Review
Review

Affiliation:
1Department of Cardiology, Zhongshan Hospital Fudan University, Shanghai Institute of Cardiovascular Diseases, Shanghai 200032, China
Affiliation:
2School of Pharmacy, University of Wyoming College of Health Sciences, Laramie, WY 82071, USA
Affiliation:
1Department of Cardiology, Zhongshan Hospital Fudan University, Shanghai Institute of Cardiovascular Diseases, Shanghai 200032, China
3Department of Laboratory Medicine and Pathology, University of Washington, Seattle, WA 98195, USA
Email: jren_aldh2@outlook.com
Explor Med. 2022;3:188–204 DOI: https://doi.org/10.37349/emed.2022.00085
Received: January 06, 2022 Accepted: February 27, 2022 Published: April 26, 2022
Academic Editor: Milan Obradovic, University of Belgrade, Serbia
The article belongs to the special issue Reactive Oxygen Species (ROS) in Pathophysiological Conditions
Cardiovascular diseases are among the leading causes of death worldwide, imposing major health threats. Reactive oxygen species (ROS) are one of the most important products from the process of redox reactions. In the onset and progression of cardiovascular diseases, ROS are believed to heavily influence homeostasis of lipids, proteins, DNA, mitochondria, and energy metabolism. As ROS production increases, the heart is damaged, leading to further production of ROS. The vicious cycle continues on as additional ROS are generated. For example, recent evidence indicated that connexin 43 (Cx43) deficiency and pyruvate kinase M2 (PKM2) activation led to a loss of protection in cardiomyocytes. In this context, a better understanding of the mechanisms behind ROS production is vital in determining effective treatment and management strategies for cardiovascular diseases.
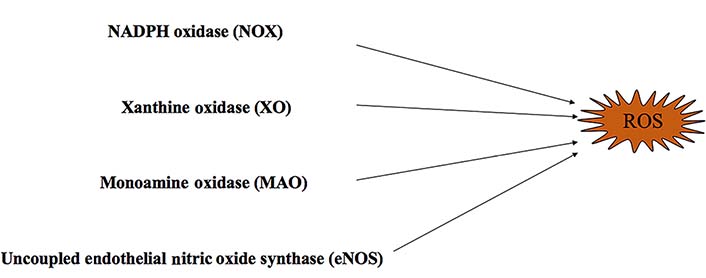
Reactive oxygen species (ROS) play an important role in the pathophysiology of cardiovascular dysfunction [1–3]. An increased production of ROS is associated with the development of an imbalance of generation and elimination in redox reactions [4]. The heart produces, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase (XO), uncoupled nitric oxide (NO) synthase (NOS), and monoamine oxidases (MAOs) by oxidant systems [5]. Oxidative stress performs different functions based on the amount produced. Under normal conditions, oxidative stress is maintained at low levels to sustain physiological metabolism [3, 5, 6]. However, excessive oxidative stress can damage the cardiovascular system. ROS have various effects on cardiovascular diseases that lead to cell regeneration defect, lipid peroxidation, protein degeneration, DNA damage, mitochondrial injury and energy metabolism disorder [5, 7–11].
After the discovery of free radicals in biological systems in 1950 [12], free radicals have been reported to be involved in diverse pathological processes. Redox signaling is an essential process of an organism. ROS include oxygen radicals, such as superoxide anion (O2–), hydroxyl radical (OH), hydrogen peroxide (H2O2), NO, peroxynitrite (ONOO) and hypochlorite (OCL–), which are highly reactive molecules [13, 14].
In nature, many oxidase enzymes contribute to ROS generation. Oxidative stress is characterized by an imbalance between the pro-oxidant and antioxidant systems, resulting in an increase of ROS production [11, 15] (Figure 1). In vivo, there are many oxidant systems such as NADPH oxidases (NOXs), mitochondrial respiratory chain enzymes, XO, MAOs, uncoupled endothelial NOS (eNOS), and lipoxygenases. These systems influence the formation and development of cardiovascular diseases through ROS production [16–21]. All these oxidases can be regulated by antioxidant systems, including superoxide dismutase (SOD), catalase, glutathione (GSH) peroxidases, paraoxonases, thioredoxin system, and peroxiredoxins [21]. In addition, mitochondrial mutations can lead to ROS production [22]. Mitochondrial DNA (mtDNA) is easily damaged because of its limited capacity to repair DNA [22]. In addition, excessive mitochondrial ROS (mtROS) generation results in increased possibility of permeability transition pore (PTP) opening and leads to cell death [23]. The increased ROS also leads to mtDNA leakage and contributes to inflammation [24]. Furthermore, ROS contributes to dysregulation of intracellular Ca2+ homeostasis, by causing mitochondrial membrane depolarization and Ca2+ release [25].
NOXs are multi-transmembrane enzyme complexes composed of a plasma membrane and cytosolic components [26, 27]. They are also the major enzymes involved in the generation of ROS that contribute to cardiovascular diseases [28] (Figure 1). NOXs consist of seven isoforms NOXs: NOX1, NOX2/gp91(phox), NOX3, NOX4, NOX5, dual oxidase 1 (DUOX1) and dual oxidase 2 (DUOX2) [26, 29]. Each oxidase expresses itself differently in cardiovascular, endothelial, and vascular smooth muscle cells [30–32]. Activated NOXs can utilize NADPH as an electron donor and transfer an electron to molecular oxygen. This reaction generates a superoxide molecule that plays an important role in the redox reaction [23, 27, 33, 34]. Several studies have identified that NOXs contribute to cardiovascular diseases, such as atherosclerosis, hypertension, heart failure, and ischemia-reperfusion injury (I/R) [30]. The increased expression and activation of NOX can contribute to ROS production [1, 35–37]. ROS signaling is vital in establishing communication between mitochondria and NOXs in the cardiovascular system [3, 13]. Further research indicates that NOX can also activate the nucleotide-binding and oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in macrophages which contributes to the progression of atherosclerosis [38, 39].
The structure of the mitochondrion was first described in 1888. Mitochondria are known as the source of chemical energy within a cell, but also participate in aerobic respiration which generates ROS [40, 41]. mtROS production has been observed in vivo at complex I and complex III in the electron transport chain (ETC) [42–44]. The process of electron transfer generates mtROS that play an important role in the intracellular redox state [38, 45]. Under physiological conditions, the loss of electrons is minimal. However, during conditions of oxidative stress, the production of mtROS is increased and causes damage to mitochondria [46]. This reaction indicates that mitochondria themselves may also be susceptible to the overexpression of ROS they produce. These effects cause damage to mitochondrial DNA and membranes, further impairing the normal activity of the ETC and generating ROS through positive feedback [47]. In addition, the changes in mtROS impact K+ and Ca2+ channels that influence cell functions [48, 49].
XO is a cytoplasmic enzyme that can be converted from xanthine dehydrogenase [38]. XO catalyzes the oxidation of hypoxanthine to xanthine by the transfer of an electron to oxygen and produces superoxide [23, 30] (Figure 1). Allopurinol is an inhibitor of XO that can be used to reduce levels of uric acid [50]. Recently, allopurinol has been found to have potential cardiovascular protection by modulating ROS and Ca2+ [50, 51].
MAOs are located at the outer membrane of mitochondria [52]. There are two isoforms of MAO namely MAO-A and MAO-B [23]. Under pathological conditions, the increased activation of MAOs generates excessive H2O2 and aldehyde, leading to dysfunction of mitochondrion [53, 54] (Figure 1). MAO is also an oxidase which has been closely associated with cardiovascular diseases such as vascular dysfunction, I/R, maladaptive hypertrophy and heart failure [55–57]. Some research indicates that the increased activation of MAOs disturbs the balance of redox and leads to excessive production of ROS that may damage cardiomyocytes [23]. Moreover, MAOs can increase the production of mtROS resulting in the activation of inflammasome [23].
NOS consists of three isoforms: eNOS, neuronal NOS (nNOS) and inducible NOS (iNOS) [30] (Figure 1). Under physiological conditions, eNOS maintains the balance of endothelial function by binding to L-arginine with the assistance of tetrahydrobiopterin (BH4) during NO synthesis [58]. BH4 is an important element for maintaining the stability of eNOS and is also the basic cofactor for NO synthesis [58]. However, during oxidative stress, BH4 will be converted to dihydrobiopterin (BH2) and promotes uncoupling by interacting with eNOS [59, 60].
ROS play an important role in the pathogenesis of cardiovascular diseases, such as I/R, vascular endothelial and atherosclerosis, hypertension, diabetic cardiomyopathy (DCM), heart failure, cardiac arrhythmias, and aortic aneurysms [3, 42].
A large number of ROS are produced during the process of I/R, due to activation of the ETC and several enzymes [54]. ROS also accelerate the loss of adenosine triphosphate (ATP) during the period of I/R [61]. In early reperfusion, the production of ROS exceeds the removal capacity of antioxidant systems leading to mitochondrial respiratory complex peroxidation [30] (Figure 2). These changes lead to oxidative damage and cardiomyocyte death [6, 61, 62].
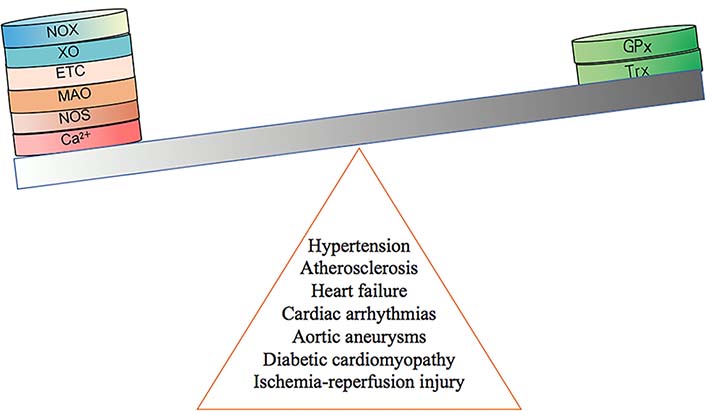
The development of cardiovascular diseases is caused by an imbalance of redox system. GPx: GSH peroxidase; Trx: thioredoxin
During the period of hypoxia/reoxygenation, the changes of intracellular calcium in aortic endothelial cells impact the uptake of Ca2+ in mitochondria [22] (Figure 2). The generation of Ca2+ can be modulated by ROS derived from the NOX [22]. Research indicates that the upregulation of receptor-interacting protein 3 (Ripk3) increases production of mtROS and cell death in I/R [63]. The increased production of mtROS is modulated by Ca2+ overload and XO in the overexpression of Ripk3 [63, 64] (Table 1, Figure 3). In combination with oxidative stress, calcium surplus leads to the opening of mitochondrial permeability transition pore (mPTP) and cardiomyocyte death [6, 22]. In addition to the Ca2+ overload, mitochondrial dysfunction and ROS-induced ROS release (RIRR) also promote cell death [61]. The release of ROS can be reduced by activation of adenosine monophosphate-activated protein kinase (AMPK)/Akt/glycogen synthase kinase-3β (GSK-3β) pathway in I/R [65] (Table 1). Inhibition of transforming growth factor-activated kinase 1 (TAK1) can also reduce ROS production in I/R [66] (Table 1). Moreover, evidence shows that signal transducer and activator of transcription 3 (STAT3) is able to control ROS production [67]. During ischemia, STAT3 overexpression reduces the production of superoxide in the heart [61, 68]. Activation of STAT3 can improve resistance in I/R and prevent cardiac remodeling by modulating interleukin-11 (IL-11) [69] (Figure 3). Additionally, nicotinamide phosphoribosyltransferase (Nampt) was proven to be effective for protection in I/R [36, 70]. Nampt inhibits apoptosis by regulating the level of nicotinamide adenine dinucleotide (NAD+) and ATP [71]. Additionally, mitochondrial connexin 43 (Cx43) modulates the production of mtROS [6, 72]. Cx43 protects the function of cardiomyocytes by casein kinase 1 (CK1) which leads to the phosphorylation of Cx43 [73]. Recently, research findings demonstrated that Cx43 deficiency can lead to a loss of protection in I/R [6, 72, 74, 75] (Figure 3). Furthermore, the citric acid cycle (CAC) is an important element of metabolism. However, it has been reported to be associated with mtROS production recently [76]. The research indicates that succinate was increased in ischemia and then oxidized by succinate dehydrogenase during the period of reperfusion leading to mtROS production [76] (Figure 3). Iron-catalysed reactions are another condition which causes increased ROS production [77]. However, the role of Fe–S clusters is an area for further research [49]. Recent evidence indicates that at the end of I/R, activation of hypoxia-inducible transcription factor (HIF) can increase levels of mitochondrial NADPH and reduce levels of mtROS which prevent cardiac fibroblast formation [78, 79] (Figure 3). Similarly, Gap junction protein Alpha 1-20 kDa (GJA1-20k) and pleckstrin homology-like domain, family A, member 1 (PHLDA1) are also two important factors capable of reducing ROS [80, 81] (Table 1, Figure 3). GPx is also found to inhibit left ventricular (LV) remodeling and improve cardiac tissue survival in I/R [36] (Figure 2, Figure 3). Glutathione exhibits protective effects in cardiomyocytes by reducing the formation of ROS [82] (Figure 3). The reduction of ROS is essential in providing protection during I/R.
Response of ROS under different mechanisms in cardiovascular diseases
| Disease types | ROS | Mechanisms | PMIDs | Publication dates |
|---|---|---|---|---|
| I/R | ↑ | Ripk3–Ca2+ overload–XO–ROS | 29502045 | 2018-02 |
| ↓ | HIF-1–NADPH–mtROS | 34763860 | 2022-02 | |
| ↓ | AMPK–Akt–GSK-3β–ROS | 28128361 | 2017-01 | |
| ↓ | TAK1↓–ROS | 32378287 | 2020-10 | |
| ↓ | GJA1-20k–ROS | 34608863 | 2021-10 | |
| ↓ | PHLDA1–ROS | 31981628 | 2020-03 | |
| Hypertension | ↓ | JMJD1A–ROS | 32461996 | 2020-05 |
| ↓ | AMPK–PAR1–ROS | 29287725 | 2018-01 | |
| ↓ | AMPK–PINK1–parkin–ROS | 29285690 | 2018-03 | |
| ↓ | AMPK–O-GlcNAC↓–ROS | 29285690 | 2018-01 | |
| ↓ | Sirt1–LKB1–MAPK–ROS | 23707558 | 2013-10 | |
| ↓ | Foxo1–SOD2–ROS | 30677512 | 2019-06 | |
| ↓ | ROS–Nrf2–ARE–ROS | 33656904 | 2020-12 | |
| ↑ | AngII–ROS | 30643968 | 2019-01 | |
| ↓ | CELF1↓–PEBP1–MAPK↓–ROS | 34669021 | 2022-01 | |
| Atherosclerosis | ↓ | IRS-1–ROS | 33000267 | 2020-11 |
| ↑ | HIF-1–ROS | 35111045 | 2021-12 | |
| ↓ | PON2–ROS | 17404154 | 2007-04 | |
| ↑ | AMPK–NOX↓–ROS | 31331111 | 2019-07 | |
| ↓ | PKM2↓–G6P–NADPH–ROS | 30222136 | 2018-10 | |
| ↓ | Sirt3–Foxo3α–MnSOD–ROS | 23665396 | 2013-10 | |
| ↓ | Sirt3–IDH2–GSH–ROS | 30455381 | 2019-01 | |
| DCM | ↑ | RAGE–NOX–ROS | 27916650 | 2017-04 |
| ↑ | PKC–NF-κB–iNOS–ROS | 27916650 | 2017-04 | |
| ↑ | NEU1–AMPKα↓–Sirt3↓–SOD2↓–ROS | 35002528 | 2022-01 | |
| Heart failure | ↑ | Sirt3–CypD–mPTP–SOD↓–ROS | 33508434 | 2021-03 |
| Vascular endothelial | ↓ | Sirt2–Foxo3α–SOD–ROS | 34028177 | 2021-07 |
AngII: angiotensin II; ARE: antioxidant response element; CELF1: cytimidine uracil guanine triplet repeat-binding protein 1; CypD: cyclophilin D; Foxo1: forkhead box protein O1; Foxo3α: forkhead box transcription factor 3α; G6P: glucose-6-phosphate; IDH2: isocitrate dehydrogenase 2; IRS-1: insulin receptor substrate 1; JMJD1A: Jumonji domain containing 1A; LKB1: liver kinase B1; MAPK: mitogen-activated protein kinases; MnSOD: manganese SOD; NEU1: neuraminidase 1; NF-κB: nuclear factor kappaB; Nrf2: nuclear factor E2-related factor 2; O-GlcNAC: O-linked N-acetylglucosamine; PAR1: protease-activated receptor 1; PEBP1: phosphatidylethanolamine binding protein 1; PINK1: phosphatase and tensin homolog-induced putative kinase 1; PKC: protein kinase C; PKM2: pyruvate kinase M2; PON2: paraoxonase-2; RAGE: receptor for advanced glycation end products; Ripk3: receptor-interacting serine/threonine-protein kinase 3; Sirt 1: sirtuin 1; PMID: PubMed ID; ↑: increase; ↓: decrease
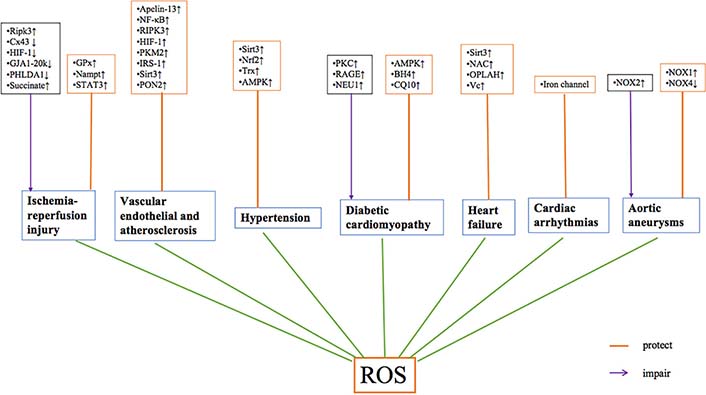
The mechanisms of ROS in cardiovascular diseases. NAC: N-acetylcysteine; OPLAH: oxoprolinase; Vc: vitamin C; ↑: increase; ↓: decrease
The process of atherosclerosis is accelerated by various factors, such as the generation of ROS, inflammatory signaling, and endothelium dysfunction [21]. However, atherosclerosis begins with endothelium dysfunction and plays an important role in vascular homeostasis [21] (Figure 2). The intima of the endothelium is formed of one layer of endothelial cells surrounded by adhesion molecules [83]. In physiological conditions, steady blood flow has little effect on vascular endothelium and increases the production of NO [21]. When the endothelium is injured, lipids deposit in the vascular wall. This causes a change in vascular blood flow and upregulates NOX, leading to oxidative stress [21]. The abnormal production of NO is caused by eNOS disorder [84]. Meanwhile, the reduction and low bioavailability of NO contributes to atherosclerosis because N-hexanoyl-D-erythro-sphingosine is activated [85]. Moreover, Sirt2 plays an important role in maintaining the function of endothelial cells. It can reduce ROS production via Sirt2/Foxo3α/SOD pathway [86] (Table 1). Therefore, the improvement of endothelial cell function could have a protective effect on vascular function [87]. Sirt3 is also the member of sirtuin family which has a protective effect in cardiovascular diseases (Figure 3). It can mediate Foxo3α/MnSOD and IDH2/GSH pathways to reduce ROS production [88–90]. HIF-1 is a heterodimeric protein which plays an important role in atherosclerosis by regulating ROS and NO production [91] (Figure 3). Moreover, novel researches demonstrated that PKM2 is related to release of ROS. The inhibition of PKM2 can activate the G6P/NADPH pathway to reduce ROS in cardiomyocytes exposed to oxygen/glucose [92, 93] (Figure 3). Apelin/APJ is a member of G protein-coupled receptors (GPCR) and is expressed on endothelial and smooth muscle cells [94]. Apelin-13 reduces lipid accumulation of foam cells through activating class III phosphatidylinositol 3-kinase (PI3K)/Beclin-1 pathway [94–96] (Figure 3). Kruppel-like factor 2 (KLF2) and KLF4 have also been reported to act as protective factors in atherosclerosis [21, 97]. Deficiencies of KLF2 and KLF4 accelerate the atherosclerotic process by inducing eNOS [98]. NF-κB can also decrease ROS accumulation by downregulation of c-Jun N-terminal kinase (JNK) [99] (Figure 3). Research has shown that loss of insulin signaling (IRS-1) in vascular endothelium leads to endothelial dysfunction and atherosclerosis [100, 101] (Figure 3). The research found that overexpression of PON2 would reduce the production of ROS by decreasing endoplasmic reticulum stress [102] (Figure 3). Furthermore, the downregulation of receptor-interacting serine/Ripk3 can reduce the activation of inflammatory processes, which has a protective effect in atherosclerosis [103]. The factors HIF-1 and AMPK also participate in ROS production in the process of atherosclerosis [91, 104, 105].
There is robust evidence that ROS production is increased in patients with hypertension [14]. Mechanisms contributing to hypertension are complex; oxidative stress is one of the most important factors [106]. Uncoupled eNOS can promote an increase in cardiomyocyte hypertrophy in response to chronic hypertension [107, 108]. Cardiomyocyte hypertrophy increases the expression of XO [27] (Figure 2). CD8+ T cells contribute to salt-sensitive hypertension by increasing ROS production and sodium retention [109]. Additionally, a reduction in Sirt3 leads to endothelial dysfunction by creating mitochondrial oxidative stress in hypertension [14] (Figure 3). The depletion of Sirt3 contributes to vascular inflammation and hypertrophy, leading to progression of hypertension [110]. The activation via Sirt1/LKB1 and CELF1/PEBP1 pathways can both reduce ROS production [111, 112]. AMPK is a key factor in reducing the production of ROS via different pathways such as AMPK/PAR1, AMPK/phosphatase and tensin homolog-induced putative kinase 1 (PINK1)/parkin and AMPK/O-GlcNAC [113–116] (Table 1). ROS can also be reduced through activation of JMJD1A and Foxo1 [117, 118] (Table 1). Furthermore, NOX4-mediated mtROS signaling is important in the response to chronic pressure overload [27]. NOX4 protects the heart from hypertrophic dysfunction by activation of HIF1a/vascular endothelial growth factor (VEGF) signaling pathway in cardiomyocytes [119]. In hypertension models, the activation of Nrf2 demonstrated an antihypertensive effect [120] (Figure 3). The activation of the Nrf2 pathway induces the production of antioxidant enzymes [120] (Table 1). Nrf2 can be modulated by the mammalian STE20-like protein kinases 1/2 (Mst1/2) to sustain the balance of cellular redox reactions [38, 121]. Thus, presence of mitochondrial antioxidants may improve vascular function [14]. Finally, the Trx system is able to reduce ROS and act as an anti-hypertrophic factor [27] (Figure 3).
Hyperglycemia causes an alteration of mitochondrial morphology, including mitochondrial fragmentation and swelling, leading to an increase in ROS production [60, 122, 123]. Increased production of ROS can also contribute to changes in mitochondrial morphology [23]. However, antioxidants may reduce the production of ROS, offering a potential therapeutic strategy for the treatment of DCM [124]. Altered mitochondrial function may inhibit insulin signaling which leads to activation of PKC [125]. PKC is from a family of kinases related to an increase in ROS production [101]. PKC activates NOX in diabetes and induces oxidative stress [59] (Figure 3). PKC can also activate NF-κB to increase ROS release [59] (Table 1). Furthermore, PKC can be up-regulated by p66shc, and then inhibit eNOS activity, creating a vicious cycle [101] (Figure 2). AMPK is also reported to reduce the production of mtROS [126] (Figure 3). Activated phosphorylated AMPK (pAMPK) can inhibit pyroptosis in DCM [127]. However, research indicates that inhibiting the expression of AMPK/p38 MAPK signaling reduces ROS production [128]. This production of ROS is different due to the distinct pathway that AMPK activates. For example, activation of NEU1 inhibits the AMPKα/Sirt3-SOD2 pathway and leads to ROS production [129] (Table 1). Moreover, advanced glycation end products (AGE) binding to the RAGE cause activation of NADPH oxidase enzymes that lead to ROS generation [59] (Table 1, Figure 3). In DCM, the increased expression of NOX2 contributes to ROS production [23]. ROS formation through NOX is associated with pathways involving sodium/glucose cotransporter 1 (SGLT1), PKCβ, and calcium/calmodulin dependent kinase II (CaMKII) [130]. Further, the decreased activation of NOX2 can reduce myocardial oxidative stress and remodeling which improves cardiac function [131]. In addition to ROS, high glucose-induced generation of NOS causes DNA damage [23, 60]. Excess glucose also induces arginase activity and upregulates eNOS activity [23, 60]. The expression of NOS is increased in diabetic hearts and leads to enhanced lipid peroxidation and peroxynitrite generation [23]. Lastly, iNOS is up-regulated in DCM, with increased levels of 4-hydroxynonenal (4-HNE) [59]. Supplementation with BH4 may be possible to reduce oxidative stress in DCM [59] (Figure 3). Coenzyme-Q10 (CQ10) mitochondria-targeted antioxidants also reduce H2O2 in hyperglycemia [59] (Figure 3).
Heart failure is a condition where the heart exhibits abnormal cardiac structure and function leading to pump failure [22, 27]. The mechanisms contributing to heart failure are complex, including mitochondrial dysfunction, redox imbalance, ion disorder, and inflammation [24]. The expressions of XO and MAO are elevated in heart failure, leading to increased ROS production [27, 36] (Figure 2). One of the major mechanisms in heart failure is an increase of mtROS due to mitochondrial stress [24] (Figure 3). Furthermore, ROS release contributes to the progression of heart failure and leads to cardiac dysfunction and ventricular remodeling [132]. In heart failure, diastolic calcium leak contributes to cytoplasmic calcium overload and diastolic dysfunction and arrhythmia [27]. In turn, ROS inhibits calcium reuptake and impacts diastolic function [133]. Additionally, ROS activates the apoptosis signal-regulating kinase-1 (ASK-1)/JNK-dependent pathway which causes apoptosis in in vivo models of heart failure [134]. Sirt3 has been shown to be downregulated in the failing heart [24] (Figure 2). Sirt3 offers heart protection by maintaining mitochondria function [135]. The activity of Sirt3 is mediated by NAD+ availability [136]. Decreased NAD+ levels suppress NAD+ dependent protein deacetylation, resulting in mitochondrial protein hyperacetylation and impaired function [24, 136]. Sirt3 can also regulate ROS production by CypD/mPTP/SOD pathway [137] (Table 1).
Several studies demonstrated that antioxidant NAC can improve GSH levels, reduce ROS production, and improve cardiac function [36, 59] (Figure 3). A number of clinical trials have demonstrated that the enhanced expression of 5-oxoprolinase (OPLAH) could improve GSH/oxidized glutathione (GSSG) ratio and benefit heart failure [36] (Figure 3). Vc may also be used as an antioxidant to improve endothelial function in heart failure [22] (Figure 3).
Atrial fibrillation (AF) is one of the common arrhythmias related to ATP deficiency and changes in Na+, K+, and Ca2+ channels [138] (Figure 3). Recently ROS was reported to play an important role in AF [139]. NOX2-derived ROS generation has been implicated in experimental and clinical AF [27]. The production of mtROS contributes to cardiac fibrosis which is a characteristic of AF [139]. Furthermore, AF causes the opening of mPTP leading to a disruption of Ca2+ homeostasis and mtDNA damage [140]. The nitroso-redox balance may sensitize cardiac ryanodine receptor (RyR2) to induce ventricular arrhythmias. This reaction leads to an imbalance of Ca2+ and increased formation of ROS [141]. Modulating ion channels is one target for arrhythmia treatment and many anti-arrhythmic medications target these channels [106, 142]. Radiofrequency ablation is used to block abnormal conduction bundles and the origin of tachyarrhythmias. All of these sustain ion homeostasis in cardiomyocytes.
Marfan’s syndrome (MFS) is a systemic disease with a high incidence of aortic aneurysm and aortic dissection. These conditions have a high mortality in MFS. ROS produce endothelial dysfunction, switch smooth muscle cell phenotype, and cause extracellular matrix remodeling, leading to the progression of MFS [143]. NOX is one of the sources of ROS production. However, NOX has a different function in the process of aortic aneurysm [144]. The lack of NOX1 has a protective effect in aortic aneurysm [145] (Figure 3). However, a deficiency of NOX2 can contribute to the development of aortic aneurysm due to activation of inflammatory processes. A low expression of NOX4 offers potential protection in aortic aneurysm by ameliorating elastic fiber [146] (Figure 3). ROS also participate in cell death which contributes to aortic aneurysm [147]. Treatment of antioxidant stress may provide a potential option for preventing aortic aneurysm.
ROS are highly reactive molecules produced by a system of oxidases which have a great impact in the progression of cardiovascular diseases. The production of ROS disrupts the function of mitochondria and intracellular Ca2+ homeostasis, leading to damage to the cardiovascular system. Several mice models demonstrate that modulation of different pathways can rescue the impaired cardiomyocytes, retard myocardial remodeling, and maintain ion homeostasis. Therapies that target the activation of antioxidant systems, such as an exogenous antioxidant supplement, may be an effective treatment option in cardiovascular diseases. Further research is needed to explore the effect of controlling ROS in cardiovascular diseases.
AF: atrial fibrillation
AMPK: adenosine monophosphate-activated protein kinase
ATP: adenosine triphosphate
BH4: tetrahydrobiopterin
Cx43: connexin 43
DCM: diabetic cardiomyopathy
eNOS: endothelial nitric oxide synthase
ETC: electron transport chain
Foxo3α: forkhead box transcription factor 3α
GSH: glutathione
H2O2: hydrogen peroxide
HIF: hypoxia-inducible transcription factor
I/R: ischemia-reperfusion injury
iNOS: inducible nitric oxide synthase
KLF2: Kruppel-like factor 2
MAOs: monoamine oxidases
MAPK: mitogen-activated protein kinases
MFS: Marfan’s syndrome
mPTP: mitochondrial permeability transition pore
mtDNA: mitochondrial DNA
mtROS: mitochondrial ROS
NAC: N-acetylcysteine
NAD+: nicotinamide adenine dinucleotide
NADPH: nicotinamide adenine dinucleotide phosphate
NF-κB: nuclear factor kappaB
NO: nitric oxide
NOS: nitric oxide synthase
NOXs: nicotinamide adenine dinucleotide phosphate oxidases
Nrf2: nuclear factor E2-related factor 2
PKC: protein kinase C
PKM2: pyruvate kinase M2
Ripk3: receptor-interacting protein 3
ROS: reactive oxygen species
Sirt1: sirtuin 1
SOD: superoxide dismutase
STAT3: signal transducer and activator of transcription 3
XO: xanthine oxidase
JF, LJD and JR contributed conception and design of the study; JF and JR wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Work in our lab has been supported by the National Key R&D Program of China (2017YFA0506000). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Yuka Ikeda ... Satoru Matsuda
Jelena Radovanovic ... Esma R. Isenovic
Ranjeet Singh, Partha Pratim Manna
Anastasija Panic ... Esma R. Isenovic
Tao Wang, Haiyan Xu
Alberto Rubio-Casillas ... Raied Badierah
Largee Biswas ... Anita Kamra Verma