Review
Review

Affiliation:
Laboratory of Immunopathology and Immunosenescence, Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, 90134 Palermo, Italy
ORCID: https://orcid.org/0000-0002-6075-0845
Affiliation:
Laboratory of Immunopathology and Immunosenescence, Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, 90134 Palermo, Italy
ORCID: https://orcid.org/0000-0003-3565-9529
Affiliation:
Laboratory of Immunopathology and Immunosenescence, Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, 90134 Palermo, Italy
ORCID: https://orcid.org/0000-0003-2593-3221
Affiliation:
Laboratory of Immunopathology and Immunosenescence, Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, 90134 Palermo, Italy
ORCID: https://orcid.org/0009-0001-0760-2587
Affiliation:
Laboratory of Immunopathology and Immunosenescence, Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, 90134 Palermo, Italy
Email: giuseppina.candore@unipa.it
ORCID: https://orcid.org/0000-0002-9966-934X
Explor Immunol. 2026;6:1003241 DOI: https://doi.org/10.37349/ei.2026.1003241
Received: July 30, 2025 Accepted: January 23, 2026 Published: March 24, 2026
Academic Editor: Giuseppe Murdaca, University of Genova, Italy
Autoimmune diseases (ADs) comprise a heterogeneous group of disorders characterised by a breakdown of immune tolerance and chronic immune-mediated tissue damage. Their onset and progression result from a complex interplay between genetic susceptibility, hormonal influences, immune regulatory mechanisms, and environmental exposures collectively referred to as the exposome. A striking feature of ADs is their pronounced sexual dimorphism, with a markedly higher prevalence in women for most conditions, alongside sex-specific differences in clinical presentation, severity, and prognosis. This review provides a comprehensive overview of the genetic and environmental determinants contributing to autoimmunity, with particular emphasis on how sex- and gender-related factors shape immune tolerance and disease susceptibility. We discuss central and peripheral tolerance mechanisms, the role of key regulators such as AIRE and FOXP3, sex chromosome-linked effects including X-chromosome inactivation, and the immunomodulatory impact of sex hormones across different life stages. Furthermore, we examine how environmental exposures, infections, and geographic variability interact with genetic background and sex-specific immune regulation to influence autoimmune disease risk. By integrating biological sex and gender-related factors within the framework of immune tolerance and the exposome, this review highlights the need for a sex-aware and personalised approach to understanding, diagnosing, and treating ADs.
Autoimmune diseases (ADs) occur when the immune system fails to correctly distinguish between self and non-self, resulting in an attack on the tissues of the body. This failure in immunological discrimination, commonly described as a loss of immune tolerance, triggers inflammation, damages tissues, and compromises organ function.
The emergence of these diseases is multifactorial, with contributions from genetic predisposition, hormonal influence, environmental exposures, and immune regulation abnormalities [1]. Indeed, most ADs have a polygenic basis, complicating the analysis of the individual contribution of each gene to disease development. Rare ADs are characterised by monogenic manifestation, such as autoimmune polyendocrinopathy syndrome type I (APS-I) or immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX), which makes it easier to define the determinants of the pathology [2]. The first one is caused by mutations in the autoimmune regulator (AIRE) gene, which is involved in the expression of auto-antigens in the medullary thymic epithelial cells (mTECs), hence in the process of central tolerance. The second one, instead, is characterised by an alteration in the forkhead box P3 (FOXP3) gene and could be correlated with different expression in genes related to the function of regulatory T cells (Tregs). Even if these diseases are well characterised under a genetic aspect, the manifestation is always variable, involving different mutational loci, as in the case of APS, and different tissue involvement [2].
Furthermore, several genes are associated with heightened susceptibility to autoimmunity manifestation (see below) [3], but inheritance alone does not fully explain disease occurrence. This is demonstrated by the case of monozygotic twins, whose concordance rates for ADs range from 12% to 67%, indicating the relevance of environmental, stochastic, or epigenetic influences in addition to genetic factors [4, 5]. For instance, genetically predisposed individuals may develop autoimmunity upon encountering specific environmental triggers, which are indicated in this review as the exposome [6].
Among the genetic contributors to the development of ADs, since its discovery nearly seven decades ago, the human leukocyte antigen (HLA) system has substantially advanced our understanding of the immune system and the pathogenesis of ADs. Genetic associations between HLA and ADs were first reported in the early 1970s, and since then, HLA typing has been routinely integrated into the diagnostic evaluation of several autoimmune conditions. To date, HLA loci consistently exhibit the strongest association signals among all genomic regions identified by genome-wide association studies (GWAS) across a broad spectrum of autoimmune and inflammatory diseases. The HLA system, corresponding to the major histocompatibility complex (MHC) in humans, is central to the presentation of intracellular and extracellular antigenic peptides and to the regulation of both innate and adaptive immune responses. Its critical role in thymic selection and the induction of peripheral T-cell anergy provides a mechanistic basis for its involvement in autoimmunity. Although the selective presentation of disease-relevant self-peptides by susceptibility-associated HLA alleles remains the most widely accepted and empirically supported mechanism, the precise biological processes underlying the HLA-autoimmunity association continue to be elucidated [7]. Moreover, significant progress has been made in elucidating the timeline of autoimmune development. Research has shown that autoantibodies can appear years before the onset of symptoms [8]. These preclinical phases have been observed in type 1 diabetes (T1DM) and other ADs.
Then, the manifestation of ADs can be organ-specific or affect the entire body, thus having a systemic involvement. In organ-specific disorders such as T1DM and myasthenia gravis (MG), immune cells recognise tissue-restricted autoantigens, e.g., pancreatic β-cell proteins or acetylcholine receptor components. Conversely, systemic ADs like systemic lupus erythematosus (SLE) or dermatomyositis involve immune responses against antigens expressed across multiple tissues, including skin, kidney, lung, joints, and bone marrow [9].
These findings underscore that persistent immune recognition of autoantigens can gradually evolve into clinical disease, with both qualitative and quantitative changes in the autoimmune response contributing to symptom manifestation [10].
This review aims to examine the mechanisms responsible for sex and gender differences in the incidence of ADs, focusing on the interplay between genetic factors and the so-called exposome in disease development.
Epidemiological evidence has consistently demonstrated that ADs tend to run in families and are frequently inherited as autosomal disorders [11]. Although the exact aetiology of ADs remains elusive and often multifactorial, as previously outlined in relation to the role of the HLA system, genetic predispositions play a critical role in both disease susceptibility and prognosis, as well as in defining the sex-specific patterns of disease manifestation. Of course, the importance of sex, and so of genetic and hormonal factors, must not be overlooked, as most of ADs are more prevalent in women [12]. In fact, the distribution, presentation, and progression of many ADs are significantly influenced by sex-linked genetic factors [13]. Simultaneously, exposome-related elements, which encompass factors such as individual lifestyle, occupational exposure, and broader socioeconomic conditions, have emerged as key contributors in shaping what are recognised as “gender differences” in the development and expression of ADs (see below).
The enhanced sex-specific disparities in the manifestation of ADs are also reflected in the increasing global burden of ADs over recent decades. Data reveal that the worldwide incidence and prevalence of ADs have risen annually by approximately 19.1% and 12.5%, respectively, across sexes [14]. ADs currently affect an estimated 8% to 10% of the global population, with a disproportionately high occurrence in women, who account for nearly 85% of diagnosed cases [15, 16]. Exceptions to this pattern exist; for example, T1DM shows a higher incidence and prevalence among males [17]. The ratio of affected women to men in specific ADs has widened significantly, now ranging from 1:1 to as high as 10:1 in some conditions (Table 1) [18].
Sex-bias and dominant immune mechanisms in autoimmune diseases.
| Autoimmune disease | Sex predominance | Female:Male ratio | Dominant immune response | References |
|---|---|---|---|---|
| SLE | Female | 8:1 | Humoral/autoantibody-mediated | [16–20] |
| SS | Female | 19:1 | Humoral/autoantibody-mediated | [16–20] |
| SSc | Female | Not specified (female-biased) | Humoral/mixed | [19] |
| RA | Female | 2–3:1 | Humoral and T-cell-mediated | [16–20] |
| MG | Female | 3:1 | Humoral/autoantibody-mediated | [16–20] |
| MS | Female | 2–3:1 | T-cell mediated (with humoral contribution) | [16–20] |
| HT | Female | 8–15:1 | Humoral/autoantibody-mediated | [16, 24] |
| T1DM | Male | Not specified | T-cell mediated | [20] |
| AS | Male | 1:2–3: most commonly, but ranges from 1:2 to 1:10 or higher in some populations | T-cell mediated | [16, 25, 26] |
| UC | Male | 3.6:1 | T-cell mediated | [16, 23] |
SLE: systemic lupus erythematosus; SS: Sjögren’s syndrome; SSc: systemic sclerosis; RA: rheumatoid arthritis; MG: myasthenia gravis; MS: multiple sclerosis; HT: Hashimoto’s thyroiditis; T1DM: type 1 diabetes mellitus; AS: ankylosing spondylitis; UC: ulcerative colitis.
A comprehensive epidemiological study examining 19 of the most prevalent ADs in the United Kingdom, such as SLE, systemic sclerosis (SSc), and Sjögren’s syndrome (SS), has confirmed an increasing female predominance in these diseases [19]. In general, women are more frequently affected by ADs like SLE (female-to-male ratio 8:1), rheumatoid arthritis (RA) (2–3:1), MG (3:1), multiple sclerosis (MS) (2–3:1), and SS (19:1) [16, 20]. Conversely, ADs such as T1DM and ankylosing spondylitis (AS) are more prevalent in men (Table 1) [20].
The observed sex bias in ADs distribution is closely linked to the type of immune response that characterises each disease. Indeed, a clear sexual dimorphism in the human immune system exists, with women generally exhibiting a stronger immune response than men. This heightened immune activity in women is observed in both innate and adaptive immunity, including more robust antibody production [21, 22].
Women are more susceptible to diseases mediated by humoral immunity and characterised by high levels of autoantibody production, such as Hashimoto’s thyroiditis (HT), SS, and SLE [16]. On the other hand, male-dominant ADs like AS and ulcerative colitis (UC) are typically associated with T-cell-mediated responses, and this last one has a strong male association with increasing age [16, 23]. These distinctions highlight the complex interplay between genetic predisposition and immunological pathways in determining disease risk, according to sex differences.
Sex-related differences in ADs go beyond prevalence alone, extending into severity, symptomatology, and mortality rates. In certain ADs, such as psoriasis and SLE, men may experience more severe disease phenotypes despite lower overall incidence rates [15]. Males are often diagnosed at a younger age and may face a more aggressive disease course. In contrast, women can exhibit either a sudden acute onset or progress through a chronic form of the disease. Specifically, in the case of SLE, sex-specific symptom patterns have been well documented: women are more likely to experience alopecia, musculoskeletal complications, oral ulcers, and psychiatric disorders, whereas men are more prone to renal involvement, peripheral neuropathy, and cardiovascular manifestations [15].
While ADs in general are associated with reduced life expectancy, sex-based disparities in survival outcomes have also been observed. For instance, women diagnosed with MS typically have a better prognosis and higher survival rates compared to men, possibly due to differences in therapeutic response and overall disease progression [27]. These outcome variations suggest that both biological sex and gender identity can shape not only susceptibility but also the clinical course and outcome of ADs.
Geographic distribution also plays a significant role in shaping the epidemiology and clinical presentation of autoimmune conditions. A growing body of literature supports the existence of geographic variability in both the incidence and severity of ADs [28]. For instance, in the United States, the incidence of psoriasis is significantly higher in women (3:1 ratio), whereas this gender difference is less evident in Southern European countries such as Italy and Spain. Conversely, celiac disease appears to be more prevalent in men in certain countries, such as Italy, the United Kingdom, and India, while Northern European countries like Denmark and Sweden report higher female prevalence rates [15]. Another striking example is SLE, which shows increased incidence among women of African descent compared to Caucasian women [29]. These differences may stem from a complex web of environmental exposures, lifestyle practices, infectious disease prevalence, occupational hazards, climate variations, and disparities in healthcare infrastructure and access.
Geographic location has also been shown to influence prognosis. Populations in Europe, for example, generally experience more favourable disease outcomes than those in North America, which may be attributable to differences in environmental exposures, healthcare systems, or socio-behavioural factors [19, 30]. Despite these regional variations, it is evident that ADs represent an escalating public health concern across both developed and developing nations. The increasing incidence, variability in clinical presentation, and enhanced diagnostic capabilities have collectively contributed to a growing awareness of ADs as leading causes of morbidity and mortality [18].
Following hematopoietic stem cell differentiation, progenitor T cells exit the bone marrow and migrate to the thymus, where they progress from double-negative to double-positive T cells. Within the thymic environment, they undergo processes such as death by negative selection and positive selection facilitated by thymic epithelial cells, which consist of the central tolerance mechanism. This process is led by mTECs and by the expression of AIRE. mTECs exhibit the unusual capacity to express a wide array of peripheral tissue-restricted self-antigens, including established autoimmune targets. The presentation of these peripheral self-antigens has been proposed to be essential for the elimination of self-reactive T cells. During this process, known as negative selection, thymocytes that recognise self-antigens are deleted, thereby preventing their release into the periphery and reducing the risk of AD. Consequently, it was hypothesised that AIRE plays a critical role in promoting the expression of peripheral self-antigens in mTECs. Mutations in the AIRE region impact the ability to present self-antigens and contribute to the migration of auto-reactive cells in the tissues, causing autoimmune manifestations [2]. Furthermore, several studies have demonstrated that the expression of AIRE and AIRE-regulated tissue-restricted antigens is greater in males than in females, both in murine models and in humans. Exposure to androgens enhances AIRE expression, and the sex-related disparity in AIRE levels disappears following castration in males. Upon activation, the androgen receptor interacts with the AIRE promoter, directly promoting its transcription in human thymic epithelial cells. This regulatory mechanism has been associated with the general reduced vulnerability of males to ADs. Consistent with this, animal models show that sex-based differences in autoimmune susceptibility are entirely dependent on AIRE, as males and females lacking AIRE exhibit comparable susceptibility to experimental autoimmune encephalomyelitis and experimental autoimmune thyroiditis [31]. Thus, self-reactive lymphocytes can arise through multiple, non-mutually exclusive mechanisms, reflecting the inherent autoreactivity present in normal T-cell receptor (TCR) and B-cell receptor (BCR) repertoires. Under physiological conditions, such autoreactivity is tightly regulated by central and peripheral tolerance mechanisms, and immune activation critically depends on the context in which the antigen is encountered, including the availability of co-stimulatory and inflammatory signals. The mechanisms through which self-tolerance can be breached can be described by: i) molecular mimicry, whereby the process involves the immune system reacting to an external antigen and, due to the structural resemblance, initiating an autoimmune response against self-antigens. Both B and T cell receptors can recognise identical or similar sequences in microbial and self-antigens, providing help for autoreactive B cells and activating autoreactive T cells; ii) dual TCR expression, arises due to incomplete allelic exclusion during T cell development, allowing some T cells to express two distinct TCRs on their surface, most commonly at the TCRα locus, providing a mechanism for tolerance breakdown. If a T cell expresses one TCR specific for a self-antigen and another for a foreign antigen, activation via the foreign antigen can lead to autoimmunity through the autoreactive TCR; iii) chimeric TCRs, generated by diverse Vα and Vβ chain pairings, enabling recognition of both self and foreign antigens; and iv) altered antigen availability, whereby tissue damage caused by pathogens (e.g., viruses or bacteria) leads to the release of normally sequestered intracellular autoantigens. In inflammatory settings, microbial components and danger-associated signals can promote antigen presentation together with enhanced co-stimulation and cytokine signalling, thereby lowering activation thresholds and facilitating the activation of autoreactive lymphocytes that would otherwise remain tolerant, ultimately contributing to autoimmune disease development [32–34].
Given the critical role B cells play in ADs, gaining deeper insight into how autoreactive B cells emerge is of considerable importance. Nonetheless, our current understanding of their developmental pathways remains limited. During early development in the bone marrow, immature B cells are formed that carry uniquely generated BCRs, assembled through a random recombination process. These B cells subsequently mature in peripheral lymphoid organs, where they become activated upon encountering their specific antigen. Once activated, B cells can differentiate into memory cells or antibody-producing plasma cells, both crucial for long-term immunity. However, due to the inherently random nature of BCR generation, a significant fraction of immature B cells produced in the bone marrow are autoreactive. To prevent harmful immune responses against self-antigens, B cells are subject to multiple self-tolerance checkpoints spanning from early developmental stages in the bone marrow to their final differentiation into plasma cells. These checkpoints include central tolerance mechanisms within the bone marrow and peripheral tolerance processes, such as those for the T cells, that work in concert to eliminate or inactivate self-reactive B cells before they become pathogenic. Thus, in individuals with autoimmune disorders, these tolerance mechanisms have been compromised [35] because of failures in processes like clonal deletion or receptor editing, which may allow these self-reactive B cells to escape into the peripheral immune system [34]. Oestrogens promote the survival and activation of autoreactive B cells, and X chromosome dosage and gene expression further modulate immune responses, resulting in a higher prevalence of ADs in females. These mechanisms are supported by observed differences in B cell frequency, antibody responses, and disease patterns between sexes. However, some ADs, such as immunoglobulin (Ig) G4-related disease, deviate from this pattern, demonstrating a male predominance and highlighting the complexity of sex effects on immune tolerance [36].
Aside from the central tolerance mechanisms, peripheral tolerance encompasses multiple mechanisms. A primary method is anergy, which refers to the functional inactivation of immune cells in the absence of essential costimulatory inputs (B7 molecules). Two crucial molecules involved in sustaining this non-responsive state are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death-1 (PD-1). When a T cell recognises a self-derived antigen, CTLA-4 competes with CD28 to bind B7 molecules on antigen-presenting cells (APCs), leading to their internalisation via clathrin-mediated endocytosis, thereby halting the costimulatory signal. PD-1, expressed on several T-cell subsets, binds to PD-ligand (PD-L)1 or PD-L2 on APCs. This engagement activates tyrosine-based motifs in the PD-1 cytoplasmic domain, which ultimately suppresses TCR signalling.
These pathways represent fundamental immune checkpoint mechanisms that regulate T-cell activation and maintain peripheral tolerance under physiological conditions. While their dysfunction can contribute to the development of ADs, these mechanisms are not specific to autoimmunity but are broadly involved in immune homeostasis and are also exploited in cancer immune evasion and immunotherapy. CTLA-4 expression is characteristic of a cell population fundamental to peripheral tolerance, namely CD4+CD25+ Treg cells, which are central to the maintenance of immune homeostasis by restraining excessive or misdirected immune responses. The development, lineage stability, and suppressive function of Treg cells are controlled by the X-linked transcription factor FOXP3, which serves as the master regulator of the Treg lineage. As previously stated, genetic defects in FOXP3 result in profound immune dysregulation and give rise to immune dysregulation, polyendocrinopathy, enteropathy, and IPEX syndrome in humans. Treg cells employ multiple, non-mutually exclusive mechanisms to mediate immune suppression. These include contact-dependent inhibition via CTLA-4, metabolic suppression through CD39-CD73-mediated adenosine production, cytotoxic activity involving granzymes and perforin, and the secretion of anti-inflammatory cytokines that promote immune tolerance. However, under inflammatory or pathogenic conditions, such as SLE and MS, Treg cells exhibit a degree of functional plasticity and may acquire effector-like characteristics. Accumulating evidence indicates that FOXP3 expression and stability are dynamically regulated by TCR signalling, inflammatory cytokines, and danger-associated molecular patterns, which collectively influence Treg-cell identity and suppressive capacity [37, 38]. Disruption in any of these peripheral tolerance checkpoints and mechanisms may contribute to autoimmune pathogenesis. Another process is clonal ignorance, wherein self-reactive T cells fail to initiate an immune response against self-antigens. This may occur because of anatomical barriers, like the blood-brain barrier, that restrict access to self-antigens or because the antigen is present at levels too low to trigger activation. In other instances, apoptotic deletion maintains peripheral tolerance. When autoreactive T cells interact persistently with self-antigens, the Fas–Fas-ligand (Fas-L) pathway is engaged. Both Fas and Fas-L are expressed on T cells, and their binding triggers apoptosis through activation of caspase enzymes. Mutations in the Fas gene may result in autoimmune manifestations and lymphoproliferative syndromes [34].
At the basis of the mechanisms suggested as breakdown of tolerance mechanisms, there are both inherited genetic variations and environmental influences, facilitating the creation, activation, and survival of autoreactive T and B cells. Variants in genes related to AIRE, HLA (see below), B and T cell signalling pathways, costimulatory and inhibitory mechanisms, and cytokine or cytokine receptor regulation have all been associated with increased susceptibility to autoimmunity [39]. These shared pathways suggest that certain therapeutic targets may be common across different ADs.
A more detailed understanding of these mechanisms could pave the way for identifying precise molecular targets, ultimately contributing to the development of novel and more effective treatment strategies for autoimmune disorders [35].
The following paragraphs outline the principal factors involved in the development of ADs, with particular emphasis on the role of gender and the exposome in shaping autoimmune pathological manifestations.
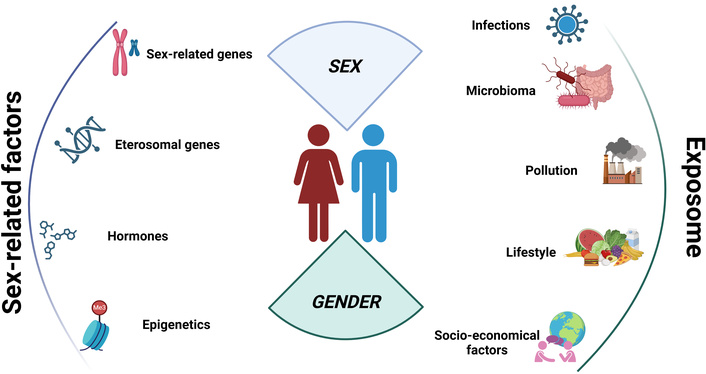
A summary of the key factors influencing the sex bias in ADs is reported in Figure 1.
In mammals, biological sex is determined by chromosomal makeup: females carry two X chromosomes (XX), whereas males carry one X and one Y chromosome (XY). The X chromosome harbours hundreds of protein-coding genes, in contrast to the significantly smaller Y chromosome, which contains a limited number of functional genes. While survival does not depend on the Y chromosome, the presence of at least one X chromosome is essential. Nonetheless, the presence of XX in females poses a potential risk of harmful overexpression of X-linked genes, unless tightly regulated [22, 29]. Although most genes involved in immune function are located on autosomes, the X chromosome plays a prominent role due to its enrichment in immune-related genes. As previously outlined, it encodes approximately 1,100 genes, far exceeding the fewer than 100 found on the Y chromosome, including some involved in male-specific inflammatory pathways. To equalize gene dosage between the sexes, female cells undergo X-chromosome inactivation (XCI), a process that transcriptionally silences one X chromosome early in embryogenesis via epigenetic modifications [29]. These modifications condense the inactivated X chromosome (Xi) into heterochromatin, suppressing the expression of most genes. However, 15%–30% of genes on Xi escape inactivation to varying degrees, depending on cell type, age, and individual genetic background. While some genes consistently evade silencing, others do so variably. This incomplete inactivation may contribute to heightened immune responsiveness and an increased risk of autoimmunity in females. Indeed, accumulating evidence indicates that in specific immune cell populations, particularly activated B and T lymphocytes, regulatory elements controlling XCI can be partially relaxed or lost, enabling selective escape from X-chromosome silencing. This phenomenon results in increased expression of X-linked immune genes, thereby enhancing immune cell activation and potentiating adaptive immune responses. Notably, a substantial number of immune-related genes are located on the X chromosome and play pivotal roles in pathogen recognition, antigen presentation, lymphocyte activation, and immune regulation, contributing significantly to sex-based differences in immune function and disease susceptibility.
An expanding repertoire of XCI-escape genes has been implicated in key immunological pathways, influencing inflammatory responses, antibody production, and the establishment of immune memory. Among the most relevant X-linked immune genes known to escape XCI are toll-like receptor (TLR)7 and TLR8, which are highly expressed in human plasmacytoid dendritic cells and critically shape CD8+ T-cell responses, T helper (Th)1 and Th17 polarisation, and B-cell activation. FOXP3, another X-linked gene, serves as the master regulator of Treg cell development and function, thereby exerting a central role in the maintenance of immune tolerance. Given that the FOXP3 gene is located on the X chromosome, X-chromosome dosage and inactivation patterns may further modulate Treg-cell function in females. Random XCI can generate mosaic populations of Treg cells expressing either mutant or wild-type FOXP3, potentially buffering against severe immunopathology such as IPEX, yet still influencing immune tolerance and disease susceptibility [37, 40].
XCI is orchestrated by the long noncoding RNA X-inactive specific transcript (Xist), which coats one X chromosome per female cell, triggering its inactivation. Xist recruits ribonucleoprotein (RNP) complexes comprising RNA-binding proteins and chromatin-modifying factors, which may also have unintended immunological effects [41].
Recent findings have identified Xist RNPs as potential contributors to female-biased autoimmunity. Chang et al. [42] proposed that these complexes act as antigenic scaffolds, triggering autoimmune responses more frequently in females. Unlike previous studies that focused on XCI escape, this research investigated the immunogenicity of the Xist complex itself. The team engineered male mice from lupus-prone and lupus-resistant strains to express an inducible Xist gene. Upon induction, Xist formed RNP structures akin to those in female cells. Notably, in lupus-susceptible strains, the expression of Xist led to autoimmune manifestations similar to those seen in females, whereas in resistant strains, disease did not develop, highlighting the influence of genetic background and environmental stimuli.
Clinicians have long observed that many autoantibodies target large nucleic acid-protein assemblies, such as chromatin and spliceosomes, in human ADs. These complexes, due to their polymeric nature, can activate immune receptors when released extracellularly during cell death. Chang’s work posits that Xist RNPs represent one such immunodominant complex exclusive to females. Every female cell expresses the 19 kb Xist RNA, which envelops the inactive X chromosome (the Barr body), forming a large antigenic structure. Upon cell death, these complexes are exposed to the immune system. In genetically resistant hosts, limited exposure to Xist leads only to subtle immunological shifts, such as altered T cell subsets and chromatin accessibility, without overt tissue damage. However, in genetically predisposed individuals, and under conditions of sustained tissue injury, exposure to Xist RNPs precipitates full-fledged organ damage and widespread immune activation [42].
Supporting this model, longitudinal analyses of serum from autoimmune patients have detected autoantibodies against proteins within Xist-associated RNPs. Some of these antibodies are specific to particular diseases and may serve as early diagnostic biomarkers; others appear across multiple conditions, suggesting a broader relevance to immune dysregulation. These insights expose a persistent bias in biomedical research: the predominant use of male-derived cell lines, which lack Xist expression, has obscured critical elements of female-specific immunology. Closing this gap is crucial for understanding sex-differentiated immune responses and for advancing diagnostic and therapeutic approaches tailored to autoimmune conditions with female predominance.
However, focusing exclusively on the contribution of X-linked genes and associated epigenetic modifications is insufficient to fully explain sex-linked dimorphism in ADs. This limitation is underscored by evidence showing that the expression and functional impact of many X-linked immune genes are themselves modulated by sex hormone levels [43].
Sex hormones significantly contribute to the observed sex-related differences in the incidence and prevalence of ADs, which, as discussed in the present review, are notably more frequent among females, who are also more prone to developing multiple concurrent autoimmune conditions, a phenomenon known as poly-autoimmunity [17]. Hormones, by interacting with transcription factors, regulate the expression, either upregulation or downregulation, of immune cell receptors. This modulation affects the strength and quality of immune responses, thereby influencing the onset and severity of autoimmune conditions [17]. Given these effects, it is unsurprising that women often experience fluctuations in AD symptoms during specific life stages marked by hormonal shifts. Puberty, for example, brings a surge in sex hormones that can influence immune function and may initiate autoimmune processes. Epidemiological data confirm a rise in the incidence of diseases like SLE and juvenile idiopathic arthritis during adolescence, highlighting the potential role of puberty in triggering immune dysregulation [44]. Beyond puberty, pregnancy, and menopause are also key periods characterised by profound hormonal changes that can modulate autoimmunity [17]. Pregnancy presents a unique endocrine state, with elevated levels of oestrogen and progesterone typically exerting an immunosuppressive effect, leading to symptom remission in many autoimmune conditions. However, this temporary improvement is often followed by postpartum disease flare-ups, coinciding with a sudden drop in hormone levels. As such, the postpartum period represents a critical window for disease relapse, necessitating close clinical surveillance [44]. Menopause, marked by a decline in oestrogen and progesterone, also influences autoimmune dynamics. In some cases, it may exacerbate existing conditions or even precipitate the onset of new ADs. While hormone replacement therapy (HRT) is commonly employed to relieve menopausal symptoms, its role in modulating autoimmune conditions remains complex and requires further investigation to clarify its therapeutic risks and benefits [44].
From an immunological perspective, progesterone and androgens are generally considered immunosuppressive and thus protective for ADs, whereas oestrogens are often described as immune-enhancing, potentially exacerbating autoimmune activity (Table 2) [45].
Sexual hormones regulate immune response and influence the sex-dimorphisms in ADs.
| Hormone | General immunological effect | Mechanism of action | Impact on ADs | Sex-specific notes | References |
|---|---|---|---|---|---|
| Oestrogens | Immune-enhancing; promotes humoral responses | Increases B cell proliferation, class switching to IgG, Th2, and Treg responses; modulates Treg transcriptional programs | Can exacerbate antibody-mediated ADs such as SLE; may protect against Th1/Th17-driven ADs (e.g., MS, RA) | Effects depend on XX chromosomal background; levels fluctuate during puberty, pregnancy, menopause; high in pregnancy, induce Th2/Treg shift; low post-menopause, induce Th1/Th17 activation | [17, 44–46, 53] |
| Progesterone | Immunosuppressive; anti-inflammatory | Induces anti-inflammatory molecules; inhibits Th1 and Th17 pathways; modulates APC activation | Generally protective; reduces autoimmune activity | High during pregnancy, inducing symptom remission in some ADs | [44, 45] |
| Testosterone | Immunosuppressive; anti-inflammatory | Reduces pro-inflammatory cytokines (IL-1β, IL-6, TNF); increases IL-10; inhibits T cell proliferation; suppresses B cells and natural killer cytotoxicity | Protective against multiple ADs; a decline in ageing men (andropause) increases susceptibility | Effects observed in hypogonadal young men; therapeutic testosterone can restore male-like immune responses | [52–56] |
| Prolactin | Immune-enhancing | Promotes B cell activation and antibody production; upregulates costimulatory molecules on APCs; modulates Th1/Th2 cytokines | Can exacerbate autoimmune activity; hyperprolactinemia linked to onset/persistence of ADs | Levels fluctuate with reproductive states (pregnancy, postpartum) | [44, 46] |
ADs: autoimmune diseases; APCs: antigen-presenting cells; Ig: immunoglobulin; Th: T helper cell; Treg: regulatory T cell; SLE: systemic lupus erythematosus; MS: multiple sclerosis; RA: rheumatoid arthritis; APC: antigen presenting cell; IL: interleukin; TNF; tumour necrosis factor.
Indeed, progesterone plays a role in immunomodulation by inducing anti-inflammatory molecules and inhibiting lymphocyte Th1 and Th17 pathways [46]. In contrast, the effects of oestrogens vary considerably across different stages of hormonal transitions. Their influence on autoreactivity and lymphocyte proliferation, and thus on the shaping of adaptive immune responses, depends largely on their circulating concentrations [17]. They are potent immunomodulators exerting context-, dose-, and receptor-dependent effects on the immune system [47]. Indeed, differential signalling through oestrogen receptors (ER) adds complexity to immune regulation: ERα generally promotes pro-inflammatory responses, whereas ERβ exerts anti-inflammatory effects. Variations in the ERα/ERβ expression ratio across immune cells, tissues, and age influence the magnitude and character of the immune response [48, 49].
Oestrogen levels are typically low during puberty and post-menopause, conditions that favour the activation of pathogenic Th1 and Th17 pathways. Indeed, during reproductive age and specific phases of the menstrual cycle, oestrogens enhance both innate and adaptive immune responses. Under these conditions, they promote the increased production of pro-inflammatory cytokines, including interleukin (IL)-1β, IL-6, tumour necrosis factor (TNF)-α, and type I interferons. These effects are mediated through the upregulation of TLR, particularly TLR7 and TLR9, enhanced activation of plasmacytoid dendritic cells, and the promotion of B-cell survival, differentiation, and Th17 polarisation [47]. Conversely, during pregnancy, oestrogen levels rise significantly, fostering a shift toward Th2 and Treg responses, enhancing FOXP3 expression, and favouring the production of anti-inflammatory cytokines such as IL-10 and tissue growth factor-β, thereby contributing to immune tolerance [50, 51]. This dualistic behaviour is particularly evident in how different ADs respond to pregnancy. For instance, SLE often worsens during pregnancy due to the Th2-skewed immune environment enhancing autoantibody production. In contrast, diseases like MS, RA, thyroiditis, and uveitis tend to improve during this period, likely due to the maternal shift from Th1 to Th2 immunity [45].
Prolactin plays a role in regulating both innate and adaptive immunity. It enhances Ig production, upregulates costimulatory molecule expression on APCs, promotes their activation, and modulates the secretion of Th1- and Th2-associated cytokines. This hormone contributes to B-cell stimulation and supports antibody generation. In the context of AD pathogenesis, elevated prolactin levels interfere with central tolerance mechanisms by impairing clonal deletion of B cells and lowering the activation threshold of anergic B cells, thereby facilitating abnormal immune reactivity. In fact, hyperprolactinemia has been implicated in both the onset and persistence of various autoimmune disorders [46].
Just as oestrogen fluctuations play a role in defining the manifestation of ADs in women, andropause also defines a different behaviour of the disease in men. Andropause, the gradual decline in testosterone levels in ageing men, contributes to immune ageing and increased susceptibility to AD. Even young males with hypogonadism show a higher prevalence of conditions like RA, SLE, and MS. Testosterone exerts predominantly anti-inflammatory and immunomodulatory effects, favouring immune tolerance and limiting excessive innate and adaptive immune responses, although its actions are highly context-dependent: castration in animal models exacerbates AD symptoms, while testosterone therapy alleviates them. Testosterone modulates immunity by reducing proinflammatory cytokines (IL-1β, IL-6, TNF) via monocytes and macrophages, boosting IL-10, and inhibiting T cell proliferation [52]. It also suppresses B-cell activity and natural killer cell cytotoxicity. Epidemiological data support these findings, showing increased AD incidence in older men, reflecting the waning immunoprotective effects of testosterone [53, 54]. These data have been confirmed by investigations into the effects of gender-affirming hormone therapy on immune cell molecular signatures in transgender men, which provide key insights. Remarkably, within just three months of starting testosterone treatment, the signalling characteristics of their immune cells began to align closely with those observed in cisgender men (male at birth). This rapid shift underscores the influence of sex hormones in shaping distinct immune responses between females and males [55].
Therefore, as affirmed before, the female bias in ADs cannot be attributed solely to X chromosome-linked factors but must also account for the broad and varied effects of sex hormones on both the immune system and target organs. These hormones influence both innate and adaptive immunity and play essential roles in shaping immune homeostasis. A typical example is given by the sex-hormone shaping of Treg-cell frequency, suppressive function, and transcriptional programs, influenced also by XCI.
Also, post-pubertal males exhibit higher frequencies of Treg cells with enhanced suppressive capacity and enrichment of anti-inflammatory signalling pathways, including PI3K-AKT signalling. Conversely, these sex-associated differences are diminished or altered in autoimmune settings such as juvenile-onset SLE, where Treg-cell suppressive function is impaired and transcriptional dimorphism is exaggerated, suggesting dysregulated hormone signalling in autoimmunity. Importantly, studies involving transgender individuals receiving gender-affirming hormone therapy further support a direct role for sex hormones in modulating Treg-cell biology, as hormone exposure induces transcriptional changes that parallel those observed between cisgender males and females [56]. The interplay between hormonal factors and environmental triggers in genetically predisposed individuals contributes to immune dysregulation and the onset of ADs.
Both sex hormones and X-linked genetic factors converge to shape immune dimorphism. A critical unresolved question remains whether genetically determined sex, through chromosomal complement and X-linked gene dosage, or hormonal influences, exerts the predominant effect in driving sex-biased susceptibility, phenotype, and progression of ADs, or whether their impact is inseparable and context-dependent. In this regard, recent work by Peckham et al. (2025) [57] provides compelling in vivo evidence that sex hormones act within a permissive chromosomal context. The authors showed that, after puberty, cisgender women (female at birth) exhibit higher frequencies of memory B cells, key mediators of immunological memory and antibody responses, compared with age-matched cisgender men. Notably, this advantage is lost after menopause, indicating a crucial role for female sex hormones. In transgender men with an XX chromosomal complement, pharmacological blockade of endogenous oestrogens resulted in a significant reduction in memory B-cell frequencies. Conversely, in transgender women with an XY background, oestrogen supplementation did not increase memory B-cell numbers (Table 3). Consistently, postmenopausal women receiving HRT displayed higher levels of memory B cells than untreated counterparts. By leveraging comparisons across prepubertal, post-pubertal, postmenopausal, cisgender, and transgender individuals receiving gender-affirming hormone therapy, this study disentangles the relative contributions of hormonal exposure and chromosomal complement in vivo. Such hormone-chromosome synergy provides a mechanistic framework for the female predominance of antibody-mediated ADs, including SLE, and highlights sex as a fundamental biological variable influencing immune tolerance, disease susceptibility, and therapeutic responsiveness.
Interaction between sexual chromosomes and oestradiol in the regulation of memory B cells [57].
| Sexual chromosomes | Oestrogen levels | Population | Frequency of memory B cells class-switched | Immunological effect |
|---|---|---|---|---|
| XX | High | Post-puberty and pre-menopausal cisgender women | Higher | Enhancement of humoral immune memory |
| XX | High | Postmenopausal cisgender women on HRT | Higher | Restoring memory compartment B |
| XY | High | Transgender women (XY) on oestrogen therapy | Not significantly increased | Oestrogen is insufficient in the absence of a double X chromosome. |
| XX | Low | Pre-puberty and post-menopausal cisgender women | Lower | Reduction in immune memory |
| XX | Low | Transgender men (XX) with pubertal blockers ± testosterone | Lower | Oestrogen dependence in the XX context |
| XY | Low | Pre- or post-puberty cisgender men | Lower | The immune profile is less oriented towards an antibody response |
| XY | Low | Transgender women (XY) with puberty blockers | Lower | Absence of effective oestrogenic stimulation |
HRT: hormone replacement therapy.
Given the divergent effects of oestrogens across various autoimmune conditions, protective in MS and RA, yet pathogenic in SLE, and the combined role of sexual chromosome and hormone fluctuation, deeper mechanistic insights are urgently needed to unravel the basis of these hormonal paradoxes influencing ADs manifestation [45].
The contribution of heritable factors in ADs is underscored by concordance rates in monozygotic twins and long-established links to HLA genes [58, 59], previously discussed. Most of ADs are polygenic, and there is frequently the involvement of overlapping genetic risk factors, suggesting convergent immunopathogenic pathways. Although pinpointing the exact genes and their downstream molecular impacts remains complex, the integration of functional assays with multi-layered genomic datasets is revealing crucial immune cells and signalling pathways involved in disease progression, with implications for future therapeutic strategies.
A wide range of ADs are classified as polygenic autoimmune disorders, including RA, SLE, SSc, T1DM, autoimmune thyroid diseases, and SS (Table 4) [60]. Polygenic ADs are associated with numerous genetic risk loci, have a later onset, and result from the interplay of multiple genetic and environmental factors [61]. For example, more than 100 risk loci have been identified for RA, accounting for approximately half of its genetic variance; GWAS have highlighted the extreme complexity of the contribution of individual genes [62]. Similarly, SLE is characterised by diverse genetic factors, many of which are located in non-coding regions of the genome [63]. Many polygenic ADs show strong associations with specific HLA alleles. For instance, RA is linked to HLA-DRB1 alleles with the “shared epitope”, accounting for a significant portion of genetic susceptibility [62]. SS is associated with the HLA-DRB10301-DQB10201 haplotype, which is linked to more severe disease and specific autoantibody profiles [64]. Many genetic loci implicated in one AD are also involved in others, highlighting shared pathogenic mechanisms and the phenomenon of poly-autoimmunity, so the presence of more than one AD in a single patient [61]. Monogenic ADs, instead, are caused by mutations in a single gene and typically present at an earlier age with more severe, multi-organ involvement [2]. A brief description of the characteristics of polygenic and monogenic ADs is presented in Table 4.
Confront among monogenic and polygenic autoimmune diseases.
| Feature | Polygenic autoimmune diseases | Monogenic autoimmune diseases | References |
|---|---|---|---|
| Genetic basis | Multiple genes (complex inheritance) | Single gene (Mendelian inheritance) | [2, 61, 65] |
| Age of onset | Usually later | Usually, early | [2, 64] |
| Prevalence | Common | Rare | [2, 61, 65] |
| Disease severity | Variable | Often severe, multi-organ | [2, 61, 65] |
| Examples | RA, SLE, T1DM, APS, MS | APS, IPEX, ALPS | [60] |
| Shared pathogenic mechanisms | Yes | Sometimes | [61] |
| Polyautoimmunity | Common | Less common | [61] |
RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; T1DM: type 1 diabetes; APS: autoimmune polyendocrinopathy syndrome; MS: multiple sclerosis; IPEX: immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome; ALPS: autoimmune lymphoproliferative syndrome.
Technological advances following the sequencing of the human genome, like GWAS, have enabled the mapping of haplotype structures and facilitated whole-genome screening in affected populations. These efforts have helped clarify a significant portion of AD heritability. Nonetheless, most risk haplotypes exert only modest effects; HLA variants usually exhibit odds ratios between 2 and 3, although exceptions exist (e.g., HLA-B27 in AS). These odds ratios tell us that a person with these variants has double or triple the risk of disease compared to someone without it. Outside the HLA, most loci show even smaller associations.
Among the sex differences in the genetically-related predisposition to ADs, a growing body of research highlights that even autosomal genes can contribute to sex-specific risk through differential expression, regulation, and interaction with biological context. One clear example comes from GWAS in RA, where an autosomal variant on chromosome 7 (rs11761231) was shown to significantly increase disease risk in women, but not in men [66]. This finding underscores that genetic variants on autosomes can exert sex-specific effects, likely due to interactions with hormonal or epigenetic factors that differ between the sexes. Further insights come from studies on SLE, a disease known for its extreme female bias. In lupus-prone mice, researchers observed higher expression of the interferon regulator factor 5 (IRF5) gene, specifically in females [67]. Interestingly, when oestrogen signalling was disrupted, IRF5 levels dropped, suggesting that even though IRF5 is not a sex-linked gene, its expression can be modulated in a sex-specific manner, contributing to disease susceptibility [68]. The observation that women exhibit a higher proportion of IRF5-positive B cells than men, along with a corresponding increase in inflammatory cytokine production, underscores a sex bias in IRF5-driven immune responses [69]. Recent studies have further shown that blocking IRF5 hyperactivation in mouse models can prevent the onset of lupus and reduce disease severity [70]. Indeed, it is specifically noted that IRF5 expression is upregulated by the female sex hormone oestrogen in immune cells [71]. Furthermore, experimental data show that deficiency of ER-α, IRF5, or STAT1 in splenic cells decreases expression of BAFF, a factor linked to SLE, further supporting the role of oestrogen and IRF5 in disease susceptibility [71]. Moreover, authors demonstrated that IRF5 inhibition after disease onset not only slowed disease progression but also helped maintain remission in mouse models. Collectively, these findings highlight IRF5 as a promising therapeutic target for the treatment of autoimmune and inflammatory disorders [69]. Transcriptomic studies of immune cells also reveal sex-specific differences in other autosomal gene expression. For example, macrophages in female mice expressed higher levels of complement-related genes such as Fcgr2b and Fcgr3a (which encode Fc gamma receptors, FcγRs), which modulate immune activation. These differences in gene expression may contribute to explaining why females often mount stronger immune responses, increasing the risk of autoimmune reactions [72].
Non-inherited HLA antigens also play a role. In women, the persistence of foetal cells carrying different HLA alleles, known as foetal microchimerism, has been implicated with increased autoimmune risk, although its exact role remains under investigation. In particular, HLA-DRB1*04 alleles associated with RA were more frequently detected in the peripheral blood of women with RA who had previously been pregnant, despite the fact that these women did not carry the HLA-DRB1*04 alleles themselves [66, 73]. This finding is consistent with the presence of persistent foetal microchimerism. Notably, this phenomenon was not observed in healthy controls, and no differences were detected between RA patients and controls with respect to alleles not associated with RA [74].
Even non-coding elements of the genome, such as microRNAs and long noncoding RNAs, show differential expression between males and females. Although many of these elements are X-linked, several are autosomal and may still contribute to sex-biased immune regulation. Their functions, ranging from fine-tuning gene expression to modulating inflammatory pathways, are increasingly recognised as part of the sex bias puzzle in autoimmunity [75].
GWAS have greatly expanded knowledge of autoimmune disease genetics, revealing extensive variant sharing across conditions and highlighting common pathogenic pathways. However, most identified risk variants lie in noncoding regions, and their functional interpretation remains incomplete, limiting direct clinical translation. While integrative and network-based approaches have begun to link genetic risk to therapeutic targets, current findings still fall short of fully explaining disease heterogeneity, sex bias, and variable treatment responses, underscoring the need for more cell-, context-, and sex-specific functional studies.
Beyond biological sex, gender-related social and environmental determinants substantially contribute to the development and expression of ADs. Gender encompasses socially constructed norms and expectations that shape roles, relationships, and power dynamics throughout the life course [76]. It represents a critical dimension of human health, as it governs behavioural patterns attributed to men and women within a given society and influences daily practices, expectations, and lived experiences. These include, but are not limited to, dietary habits [77], perceived stress, tobacco use [78], and levels of physical activity, all of which can modify disease risk and health outcomes. Importantly, the distribution of gender-associated characteristics within male and female populations may affect health trajectories in ways that differ from biological sex alone [79]. Gender norms also shape access to healthcare services, health-seeking attitudes, and interactions with healthcare systems. Furthermore, gender-driven behaviours, such as smoking patterns, lifestyle choices, stress perception, pain reporting, and nutritional practices, can induce epigenetic changes that influence gene regulation and biological phenotypes. In this way, gender-linked behaviours modulate exposure to risk factors as well as engagement in preventive strategies across a wide range of diseases.
Gender further influences both the nature and frequency of exposures encountered over the lifespan. These exposures collectively constitute the exposome, defined as the cumulative burden of environmental, lifestyle, and endogenous influences, including the microbiota, from conception onward [76]. The exposome operates at the intersection of biological and social determinants to shape health outcomes and immune function. Environmental exposures are highly heterogeneous and encompass both physicochemical agents and social-structural stressors, continuously encountered across multiple settings such as homes, schools, workplaces, and indoor and outdoor environments.
Physicochemical exposures arise from both synthetic and naturally occurring sources, including personal care products, food and food-contact materials, household chemicals, and environmental pollutants. Patterns of exposure may differ markedly according to biological sex and gender identity, and their biological impact may be sex-specific through interactions with hormonal and other physiological pathways. Exposure profiles are, indeed, shaped by sex-related biological needs as well as by cultural expectations, social norms, and pressures to conform to traditional gender roles. As an example, biological sex can influence the use of personal care products, such as menstrual and intimate care products and hormonal contraceptives [76, 80].
Overall, the interplay between biological sex and gender-driven exposures emerges as a key determinant of immune dysregulation in ADs, with important implications for risk stratification, prevention strategies, and the development of sex- and gender-informed therapeutic approaches [81]. Based on this, the rising incidence of ADs in industrialised and highly globalised regions strongly supports a central role for the exposome in disease pathogenesis [82, 83]. The cumulative exposures to microbial encounters, diet, lifestyle factors, pollutants, medications, and environmental toxins [76] contribute to long-term “immune training” shaping immune development and responsiveness and potentially triggering autoimmune manifestations.
The relevance of environmental factors is underscored by epidemiological observations, which show that populations with similar genetic backgrounds exhibit markedly different AD prevalence rates depending on their environmental context. Indeed, twin studies about MS incidence demonstrate that genetically identical individuals can develop the disease with divergent severity, highlighting the influence of non-genetic exposures, and potentially gender-modulated exposures, such as lifestyle, stress, and healthcare access [84, 85]. Comparable patterns are observed in other ADs, such as pemphigus foliaceus, whose incidence varies geographically despite shared ancestry, further suggesting that context-dependent and gender-linked environmental factors contribute to disease risk [86]. Importantly, many of these environmental exposures are unequally distributed between genders as a result of socially constructed roles, behaviours, and occupational patterns. In this context, environmental chemical exposures constitute a critical dimension of the exposome. Endocrine-disrupting chemicals (EDCs), commonly found in cosmetics, plastics, pesticides, and personal care products, can interfere with immune regulation by altering gene expression, epigenetic programming, and hormone signalling pathways. By interacting with oestrogenic and androgen receptors, EDCs can influence immune cell differentiation, proliferation, and activation, thereby promoting immune dysregulation and autoimmunity. These effects are particularly relevant to female-predominant diseases such as SLE. In SLE, genes involved in developmental programming, cell-cycle regulation, and epigenetic control appear to act as molecular interfaces between genetic susceptibility and EDC exposure, including bisphenol A, bisphenol S, and endosulfan [87]. Furthermore, epidemiological studies have linked cosmetic product use with increased risk of SLE, primary biliary cholangitis, and RA, although findings remain heterogeneous and limited to a few reports [66, 67, 88–90]. Importantly, the gender-specific patterns observed in these studies largely reflect differences in the exposome, which is shaped by sex- and gender-related factors, including occupational roles (e.g., silica exposure), sex-associated behaviours (e.g., cosmetic use), and unconventional therapeutic or cosmetic practices (e.g., mercury exposure).
Furthermore, sociocultural determinants linked to gender shape behavioural patterns, stress exposure, healthcare access, and symptom reporting, all of which can influence immune function and disease trajectories. Occupational exposures, lifestyle practices, psychosocial stress, and preventive behaviours vary substantially by gender and intersect with biological sex to modulate autoimmune risk. An example is given by reduced microbial diversity in highly industrialised societies, consistent with the hygiene hypothesis, which has been associated with increased AD prevalence due to impaired immune education and reduced Treg cell induction [91]. Certain infections may either protect against or trigger autoimmunity, depending on context, timing, and host factors, as illustrated by associations between specific pathogens and diseases such as rheumatic fever, Guillain-Barré syndrome, T1DM, and MS [86, 91]. In SLE, for instance, certain infections modulate immune responses beneficially, partly through the regulation of Tregs [82]. Protozoa and fungi may offer similar protection [82].
These findings emphasise that genetic susceptibility alone is insufficient to explain disease onset without accounting for environmental and social modifiers.
Among exposome components, diet represents a key determinant of immune homeostasis. Deficiencies in macro- and micronutrients can impair immune regulation and promote autoimmune responses [91]. Diets characterised by a high dietary inflammatory index, such as Western dietary patterns, are associated with increased prevalence of ADs, including RA [92]. These dietary effects are mediated not only through systemic inflammation but also via modulation of the gut microbiota, a central regulator of immune tolerance. Thus, alterations in microbiota composition and function, along with loss of gut barrier integrity and inflammation, have been observed in various ADs such as T1DM, MS, SLE, and RA, all of which show higher prevalence in females. The microbiota represents a dynamic and biologically integrated component of the exposome, influencing immune balance and tolerance across the lifespan. Microbial composition varies with age, sex, hormonal status, and environmental exposures [21]. Pregnancy induces profound shifts in gut microbiota, immune regulation, and metabolism, which are essential for foetal development and immune tolerance but may also increase maternal susceptibility to ADs and influence offspring immune outcomes [93–95]. Sex hormones further modulate host-microbiota interactions; for instance, 17β-oestradiol enhances TNF-α production and alters gut microbial composition, promoting immune dysregulation [96]. Post-pubertal divergence in microbiota composition between opposite-sex twins further highlights the combined influence of hormones and environmental factors [97]. Experimental models illustrate the sex-specific immunomodulatory role of the microbiota: transfer of male microbiota to female non-obese diabetic mice increases Treg cell levels and confers protection against T1DM, an effect associated with elevated testosterone [97]. In addition, microbiota dysbiosis has been consistently reported across multiple ADs, including SLE, T1DM, MS, RA, psoriasis, Crohn’s disease, and Behçet’s disease [91, 96]. These findings underscore the bidirectional relationship between sex hormones, microbial composition, and immune regulation.
Collectively, these observations highlight that AD pathogenesis emerges from the complex interplay between genetic susceptibility, biological sex, gender-related behaviours, and cumulative environmental exposures. Integrating sex- and gender-sensitive approaches into exposome research is therefore essential to better understand disease mechanisms, explain sex-biased incidence, and inform more precise preventive and therapeutic strategies.
Key exposome and gender-related risk factors in ADs are reported in Table 5.
Key exposome and gender-related risk factors in ADs.
| Category | Specific factor | Mechanism/effect | Gender differences | References |
|---|---|---|---|---|
| Environmental | Industrialised lifestyle (urbanisation, pollution) | Alters immune responses; increases risk of ADs | Affects both sexes; potentially greater impact on women | [76, 82, 83] |
| Environmental | Pollutants, chemicals | Disrupt immune homeostasis | May affect hormonal metabolism differently | [76] |
| Nutrition | Western diet (refined carbs, fats, red meat) | Promotes systemic inflammation; alters microbiota | Greater effect in women with economic hardship | [91, 92] |
| Nutrition | Micronutrient deficiency | Impairs immune regulation; breaking of self-tolerance | More common in women, especially in low-income settings | [21, 91] |
| Nutrition | Anti-inflammatory foods (polyphenols, omega-3) | Improve immune tolerance by reducing AD risk | Intake varies by culture and gendered dietary patterns | [92, 98] |
| Microbiome | Dysbiosis (imbalance of gut flora) | Reduces tolerance to self-antigens; increases inflammatory responses | Sex-specific patterns are linked to hormones and life stages | [21, 96, 97] |
| Microbiome | Early-life microbial exposure | Shapes long-term immune responses | Influenced by birth method, breastfeeding, and hygiene habits | [99, 100] |
| Microbiome | Pregnancy-associated microbiome shifts | Alters the immune landscape; may trigger ADs | Unique to women | [96] |
| Infections | Reduced exposure to microbes (hygiene hypothesis) | Impaired immune training; increased risk of hypersensitivity and autoimmunity | More pronounced in high-income countries | [82, 92] |
| Infections | Molecular mimicry (e.g., EBV, Coxsackievirus) | Cross-reactivity between pathogen and self-antigens triggers ADs | Gender-biased susceptibility in some diseases | [96] |
| Infections | Protective infections (protozoa, fungi, commensals) | Induce regulatory pathways; suppress autoimmune responses | Under study, the gender effect is less defined | [82] |
| Socioeconomic | Food and water insecurity | Increases malnutrition, infection risk; impairs microbiome | Women and children are most affected | [21, 100] |
| Socioeconomic | Health care access and education | Affects exposure, treatment, nutrition, and microbial environment | Gender gap often presents in access and outcomes | [21, 41] |
ADs: autoimmune diseases; EBV: Epstein-Barr virus.
Despite significant advances in the understanding of ADs, the mechanisms underlying their marked sex bias remain only partially resolved. While genetic susceptibility, sexual chromosomes, and hormonal influences clearly contribute to immune dysregulation, none of these factors alone is sufficient to explain the striking female predominance and the heterogeneous clinical expression observed across different autoimmune conditions. Much of the current evidence remains associative rather than mechanistic, and the relative contribution of sex versus gender-related factors is often difficult to disentangle.
The available data highlight that immune tolerance is modulated by a multi-layered network involving central and peripheral checkpoints, hormonal signalling, and X chromosome–linked regulation. However, the precise hierarchies among these mechanisms and their disease-specific relevance are still unclear. In particular, findings derived from animal models or selected patient cohorts may not fully capture the complexity of human autoimmunity, limiting their generalizability. Moreover, the same biological factor, such as oestrogen signalling or X-linked gene dosage, can exert opposing effects depending on disease context, immune compartment, and life stage, underscoring the need for caution when extrapolating broad conclusions.
Similarly, although the exposome is increasingly recognised as a key driver of autoimmune risk, its integration into mechanistic models of disease remains fragmented. Environmental, lifestyle, and socioeconomic exposures are often incompletely quantified, and their interactions with sex-specific immune pathways are insufficiently addressed in current research. This gap hampers the development of predictive models capable of explaining why only a subset of genetically predisposed individuals ultimately develop disease.
Taken together, these limitations underscore the necessity for prospective longitudinal cohort studies incorporating serial assessments of biological and clinical parameters, with sex included as a biological variable and gender-related factors modelled as modifiers of environmental exposures. Future research should move beyond descriptive associations toward mechanistic and systems-level approaches, combining genomics, epigenetics, immunophenotyping, and exposomic data. Only through such critical and multidimensional frameworks will it be possible to disentangle the complex determinants of ADs and translate sex- and gender-based insights into truly personalised diagnostic and therapeutic strategies.
ADs: autoimmune diseases
AIRE: autoimmune regulator
APCs: antigen-presenting cells
APS: autoimmune polyendocrinopathy syndrome
AS: ankylosing spondylitis
BCR: B-cell receptor
CTLA-4: cytotoxic t-lymphocyte-associated protein 4
EDCs: endocrine-disrupting chemicals
ER: oestrogen receptor
Fas-L: Fas-ligand
FOXP3: forkhead box P3
GWAS: genome-wide association studies
HLA: human leukocyte antigen
HRT: hormone replacement therapy
HT: Hashimoto’s thyroiditis
Ig: immunoglobulin
IL: interleukin
IPEX: immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome
IRF5: interferon regulator factor 5
MG: myasthenia gravis
MHC: major histocompatibility complex
MS: multiple sclerosis
mTECs: medullary thymic epithelial cells
PD-1: programmed cell death-1
RA: rheumatoid arthritis
RNP: ribonucleoprotein
SLE: systemic lupus erythematosus
SS: Sjögren’s syndrome
SSc: systemic sclerosis
T1DM: type 1 diabetes
TCR: T-cell receptor
Th: T helper
TLR: toll-like receptor
TNF: tumour necrosis factor
Tregs: regulatory T cells
UC: ulcerative colitis
XCI: X-chromosome inactivation
Xi: inactivated X chromosome
Xist: X-inactive specific transcript
AC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GA: Writing—review & editing. AA: Writing—review & editing. RZ: Investigation, Writing—original draft, Writing—review & editing. GC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. All authors read and approved the submitted version.
Giuseppina Candore, who is the Editorial Board Member of Exploration of Immunology, had no involvement in the decision-making or review process of this manuscript. The other authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 5211
Download: 51
Times Cited: 0