 Review
Review

Affiliation:
1Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
Email: tmatsuda@pharm.hokudai.ac.jp
ORCID: https://orcid.org/0000-0002-3089-3757
Affiliation:
1Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
ORCID: https://orcid.org/0000-0002-3243-9777
Affiliation:
1Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
Affiliation:
1Department of Immunology, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan
ORCID: https://orcid.org/0000-0002-5019-5243
Affiliation:
2Department of Cell Biology, Kyoto Pharmaceutical University, Kyoto 607-8412, Japan
ORCID: https://orcid.org/0000-0002-2861-603X
Affiliation:
3Department of Life Science, Faculty of Pharmaceutical Sciences, Hokkaido University of Science, Sapporo 006-8585, Japan
ORCID: https://orcid.org/0000-0002-7863-6696
Affiliation:
4Department of Hematology, International University of Health and Welfare, Narita, 286-8686, Japan
ORCID: https://orcid.org/0000-0002-5571-2457
Explor Immunol. 2022;2:771–782 DOI: https://doi.org/10.37349/ei.2022.00082
Received: August 08, 2022 Accepted: October 27, 2022 Published: December 27, 2022
Academic Editor: Noah Isakov, Ben-Gurion University of the Negev, Israel
The article belongs to the special issue The Role of Adaptor Proteins in Lymphoid Cell Signaling
Immune responses are orchestrated by controlling the initiation, magnitude, and duration of various signaling pathways. Adaptor proteins act as positive or negative regulators by targeting critical molecules of signaling cascades. Signal-transducing adaptor protein-2 (STAP-2) contains typical features of adaptor proteins, like a pleckstrin homology (PH) domain in the N-terminal region and a Src homology 2 (SH2) domain in the central region. STAP-2 binds to a variety of signaling or transcriptional molecules to control multiple steps of inflammatory/immune responses. STAP-2 enhances T-cell receptor (TCR)-mediated signaling via the association with TCR-proximal CD3ζ immunoreceptor tyrosine-based activation motifs (ITAMs) and lymphocyte-specific protein tyrosine kinase (Lck). STAP-2 decreases adherence of T-cells to fibronectin (FN) through an association with focal adhesion kinase (Fak) and Casitas B-lineage Lymphoma (c-Cbl), and increases chemotaxis of T-cells toward stromal cell-derived factor-1α (SDF-1α) through interactions with Vav1 and Ras-related C3 botulinum toxin substrate 1 (Rac1). STAP-2 positively regulates activation-induced cell deathrough the association with Fas and caspase-8. This review describes the current knowledge of the roles of STAP-2 in T-cell-dependent immune responses and the possible clinical utility of STAP-2-targeting therapies.
Adaptor proteins associate with various intracellular signaling molecules, leading to the modification of their functions [1–3]. Adaptor proteins can modulate signals by linking functional catalytic enzymes, although they generally have no intrinsic catalytic activity. Some adaptor proteins contribute to the coordination of intracellular signaling for cell proliferation, differentiation, and activation, as well as cell migration [1–3].
T-cells, the major regulator of adaptive immune responses, are essential to eliminate invading pathogens. T-cell receptor (TCR) associating with an antigenic peptide shown by major histocompatibility complex (MHC) components, initiates various developmental and/or functional events [4], such as the selection, differentiation, and proliferation of T-cells, as well as cytokine production, to provide optimal responses against invasive pathogens.
Signal-transducing adaptor protein-2 (STAP-2) was first identified as a c-Fms/macrophage colony-stimulating factor receptor (M-CSFR)-binding protein, and was subsequently reported to act as an adaptor protein for signaling and transcription factors [5]. STAP-2 has high sequence and structural similarities to STAP-1, which was identified as a c-Kit-binding protein [6, 7]. STAP-2 can modulate the transcriptional activity of the signal transducer and activator of transcription 3 (STAT3) and STAT5 [5, 8–11], as well as high-affinity immunoglobulin E (IgE) receptor (FcεRI)- [12, 13], M-CSFR- [14, 15], and Toll-like receptor (TLR)-mediated signals [16]. Furthermore, our recent studies have proposed the contribution of STAP-2 to T-cell-related signaling, such as TCR- [17], integrin- [18], chemokine- [19], and Fas-mediated signals [20]. Therefore, STAP-2 expression is likely to regulate various T-cell functions at multiple stages of immune responses. This review describes current knowledge regarding the roles of STAP-2 and how STAP-2 functions in T-cells.
Members of the STAP family contribute to various steps of immune responses and tumorigenesis. The STAP family consists of STAP-1 and STAP-2. STAP-1, which is also called as B-cell antigen receptor downstream signaling 1 (BRDG1), was identified as a protein tyrosine-phosphorylated by Tec kinase [6]. By yeast two-hybrid system with a hematopoietic stem cell library, STAP-1 was also identified as a c-Kit-binding protein [7]. STAP-2 was originally identified as a c-Fms-binding protein [5] and a murine homolog of an adaptor molecule, BKS, a substrate of breast tumor kinase (Brk) [21], which is distantly related to Src family tyrosine kinase [22]. STAP-2 interacts with Brk via its pleckstrin homology (PH) domain and is involved in the induction of robust STAT3 activation [8, 9]. STAP-1 and STAP-2 show an overall 33% amino acid (aa) identity. Both of them consist of a PH domain in their N-terminal region and a Src homology 2 (SH2) domain in their middle region [5] (Figure 1A). The aa sequence identity of the PH domains is 36% between STAP-1 and STAP-2. The aa sequence identity of the SH2 domain of STAP-2 is 40% with that of STAP-1, and 29% with the SH2 domain of phospholipase C (PLC)-γ2 [5]. STAP-2, but not STAP-1 contains a proline-rich region with a Tyr-X-X-Gln (YXXQ) motif that binds to STAT3 in the C-terminal region [5].
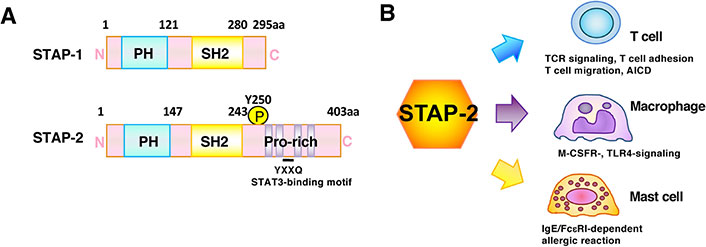
Structural and functional features of STAP-2 adaptor protein. (A) Structural characteristics of STAP-1 and STAP-2. Both consist of an amino N-terminal PH domain and a central SH2 domain. STAP-2, but not STAP-1, contains a carboxy C-terminal proline-rich domain and a STAT3-binding YXXQ motif. STAP-2 Tyrosine 250 (Y250) is phosphorylated by various protein tyrosine kinases; (B) STAP-2 modulates various immune signaling pathways. In T cells, STAP-2 modulates TCR-, integrin-, chemokine-, or Fas-mediated signals. In macrophages, STAP-2 modulates the M-CSFR- or TLR4-mediated signals. In mast cells, STAP-2 modulates the IgE/FcεRI -mediated signals. Thus, STAP-2 plays a physiological role in immune responses via these interactions. AICD: activation-induced cell death
Note. Adapted from “Possible therapeutic applications of targeting STAP proteins in cancer,” by Matsuda T, Oritani K. Biol Pharm Bull. 2021;44:1810–8 (https://www.jstage.jst.go.jp/article/bpb/44/12/44_b21-00672/_article). CC BY.
STAP-1 shows restricted expression largely in hematopoietic cells [5, 6], but its expression is upregulated in pro-inflammatory macrophages and microglia cells that contribute to neuronal apoptosis and degeneration [23]. STAP-1 gene mutations have been shown in some patients with autosomal dominant hypercholesterolemia [24, 25]; however, the STAP-1’s functional role in cholesterol homeostasis remains questionable [26, 27]. STAP-1 acts to control the maintenance and activation of invariant natural killer (NK) T-cells and is involved in the pathogenesis of autoimmune hepatitis [28]. STAP-1 also plays a critical role in the maintenance of chronic myeloid leukemia leukemic stem cells [29]. STAP-2 is expressed in many types of cells and tissues, such as lymphocytes, dendritic cells, macrophages, and hepatocytes [30]. The ubiquitous expression pattern of STAP-2 suggests that it broadly contributes to various signaling and transcriptional activities (Figure 1B). In T-cells, STAP-2 regulates STAT5- and STAT3-mediated expression of cytokine-responsive genes and promotes activation of the Fas-mediated caspase cascade [30]. In dendritic cells and macrophages, STAP-2 promotes TLR-mediated signals and downregulates FcεRI-mediated signals [30]. Therefore, STAP-2 is now believed to adequately control both immune and inflammatory responses.
TCR stimulation triggers fundamental events in T-cell responses, such as selection, differentiation, proliferation, and cytokine production. TCRs are composed of TCRα/β heterodimers and three dimers of subunits of the invariant signaling protein CD3. Intracellular signaling downstream of TCR is stimulated by tyrosine-phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in the cytoplasmic domain of CD3 [4, 31]. Phosphorylation of the tyrosine residues in ITAMs by Src family protein tyrosine kinases, such as lymphocyte-specific protein tyrosine kinase (Lck) and Fyn, leads to the recruitment of 70 kDa zeta-chain associated protein (ZAP-70) via its tandem SH2 domain [32, 33], followed by amplification of TCR-mediated signals via phosphorylation of additional intracellular substrates, including PLC-γ, Cbl, Vav, Linker for activation of T-cells (Lat), and SH2 domain-containing leukocyte protein of 76 kDa (SLP-76) [34–37]. Tyrosine phosphorylation of PLC-γ promotes its enzymatic activity, which leads to activation of the inositol phospholipid pathway, followed by activation of protein kinase C and an increase in intracellular calcium. Calcineurin, a Ca2+-induced phosphatase, is then activated, followed by induction of nuclear factor of activated T-cells (NFAT) activation, which finally induces transcription of the interleukin 2 (IL-2) gene [38]. Thus, phosphorylation and dephosphorylation of downstream tyrosine residues determine the strength of TCR-mediated signals [4].
It has recently been demonstrated that STAP-2 can modulate TCR-mediated signals [17]. TCR-mediated signals are impaired in STAP-2-deficient T-cells but are enhanced in STAP-2-overexpressing T-cells. Concerning its molecular mechanisms, STAP-2 constitutively binds to CD3ζ ITAM and acquires Lck-binding capacity after TCR engagement (Figure 2A). The formation of a trimolecular complex of TCR with a MHC and CD4/CD8 co-receptor occurs in two major steps. In the first stage, the binding of MHC bearing a peptide antigen to corresponding TCRs leads to ITAM phosphorylation by free Lck [39]. In the second step, subsequent MHC-co-receptor binding recruits co-receptor-associated Lck, leading to an interaction between Lck and CD3ζ ITAM [39–41]. However, detailed molecular mechanisms of how CD3ζ ITAM recruits Lck and how Lck initiates CD3ζ ITAM phosphorylation remain to be resolved. The STAP-2 PH domain binds to Lck, and the STAP-2 C-terminal region binds to CD3ζ ITAMs. Additionally, Lck-STAP-2 interactions are dependent on the phosphorylation of STAP-2 at Tyr250. Upon TCR stimulation, STAP-2 seems to act as a scaffold protein to enhance binding involving CD3ζ ITAM and Lck in a phosphorylation-dependent manner. Otherwise, STAP-2 in T-cells is essential for obtaining full TCR signaling activity. The binding of STAP-2 to CD3ζ ITAM and phosphorylated Lck may provide an answer. STAP-2 enhances interactions between CD3ζ ITAM and Lck only when TCR is engaged. Alternatively, STAP-2 may strengthen and prolong their binding.
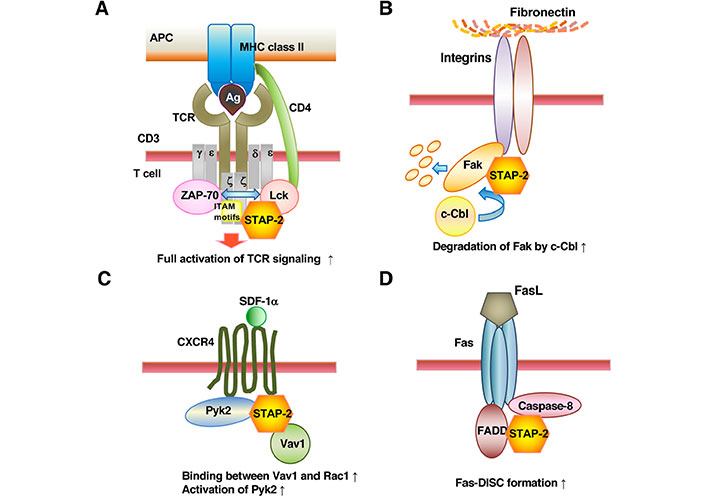
STAP-2 modulates T-cell functions through interaction with individually unique signaling molecules. STAP-2 can modulate (A) TCR-, (B) integrin-, (C) chemokine-, or (D) Fas-mediated signals as illustrated. APC: antigen-presenting cell; Ag: antigen; CXCR4: C-X-C motif chemokine receptor 4; Fas-DISC: Fas-death-inducing signaling complex; FADD: Fas-associated protein with death domain; FasL: Fas ligand; Pyk2: protein tyrosine kinase 2; SDF-1α: stromal cell-derived factor-1α; ↑: up regulation
Note. Adapted from “STAP-2 adaptor protein regulates multiple steps of immune and inflammatory responses,” by Matsuda T, Oritani K. Biol Pharm Bull. 2021;44:895–901 (https://www.jstage.jst.go.jp/article/bpb/44/7/44_b21-00224/_article). CC BY.
STAP-2 expression levels affect the onset and severity of CD4+ T-cell-related autoimmune diseases, such as Propionibacterium (P.) acnes-induced granulomas and experimental autoimmune encephalomyelitis (EAE). Granulomas are formed to compartmentalize intracellular bacteria and to limit their infection within a restricted area [42]. However, excessive granulomatous responses impair normal organ functions, resulting in tissue damage. Certain human autoimmune disorders, such as sarcoidosis, Crohn’s disease, and Wegener’s granulomatosis, are characterized by pathologic granulomatous inflammation [42–44]. P. acnes injection promotes splenomegaly and liver granuloma formation as a result of activation of T-helper 1 (Th1) or Th17 cells [45]. Of note, granuloma formation is reduced in STAP-2-knockout (KO) mice but is augmented in STAP-2-transgenic (Tg) mice [17]. Additionally, P. acnes-induced expression of IL-2 and IFN-γ messenger RNAs (mRNAs) in the liver is lower in STAP-2-KO mice and higher in STAP-2-Tg mice. EAE, an animal model for human multiple sclerosis, culminates in nerve demyelination, axonal damage, and paralysis as a result of autoimmune responses against central nervous system (CNS) structures [46–48]. After myelin oligodendrocyte glycoprotein (MOG)35–55 immunization, STAP-2-KO mice exhibit lower EAE clinical scores, whereas STAP-2-Tg mice display more severe EAE phenotypes [17]. In addition, IL-17A-expressing CD4+ cells heavily infiltrate the CNS in STAP-2-Tg mice. Therefore, STAP-2 in T-cells is likely to influence the severity of autoimmune and/or inflammatory diseases.
Adhesion and interaction of T-cells with other immune cells are important events in immune responses. Integrins on T-cells and their ligands, such as fibronectin (FN), are key players in lymphocyte migration and adhesion [49]. After their interactions, focal adhesion kinase (Fak) plays a central role in integrin-mediated signals. Indeed, cells derived from Fak-KO embryos display severely impaired capacity in terms of migration and adhesion [50]. Conversely, Fak overexpression enhances cell motility and survival in an anchorage-independent manner [51, 52]. Mechanistically, ligand binding to integrins induces catalytic activation of Fak and its autophosphorylation at Tyr397, which is presented as a recognition site for Src family kinases. In addition, Fak also interacts with the adaptor protein Growth factor receptor-bound protein 2 (Grb2) to activate the pathway [53].
We have published results demonstrating that STAP-2 can modulate integrin-mediated signals [18]. STAP-2-KO T-cells have high cell adhesion ability to FN by phorbol myristate acetate-treatment. Of note, STAP-2-KO T-cells contain dramatically increased Fak protein levels, while STAP-2 overexpression promotes decreased Fak protein abundance and impaired integrin-induced adhesion to FN. Regarding molecular mechanisms, STAP-2 binds to Fak and recruits Casitas B-lineage Lymphoma (c-Cbl), an endogenous E3-ubiquitin ligase, to Fak, leading to the enhancement of Fak degradation by c-Cbl. In addition, STAP-2 colocalizes with Fak at focal-adhesion sites when integrins are activated. Therefore, STAP-2 expression downregulates integrin-mediated signaling in T-cells, in which STAP-2 negatively regulates Fak protein content.
The protein 4.1, ezrin, radixin, moesin (FERM) homology domain in the N-terminal region of Fak can interact with both STAP-2 and c-Cbl. These interactions seem to allow STAP-2 to recruit c-Cbl to Fak, thereby following the enhanced Fak ubiquitination. This possibility is partly supported by data showing that Fak protein content in Jurkat/STAP-2 cells is raised by treatment with proteasome inhibitors and that c-Cbl-overexpression reduces and c-Cbl-knockdown raises Fak protein content.
The N-terminal region of c-Cbl consists of a tyrosine kinase binding (TKB) domain and a RING finger domain, both of which comprise a basic functional unit of ubiquitin ligase [54, 55]. The TKB domain recognizes a consensus phosphotyrosine motif, N/DXpYXXXf, in which pY represents a phosphorylated tyrosine residue and f represents a hydrophobic residue [56]. The aa sequence DDYAEII, surrounding Fak Tyr397 completely coincides with the consensus phosphotyrosine motif for c-Cbl-recognition. Thus, c-Cbl negatively controls integrin/Fak-induced signals by stimulating focal-adhesion dissociation. Therefore, the STAP-2 protein level seems to determine the focal-adhesion turnover by regulating the levels of Fak proteins (Figure 2B).
Chemokines, a group of chemotactic cytokines, play a critical role in chemotaxis and transendothelial migration of immune cells in immune and inflammatory responses [57]. SDF-1α (also called C-X-C motif chemokine 12 (CXCL12)) belongs to the CXC chemokine family, which interacts with the seven-transmembrane G protein-coupled receptors, CXCR4 and CXCR7 [57–59]. Ligation of SDF-1α to CXCR4 triggers intracellular Ca2+ influx and the activation of Extracellular signal-regulated kinase (Erk), phosphatidylinositol-3 kinase (PI3K), nuclear factor-κB (NF-κB), the Rho family proteins [RhoA, Ras-related C3 botulinum toxin substrate 1 (Rac1), and Cdc42], and Pyk2. The binding of SDF-1α to CXCR4 promotes essential signals for leukocyte trafficking and surveillance, as well as the homing of hematopoietic stem cells into bone marrow and their retention within a supportive niche. Both SDF-1α and CXCR4-KO mice have significantly reduced numbers of lymphocytes and myeloid progenitors [60]. In addition, SDF-1α promotes strong adhesion of rolling immune cells to endothelial cells, followed by their further transendothelial migration.
It has been demonstrated that STAP-2 can modulate chemokine-mediated signals [19]. STAP-2 overexpression upregulates, whereas STAP-2-deficiency downregulates T-cell migration toward SDF-1α (Figure 2C). In terms of molecular mechanisms, STAP-2 binds to not only Vav1, a guanine-nucleotide exchange factor for Rac1, but also the Rho family protein Rac1. Constitutive interaction of STAP-2 with both Vav1 and Rac1 promotes tight binding between Vav1 and Rac1, leading to the enhancement of downstream Vav1/Rac1 signaling [61]. The other possible involvement of STAP-2 is in the control of Pyk2, which comprises signaling complexes together with paxillin, CT10 regulator of kinase (Crk), and p130 Crk-associated substrate (Cas) [62]. STAP-2 directly binds to Pyk2 and enhances its phosphorylation. Of note, SDF-1α-induced T-cell chemotaxis is controlled by Pyk2 small interfering RNA (siRNA) treatment or a Pyk2 inhibitor in Jurkat T-cells expressing STAP-2 [63]. Therefore, STAP-2 expression contributes to chemokine-mediated lymphocyte migration and homing through upregulating the functions of Pyk2 and Rho family proteins.
In response to infection or immunization, T-cells expressing antigen-specific TCRs go into an activated and proliferative phase and some differentiate into effector cells through the TCR signaling described above. Activated T-cells then produce cytokines, which coordinate the immune response to eliminate pathogens [4]. Clearance of the antigen is accompanied by the shutdown of T-cell immune responses and involves apoptosis of a large fraction of antigen-activated T-cells. This avoids the accumulation of no-longer-needed and potentially dangerous effector cells to preclude immunopathology through mitochondrial apoptotic and/or Fas-mediated signaling pathways (termed AICD) [64]. Fas oligomerization results in Fas-DISC formation, in which caspase-8 activation occurs. The recruitment of FADD to Fas triggers dimerization and conformational changes of caspase-8, leading to its full enzyme activity. Activated caspase-8 then undergoes autoproteolytic processing and leaves Fas-DISC to access its substrates. Indeed, mice with naturally occurring mutations involving the Fas or FasL genes exhibit severe lymphocytosis and frequently experience autoimmune diseases [65].
It has been demonstrated that STAP-2 can modulate Fas-mediated signals [20]. STAP-2 expression in T-cells promotes Fas-induced apoptosis. STAP-2 directly binds to both caspase-8 and Fas, thereby enhancing interactions between caspase-8 and FADD in Fas-DISCs (Figure 2D). STAP-2 interacts with the death effector domain of caspase-8, even under steady-state conditions, and upregulates the activation and aggregation of caspase-8 during Fas-signaling. It is noteworthy that the C-terminal domain of STAP-2 has a consensus cleavage aa sequence, for caspase-8, Val-Glu-Ala-Asp (VEAD), and cleavage of STAP-2 protein by caspase-8 is essential for maximum induction of apoptosis. Indeed, STAP-2-KO mice display impaired AICD and superantigen-induced T-cell depletion, showing the physiological roles of STAP-2 in Fas-mediated signaling. Therefore, STAP-2 plays as a new member of Fas-DISC and contributes to AICD.
As described in this review, the adaptor protein STAP-2 influences multiple T-cell functions, such as TCR signaling, adhesion to FN, chemotaxis toward SDF-1α, and AICD, through individually unique molecular mechanisms. It has been speculated that diverse effects may come from the wide binding capacity of STAP-2 in each domain to a variety of signaling and transcriptional molecules. Indeed, the STAP-2 PH domain interacts with Brk, c-Fms, and Pyk2 [9, 14, 63]. The STAP-2 SH2 domain interacts with Myeloid differentiation primary response protein 88 (MyD88), IκB kinase β (IKK-β), FAK, Pyk2, Cbl, Vav and PLC-γ1 [12, 16, 18, 19, 63, 66]. The STAP-2 C-terminal interacts with Vav and PLC-γ1 [12, 19]. Through comprehensive protein-protein interactions between STAP-2 and key signaling molecules, STAP-2 expression in T cells contributes to immune and inflammatory responses at multiple steps in vivo and depends on its expression levels in the situation. Thus, good manipulation of STAP-2 seems to have the potential for clinical applications.
Chimeric antigen receptor T-cells (CAR-T) is a new technology to treat patients with malignancies. CD19-targeting CAR-T cells, which is a therapy approved for refractory or relapsed B-cell acute lymphoblastic leukemia and lymphoma, exhibit high response rates and tolerability [67, 68]. Many researchers have sought to augment CAR-T cell functions by genetically modifying its structure, as well as the activation and proliferation of T-cells by immune cytokines. Usually, early CARs have antibody single-chain variable fragments fused through a transmembrane domain to the cytoplasmic domain of CD3ζ, although the addition of costimulatory signaling domains is required to perform optimal clinical efficacy. Next-generation CAR constructs are being developed by adding additional signaling motifs, including not only TCR signaling but cytokine signals. Thus, novel signaling motifs are attractive to create new types of CARs. Because STAP-2 overexpression can enhance TCR-mediated signaling [17], alter T cell migration and adhesion [18, 19], and regulate survival of activated T cells [20], manipulated ectopic expression of STAP-2 in CAR-T cells may be a powerful and promising method to improve the therapeutic efficacy of CAR-T therapy.
The onset and development of autoimmune diseases are largely caused by dysregulated immune systems. Some available treatment options inhibit key events during immune responses. Because STAP-2 enhances TCR-mediated signaling and is required for their maximum activity, STAP-2 inhibition may improve clinical symptoms of autoimmune diseases. Thus, the development of peptides or low molecular weight compounds to inhibit STAP-2 proteins is desirable in clinical fields. As shown above, the binding capacity of each STAP-2 functional domain to various key molecules during signaling and inflammatory responses has been identified. The specific blockage of interactions of STAP-2 with these signaling molecules seems to regulate each signaling. Although many binding partners may be weak points for a new drug because of the increasing possibility of off-target effects, we are trying to determine the detailed aa sequence responsible for their interactions, and utilized the synthetic peptide for their sequences with octa-arginine sequences as cell-permeable interfering peptides for each signaling. Interfering peptides, which target protein-protein interactions, became promising therapeutics. Such interfering peptides to inhibit important protein-protein interactions for cellular events have been reported, and some are now tested in clinical trials [69]. For example, a cell-permeable peptide inhibitor, estrogen receptor-α (ERα) activity-regulator peptide (ERAP), which specifically disrupts the brefeldin A-inhibited guanine nucleotide exchange protein 3 (BIG3)-prohibitin 2 (PHB2) interaction, suppresses tamoxifen resistance and enhances tamoxifen responsiveness in ERα-positive breast cancer cells by regulating multiple ERα-signaling pathways driving breast cancer cell growth via reactivating tumor-suppressive activity of PHB2 [70]. Of note, fewer side effects of STAP-2 inhibitors can be predicted, since STAP-2-deficient mice show no severe phenotype under steady-state conditions.
In conclusion, STAP-2 regulates T-cell functions at multiple events during immune responses. Manipulation of STAP-2 may be a powerful strategy in CAR-T therapy against malignancies, as well as establishing new drugs for immune-related diseases.
aa: amino acid
AICD: activation-induced cell death
Brk: breast tumor kinase
CAR-T: chimeric antigen receptor T
c-Cbl: Casitas B-lineage Lymphoma
CXCR4: C-X-C motif chemokine receptor 4
EAE: experimental autoimmune encephalomyelitis
ERα: estrogen receptor-α
FADD: Fas-associated protein with death domain
Fak: focal adhesion kinase
Fas-DISC: Fas-death-inducing signaling complex
FN: fibronectin
IL-2: interleukin 2
ITAM: immunoreceptor tyrosine-based activation motif
KO: knockout
Lck: lymphocyte-specific protein tyrosine kinase
M-CSFR: macrophage colony-stimulating factor receptor
MHC: major histocompatibility complex
P.: Propionibacterium
PH: pleckstrin homology
PLC: phospholipase C
Pyk2: protein tyrosine kinase 2
Rac1: Ras-related C3 botulinum toxin substrate 1
SDF-1α: stromal cell-derived factor-1α
SH2: Src homology 2
STAP: signal-transducing adaptor protein
STAT: signal transducer and activator of transcription
TCR: T-cell receptor
Tg: transgenic
TLR: Toll-like receptor
We would like to thank Editage (www.editage.com) for English language editing.
TM: Writing–Original & Final Draft. Y Sekine, JK and KO: Conceptualization & Design, Editing–the Section of the Paper. Y Sasaki, KK and KS: Writing–Review & Editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was supported in part by Grant-in-Aid for scientific research [19H03364, 22H03544, and 21K08451] from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Mari Hikosaka Kuniishi ... Takanori So
Laura E. McMillan, Christoph Wülfing
Shulamit Katzav
Xiaoyu Jiang, Izidore S. Lossos
