversely, reducing the expression of these genes in model systems has been shown to enhance microglial function and mitigate cellular aging (Figure 4).
RIPK4 increases TNF-α production and promotes cytotoxicity via TNFR1 activation [170]. CXADR overexpression is associated with neuroinflammation and M1 macrophage activation in DS [161]; however, its expression decreases in response to AD-related or systemic inflammation. The overexpression of immune-related genes on chromosome 21 leads to persistent dysregulation of both innate and adaptive immunity. These changes likely amplify neuroinflammation and neurodegeneration and may serve as early biomarkers for dementia in DS.
Synaptic loss strongly correlates with cognitive impairment in AD. In DS, several Hsa21 genes impair synaptic integrity through various mechanisms. Synaptojanin 1 (SYNJ1) regulates synaptic vesicle recycling and phosphoinositide metabolism. It is overexpressed in DS and linked to synaptic deficits and cognitive decline [171]. Genetic variants in SYNJ1 have also been associated with altered AD onset and memory impairment [172, 173]. In mouse models, SYNJ1 overexpression impairs the function of hippocampal place cells, contributing to memory deficits [174]. Other key genes, such as RCAN1, DYRK1A, ITGB2, and GABPa, also impair both synaptic plasticity and synaptic transmission via inflammatory and metabolic pathways, mitochondrial stress and epigenetic dysregulation (Figure 4) [149]. GABPa triplication may also impair the regulation of genes essential for neuronal communication [148].
ADAMTS1 overexpression affects the composition of the extracellular matrix in the hippocampus, disrupting inhibitory neurotransmission [175]. CBS, which is involved in homocysteine metabolism, produces excess hydrogen sulfide (H2S). This impairs synaptic signaling and increases the risk of AD later in life for individuals with DS [176].
Neuronal apoptosis is a convergence point of multiple AD-related pathological processes. RCAN1 enhances Aβ-induced cytotoxicity and mitochondrial stress, triggering apoptotic pathways [159]. RUNX1 dysregulates the PI3K/Akt pathway, thereby increasing neuronal susceptibility to degeneration [167]. RIPK4, another Hsa21 gene, activates nuclear factor kappa B (NF-κB) signaling and promotes cell death via the TNFR1 pathway [169].
Interestingly, COL6A1 and COL6A2, which encode collagen VI subunits, are upregulated in response to Aβ exposure and may exert neuroprotective effects by inhibiting apoptosis [177]. However, dysregulation of these genes may paradoxically contribute to neurodegeneration.
In addition to protein-coding genes, chromosome 21 contains several non-coding RNAs, including microRNAs (miRNAs), that play a crucial role in regulating early molecular events involved in the pathophysiology of DS and AD [148]. Circulating chromosome 21-encoded miRNAs, such as miR-155 and let-7c, have been associated with cognitive impairment and dementia in Mexican adults with DS [178]. Let-7c promotes neurodegenerative processes by inducing the production of pro-inflammatory molecules and activating caspase-3, ultimately leading to neuronal cell death [179].
Other studies have demonstrated that miR-155, miR-99a, and miR-125b, which are also encoded on chromosome 21, are inducible and regulated by NF-κB, a central mediator of inflammatory signaling. The upregulation of these miRNAs may suppress the expression of anti-inflammatory and innate immune regulatory genes, contributing to AD pathogenesis [180].
In DS mouse models, reduced levels of miR-17, miR-20a, miR-101, and miR-106b are associated with increased expression of the APP, while decreased levels of miR-199b correlate with elevated levels of DYRK1A. These miRNA expression alterations contribute to Aβ accumulation and tau hyperphosphorylation, hallmark pathological features of AD in individuals with DS [181].
A related area of interest involves chromatin remodeling, a crucial nuclear process that regulates gene expression. Recent studies have revealed key mechanisms by which chromatin remodeling is altered in DS. For instance, overexpressing DYRK1A disrupts REST/NRSF levels and impairs the SWI/SNF chromatin remodeling complex. This leads to the dysregulation of genes involved in dendritic development [182]. Similarly, triplication of HMGN1, another Hsa21-encoded gene, interferes with PRC2 activity and contributes to abnormal neuronal phenotypes [183]. Additionally, elevated BRWD1 expression, encoded by another Hsa21 gene, has been found to misdirect the BAF complex, thereby altering gene expression in DS neurons [184]. Restoring BRWD1 gene dosage in trisomic mouse models has been shown to improve cognitive deficits and normalize BAF complex localization [184]. These findings underscore the pivotal role of chromatin remodeling in DS neurodevelopmental and neurodegenerative pathologies, suggesting promising avenues for targeted therapeutic interventions. Advances in genetic engineering and nanotechnology further expand the potential for novel strategies to modify DS [185].
Research on DS has revealed significant epigenetic alterations that contribute to accelerated aging, cognitive decline, and an increased risk of AD. Trisomy 21 induces broad changes in epigenetic regulation, including DNA methylation, histone modifications, and chromatin remodeling [85, 86, 186]. Horvath et al. [86] demonstrated that individuals with DS have an increased biological age, averaging 6.6 years older than their chronological age, while Jones et al. [187] identified distinct DNA methylation patterns linked to trisomy 21 and cognitive impairment. Furthermore, Do et al. [188] reported that the extra chromosome exerts widespread trans-acting epigenetic effects, including differential CpG methylation on Hsa21 genes. These changes can disrupt normal gene expression, reactivate transposable elements, and induce genomic instability [189].
These epigenetic alterations are closely tied to the dysregulation of genes involved in transcription and chromatin architecture, which may intensify neurodegeneration. In AD, aberrant epigenetic states characterized by DNA hypermethylation and histone deacetylation have been associated with impaired synaptic plasticity, elevated oxidative stress, neuroinflammation, and reduced neurogenesis [190, 191]. Epigenetic modifications are notably dynamic and responsive to environmental influences, such as diet and stress. This offers promising avenues for therapeutic intervention.
Genome-wide methylation studies further highlight the extent of epigenetic disruption in DS. Haertle et al. [192] identified 2,716 differentially methylated sites and regions in blood samples from a French cohort that distinguished individuals with DS from controls. Among these were nine methylation differences specifically linked to AD dementia. One of these was the ADAM10 gene, which encodes a protein that prevents amyloid plaque formation. Similarly, analyzing buccal epithelial cells from Canadian adults with DS revealed over 3,300 differentially methylated CpG sites, including the TSC2 gene. Hypomethylation of the TSC2 gene has been associated with reduced protein expression, leading to dysregulated mTOR signaling and tau accumulation. These are both key processes in AD pathogenesis [190].
Importantly, epigenetic modifiers encoded on chromosome 21 may play a direct role in neurodegeneration. DNMT3L, a gene involved in establishing DNA methylation patterns, is overexpressed in fetal cortex tissue from DS pregnancies and may contribute to gene-specific hypermethylation [193]. Hypermethylation of TSPYL5 can increase neuronal apoptosis, a hallmark of AD [194]. TSPYL5 regulates the TP53 pathway. Furthermore, DNMT3L overexpression has been associated with increased APP expression, which promotes the formation of Aβ plaques and reinforces its role in Alzheimer-like pathology.
mtDNA methylation is also emerging as a significant epigenetic factor in DS-related neurodegeneration. Altered methylation patterns in the D-Loop and coding regions of mtDNA have been observed in individuals with DS, leading to dysregulated mitochondrial gene expression [106]. These changes may amplify oxidative stress and proinflammatory responses, impairing neuronal function and survival and increasing susceptibility to early-onset dementia (Figure 4).
Together, these findings underscore the pivotal role of epigenetic mechanisms in linking the molecular pathology of DS and AD. They also provide new insights into therapeutic strategies that target gene expression and slow disease progression.
Various pharmacological and non-pharmacological interventions have been proposed to mitigate cognitive decline and reduce the risk of developing AD in individuals with DS. Non-pharmacological approaches, such as structured physical activity [195, 196] programs including Tai Chi [197] and creative adaptations of GameSquad [198] have demonstrated effectiveness in enhancing cognitive and physical health. These interventions can be delivered in natural environments or remotely via online platforms [199] and are suitable for children, adolescents, and adults with DS. They support executive functions, such as attention, working memory, cognitive flexibility, and inhibitory control [200]. Additionally, these programs improve physical health by reducing blood pressure, enhancing balance, lowering adiposity, and decreasing weight and body mass index. Importantly, these programs also contribute to the psychological well-being of caregivers, highlighting their value as holistic interventions. Music-based therapies also show promise, offering engaging, low-risk strategies for promoting cognitive function and emotional well-being [201].
In terms of pharmacology, several compounds have been investigated for their ability to enhance cognitive function and counteract neurodegeneration in individuals with DS. These include acetylcholinesterase inhibitors, DYRK1A inhibitors, and GABA and NMDA receptor modulators [202]. Other promising agents include flavonoids, such as epigallocatechin-3-gallate (EGCG), fluoxetine, and certain fatty acids [203–207]. These interventions target mechanisms such as neurotransmitter regulation and the reduction of oxidative stress and neuroinflammation. These mechanisms are altered in DS and contribute to AD pathology. Additionally, various plant-derived compounds could be used to attenuate symptoms and decelerate the progression of AD in adults with DS. In particular, diadzein and formononetin have been identified as modulators of key signaling pathways involved in inflammatory processes and the aggregation of Aβ [208, 209]. Furthermore, these compounds could induce neuroprotective mechanisms by reducing oxidative stress, increasing the activity of antioxidant enzymes such as SOD, catalase, and reduced glutathione, as well as improving mitochondrial function and decreasing neuronal apoptosis [208, 209]. These findings support the potential of diadzein and formononetin as therapeutic agents in the context of AD associated with DS.
Despite these advancements, managing AD in individuals with DS remains a significant challenge with far-reaching implications for families, caregivers, and healthcare systems. The convergence of age-related neurodegeneration and the preexisting neurodevelopmental profile of DS creates complex needs that are often poorly understood. In many group care settings, staff rely on improvised trial-and-error approaches to manage AD progression, reflecting a lack of specialized training and systemic preparedness [210]. Caregivers, especially younger ones, report high levels of anxiety about the potential onset of AD [26]. Families often face a fragmented service landscape that emphasizes long-term disability care rather than responsive dementia care strategies [211, 212]. This mismatch can lead to emotional distress, confusion, and a pervasive sense of helplessness, underscoring the urgent need for integrated, coordinated care models [211].
In this context, developing inclusive, person-centered health policies is crucial to supporting individuals with DS and their families throughout their lives. Early intervention services beginning in childhood should promote holistic development and be tailored to individual needs [213]. Educational systems must adopt inclusive strategies that accommodate language and communication challenges, foster collaboration with families, and ensure smooth transitions between stages of schooling [214]. However, disparities in regional service availability hinder access to high-quality care, contributing to unequal outcomes and reduced quality of life [215]. Therefore, public health strategies must prioritize training professionals, providing psychosocial support, and implementing stress management programs. Particular attention must be paid to the compounded challenges posed by AD in individuals with DS.
Finally, the NIH INCLUDE initiative has played a transformative role in enhancing DS research infrastructure, supporting participant recruitment, and promoting clinical trial engagement [216]. Previously underfunded and narrowly focused, DS research is evolving toward more robust trial designs, the validation of reliable biomarkers, and integrative strategies combining pharmacological treatments with cognitive interventions [216, 217]. These ongoing efforts aim to refine therapeutic strategies and improve clinical outcomes for individuals with DS who are at risk for or affected by AD.
Individuals with DS are uniquely predisposed to developing AD due to genetic, molecular, and physiological alterations associated with trisomy 21. The overexpression of Hsa21 genes, chromatin remodeling disturbances, mitochondrial dysfunction, and widespread epigenetic changes collectively accelerate neurodegeneration. These mechanisms are further amplified by chronic inflammation and synaptic dysregulation, contributing to the early and aggressive onset of dementia. While there is currently no cure for AD, emerging therapeutic strategies, including gene-targeted treatments, anti-inflammatory agents, lifestyle interventions, and inclusive support systems, offer hope for delaying cognitive decline and improving quality of life. Moving forward, a multidisciplinary and individualized approach is essential to address the complex clinical needs of individuals with DS and reduce the burden of AD across this vulnerable population.
AD: Alzheimer’s disease
APOE: apolipoprotein E
APOEε4: apolipoprotein E epsilon4
APP: amyloid precursor protein
Aβ: amyloid-β
CAA: cerebral amyloid angiopathy
DS: Down syndrome
DYRK1A: dual-specificity tyrosine-phosphorylation-regulated kinase 1A
FDG: fluorodeoxyglucose
Hsa21: human chromosome 21
miRNAs: microRNAs
MRI: magnetic resonance imaging
mtDNA: mitochondrial DNA
NfL: neurofilament light chain
NFTs: neurofibrillary tangles
NF-κB: nuclear factor kappa B
PET: positron emission tomography
RCAN1: regulator of calcineurin1
ROS: reactive oxygen species
SOD1: superoxide dismutase 1
SYNJ1: synaptojanin 1
TSP-1: thrombospondin-1
OG: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. EDdlC, IGLM, JAGC, and JAVP: Investigation, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by PAPIME [PE302323] (OG). IGLM and JAGC received a CONAHCYT scholarship for master’s studies. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Atif Salim Khatib ... Daniya Tasnim
Graziella Martins Guimarães ... Márcia Maria de Souza
Zsolt G. Venkei, Masha G. Savelieff
