 Review
Review

Affiliation:
1Department of Biochemistry and Molecular and Structural Biology, Jožef Stefan Institute, 1000 Ljubljana, Slovenia
2Jožef Stefan International Postgraduate School (IPS), 1000 Ljubljana, Slovenia
Email: eva.zerovnik@ijs.si
ORCID: https://orcid.org/0000-0002-9793-8200
Explor Neurosci. 2026;5:1006136 DOI: https://doi.org/10.37349/en.2026.1006136
Received: February 13, 2026 Accepted: March 30, 2026 Published: May 08, 2026
Academic Editor: Jinwei Zhang, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China
The article belongs to the special issue Advances in Epilepsy Research
For this review paper, data on protein misfolding and aggregation in progressive myoclonus epilepsies and some developmental encephalopathies are gathered. There is evidence that in some cases of monogenic epilepsies, misfolding of the mutated protein takes place, often leading to protein aggregation. On one hand, protein aggregation reduces the amount of protein and its activity; on the other, it exerts generic toxicity to neurons. Understanding the molecular causes due to loss of normal function and gain of toxic function of the mutated aggregate-prone proteins is important to obtain new therapies. By observing the symptomatology of progressive and developmental epileptic syndromes, one can derive some conclusions about the relevance of protein misfolding and aggregation in the picture. A plausible view seems that the most severe symptoms of dementia, behavioral and psychiatric symptoms, are linked to protein aggregation and downstream effects on cellular degradation and energy systems. Finally, I discuss the potential of targeting the proteostasis network to develop novel anti-seizure and neuroprotective therapies.
The aim of this review is to bring to light the cases of protein aggregation in inherited epileptic syndromes. The mutated proteins are prone to aggregate spontaneously or are indirectly involved in regulating proteostasis, such as through chaperone function or ubiquitin proteasome system (UPS)-mediated degradation. An important epiphenomenon of protein aggregation is excitotoxicity. Excitotoxicity and protein aggregation function as converging, self-reinforcing pathogenic mechanisms, rather than strictly parallel processes. They create a vicious cycle: protein aggregates trigger excitotoxicity by interfering with membrane integrity and glutamate homeostasis, while excitotoxicity causes mitochondrial dysfunction and oxidative stress that further accelerates protein misfolding and aggregation [1, 2].
Glutamate is the major excitatory neurotransmitter of the nervous system [3]. With its receptors and transporters, it shapes the fate of the excitatory synapse [3]. Jeffrey Watkins and co-workers (already in 1970) contributed to the field of excitatory glutamate receptors by developing agonists that could distinguish between different glutamate receptor subtypes. Four agonists were found: N-methyl-D-aspartate (NMDA), amino-3-hydroxy-5-methylisoxazolepropionic acid (AMPA), kainate, and quisqualate, which bind to different glutamate receptors named after the agonist [4, 5]. After various central nervous system (CNS) injuries, such as ischemic stroke, head injury leading to perfusion or low-grade hydrocephalus, Alzheimer’s disease (AD), and other neurodegenerative, epileptic and neuropsychiatric diseases, glutamate levels increase, activating the ionotropic glutamate receptors, causing intracellular Ca2+ overload, oxidative stress, mitochondrial dysfunction, and cell death [6].
Dong et al. [7] explain the signaling cascades triggered by excitotoxic insults in neurodegenerative diseases, which result in elevation of intracellular Ca2+, accumulation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), and impairment of mitochondria. Furthermore, the chronic neuroinflammation observed in glial-activated brain leads to autophagy failure, which increases the accumulation of protein aggregates, which, in a positive feedback loop, causes more activated glia. Eventually, neuronal death happens [8]. In their paper, the authors Michalska P. and León R. discuss what could be the protective measures against such aberrations [8]. It is well recognized that various brain insults, including epilepsy, lead to increased protein aggregation and markers of protein deposits, such as neurofilament light chain (NfL) [9], that can be detected in the blood or cerebrospinal fluid.
In this contribution, I explore the notion that abundant protein aggregates, which appear after various excitotoxic insults, increase oxidative stress by interacting with metal ions, thus increasing ROS via the Fenton-like reaction. On the other hand, the protein aggregates act as damage-associated molecular pattern (DAMP) signals [10] on microglial cells, triggering the inflammatory response. The possible microglia-targeted interventions and treatments against neurodegenerative diseases are of common interest to many neurodegenerative and mental state diseases, as well as epilepsies [11]. It has been suggested that modification of tau, amyloid-β, neuroinflammation, mTOR, and AMPA pathways is a plausible target for the development of epilepsy therapies [12].
As well, typical symptoms of cognitive decline, impaired memory, behavioral difficulties with aggressive traits, as observed in AD, have also been documented for drug-resistant epilepsy. To get a possible connection between cognitive decline and protein aggregation, Aroor et al. [13], determined the amount of p-tau and Aβ in the resected brain samples of patients with refractory epilepsy.
In the following, available data on epileptic syndromes, where protein aggregation of a mutated protein or other proteins relevant to proteostasis are reviewed. Apart from developmental cortical dysplasia, two of the progressive myoclonus epilepsies (PMEs) are considered next: EPM1 and EPM2, also known as Unverricht-Lundborg disease (ULD) and Lafora disease (LD), respectively. Dravet syndrome and familial encephalopathy with neuroserpin inclusion bodies (FENIB) are described next, along with the structure and morphology of the aggregated/mutated proteins, for which data exist. The review concludes with two more sections: Perspective–implications for therapy and Conclusions.
Madhamanchi et al. [14] examined the epileptic brain samples of the focal cortical dysplasia (FCD) patients, looking into the unfolded protein response (UPR) and autophagy changes by using Western blots and Thioflavin T assays. Of note, the epileptic patients, who were seizure-free, were found to have few protein aggregates. On the other hand, patients with seizure recurrence (classified as class 2 and class 3) had an increased amount of protein aggregates. It was assumed that impairment in the endoplasmic reticulum (ER) stress response may contribute to seizure recurrence in some epilepsy patients. Indeed, in these classes of patients, they found significant upregulation of the apoptosis initiation factor CHOP as well as Bip/GRP78, p-IRE1α, XBP1, and p-eIF2α, all involved in ER stress. A significant increase in Beclin1, Atg7, Atg12 + 5, Atg16L1, p62, and LC3B, all related to autophagy, was also detected in the FCD samples belonging to classes 2 and 3 [14]. Of note, FCD patients present with mTOR hyperactivation alongside the presence of Aβ and tau pathology [13].
The next case of epileptic syndromes with protein aggregation as an important feature is PMEs. One of the PMEs is LD; another is EPM1, also known as ULD. LD is an autosomal recessive PME. Two genes were identified causing this disease: EPM2A is located on chromosome 6q24, and EPM2B is located on chromosome 6p22.3. They encode laforin, a dual-specificity phosphatase, and malin, a ubiquitin E3 ligase, respectively. Both of the proteins are involved in glycogen metabolism, leading to accumulation of Lafora bodies (LBs), which consist of deposits of polyglucosans, fibrillary glucose polysaccharides. A plausible hypothesis put forward by Mitra et al. [15] is that laforin binds to insoluble glycogen bodies and attracts malin, which ubiquitinates LBs for autophagy.
There are several reports of protein aggregation in LD. Rao et al. [16] have shown that malin is an aggregate-prone protein. Most malin mutations form aggregates both in the cytoplasm and the nucleus. Additionally, aggresomes of malin sequester chaperones and thus increase protein aggregation of other proteins [17]. It is possible that loss of function (loss of proteasomal degradation) and gain in toxic function by the toxicity of the protein aggregates result in the observed phenotype [16]. An interesting paper is also an earlier paper by Garyali et al. [18], who stated that laforin and malin form a complex with Hsp70 in preventing the toxicity of misfolded proteins, recruiting them to UPS.
Even though previously no intracellular inclusion bodies or storage material had been reported in ULD patientsʼ cells or tissue [19, 20], there is a report by Cohen et al. [21], on cytoplasmic and nuclear inclusions of proteins characteristic of neurodegeneration, FUS and TDP-43, in addition to some lysosomal proteins. In the majority of ULD cases with cystatin B mutations, 2 alleles with dodecamer repeats in the gene promoter are observed, leading to much lower expression of the protein [22]. However, in some cases of cystatin B missense mutations, protein aggregation has been observed, both in vitro and in patients. If the mutation in cystatin B is aggregate-prone, usually one allele still codes for dodecamer repeats. Due to complete loss of function and inherent toxicity of the aggregates, the severity of symptoms in such cases becomes higher [23].
On the basis of our in vitro stability and folding/aggregation studies [24] as well as cellular studies of the EPM1 stefin B (cystatin B) mutants, a gain in toxic function of the aggregates was proposed [25–27]. Proneness to aggregate in vitro and in the cell increases from G4R to G50E and Q71P [24, 26, 27], while the fragment R68X aggregates the fastest and forms the largest aggregates, deposited as aggresomes perinuclearly [28].
Similarly to LD, we observed that the lack of stefin B protein in stefin B knock-out murine macrophages led to more inclusions of other proteins [29], likely due to impairment in protein aggregate clearance by autophagy. Therefore, we proposed a hypothesis that, similarly to complex laforin, malin, and Hsp70 roles in UPS, cystatin B could possess an alternative function as a co-chaperone [30, 31]. Indeed, that cystatin B in cells is part of a multiprotein complex was shown already by Di Giaimo et al. [32], who later suggested that this protease inhibitor may possess alternative functions, related to cytoskeleton/plasma membrane interaction [33]. Yet another function might be, as Rispoli et al. [34] suggested, redox sensing. Namely, they report that the monomeric protein requires Cu2+ for chaperone Hsp70-dependent polymerization [34]. They also suggest that Cu2+ interacts with at least two conserved histidines, at positions 72 and 95, which is in accordance with our in vitro studies, where two strong binding sites were detected, as shown by Zerovnik et al. [35]. The Cu2+/Hsp70-dependent polymers were found to be sensitive to reducing agents. If the cellular environment has increased ROS, the presence of Cu2+/Hsp70-stefin B complex could form oligomers and interact with partially folded or amyloid-forming proteins, such as Aβ, acting as an amateur chaperone. In accordance, the interaction of stefin B tetramers with Aβ was detected in vitro and in cells [30].
There are more evident cases of protein aggregation in PMEs. FENIB [36] is a proteinopathy, characterised by neuronal inclusions composed of polymerized neuroserpin (NS). SERPINI1 gene, located on chromosome 3q26, encodes for a serine protease inhibitor, NS. FENIB is a serious disease presenting with PME and progressive dementia.
The inclusion bodies, the so-called Collins bodies, are composed of polymerized, mutated NS that accumulates within the ER of neurons. No typical amyloid fibrils were detected with NS aggregates; rather, they consist of entangled fibrils and short-chain filaments. Several hypotheses for the mechanism of serpin polymerization are available. Serpin polymers in vitro were shown to form by a mechanism of loop insertion, where the loop of one monomer inserts into the β-sheet A of another, creating a chain-like, “beads on a string” linear polymer [37, 38].
Function-wise, the serine protease inhibitors (serpins) are a super-family of proteins that inhibit a wide range of serine proteases. NS inhibits tPA (tissue-type plasminogen activator; converts plasminogen into plasmin, the major enzyme responsible for clot breakdown) and exerts a role in the development of the CNS, in learning, and memory. NS also regulates the sensitivity of neurons to glutamatergic excitatory neurotransmission by the NMDA receptors. The epileptic phenotype of FENIB may thus be at least partially due to dysfunction of the NS/tPA pathway [39]. Of interest, similarly to transthyretin, NS might exert a chaperone action [40]. We have proposed for cystatin B (stefin B) a similar alternative function [30, 31].
FENIB patients show, apart from myoclonic epilepsy, cognitive decline, deficits in attention and concentration, disinhibitory behaviors, and impaired visuospatial skills. Memory is also impaired. In adulthood, generalized seizures and even status epilepticus can occur. Slow speech, diplopia, nystagmus, dysarthria, and myoclonus were also described. For the final stages of the disease, loss of self-sufficiency and dementia were reported [39].
Genotype-phenotype correlations for the NS mutants are remarkably strong, with clinical features increasing in severity (S49P, S52R, L47P, H338R, G392E to G392R, from the least to the most severe). Clinical severity refers to an earlier onset of the symptoms and an increasing contribution of the epileptic component of the syndrome [41].
Mutations render serpins aggregate-prone, and the intraneuronal accumulation of NS aggregates (polymers) leads to a gain in toxic function. Belorgey et al. [39] reported on 2 unrelated Caucasian families, carrying 2 different heterozygous mutations (S49P and S52R). The rate of aggregation of the least clinically-aggressive mutant, S49P, is more than 10 times higher than that of the wild-type protein, whereas the association rate constant for tPA is essentially unchanged [39]. The next most severe mutation, S52R, results in a further tenfold increase in the polymerization rate and the complete loss of tPA inhibition [42]. NS polymers gradually become entangled in the neuronal ER and form inclusions, known as Collins bodies.
A few more case studies appear in the literature. Davis et al. [43] reported two American patients with the disorder, exhibiting PME, dementia, tremor, and dysarthria. L47P mutation of NS was reported by Hagen et al. [44], detected in an adult patient with PME and declining mental state. Generalized seizures occurred early with myoclonus and progressive gait disturbances. Neuroimaging revealed mild atrophy and multiple periventricular white matter lesions, consistent with demyelination. Patients with the S52R, L47P, and H338R variants demonstrated larger and more numerous intraneuronal inclusions and presented with progressively earlier onset of symptoms, during early adulthood (S52R, L47P) or adolescence (H338R) [44]. Of note, the most severe cases were caused by the most aggregate-prone mutations, G392R and G392E. The patients exhibited the earliest onset of symptoms already during childhood, with severe intellectual decline and aggressive behaviour, uncontrolled epilepsy, psychic seizures, and subtle seizures with eyelid myoclonus [39].
Dravet syndrome is a severe epilepsy condition, a developmental encephalopathy, that begins in infancy or early childhood. This rare genetic disorder occurs in one in every 30,000 births, according to the Dravet Syndrome Foundation. Treatment options are very limited and seizures remain difficult to control; they are mostly drug resistant. It was demonstrated that the GABA(A) receptor γ2 subunit gene Q351X mutation contributes to loss of the receptor function. Already in 2010, Kang et al. [45] reported that the mutant γ2(Q351X) of the GABA receptor γ2 subunits were degraded more slowly than the wild-type protein. The mutated versions formed irreversible aggregates in multiple cell types, including neurons. Using truncated subunits, they demonstrated that aggregate formation was a general phenomenon for truncated γ2S subunits and that the Cys-loop cysteines were involved in aggregate formation [45]. The same group by Kang et al. [46] developed a model of a severe human genetic epileptic encephalopathy, the mutant GABAA receptor-γ2 (Q390X) knock-in mouse. The mutant subunits aggregated intracellularly, causing widespread, age-dependent neurodegeneration [46]. The properties and morphology of these aggregates have not been determined; therefore, it is hard to judge whereas they form amyloid fibrils.
Following protein aggregate accumulation, ER stress occurs, followed by Ca2+ imbalance. One should distinguish between the general pathology caused by gain in toxic function of the accumulated protein aggregates and loss of specific function of the voltage-gated channel. In accordance, studies by Kang et al. [46, 47] revealed that the knock-in mice whose mutation did not cause ER stress, only manifested mild seizures and had normal cognition, while the mice with the mutation-causing ER stress due to protein aggregation manifested more severe epilepsy and impaired cognition.
Other mutations were reported to cause Dravet syndrome, such as mutation of sodium voltage-gated channel beta1 subunit gene (SCN1B), the GABA receptor alpha-1 subunit gene (GABRA1), and STXBP1. The STXBP1 gene, which translates into protein Munc18-1, is essential for neurotransmitter release via regulation of syntaxin. Understanding the signaling pathways disrupted upon mutant subunit aggregation will help reveal the pathological effects of the aggregates in the pathogenesis of such epilepsy syndromes. Of note, protein misfolding and aggregation often accompany the most severe cases of PMEs and refractory epilepsies.
In many cases of monogenic epilepsies, the mutations in the causing gene lead to protein misfolding and aggregation. Especially clear is the situation in FENIB, due to polymerization and aggregation of NS. The second clear proteinopathy is Dravet syndrome.
The epileptic syndromes all have a neurodegenerative side. Neurodegeneration in ULD is not that apparent in cases of diminished activity of cystatin B (which is the case for the dodecamer repeat extension in the gene, which leads to much lower expression of this protein). However, with increased protein aggregation of some missense mutants, the severity of symptoms increases.
It should be kept in mind that protein aggregation is one of the initiating factors for microglia activation [48]. Activated microglia are found in the vicinity of the insoluble protein aggregates, triggering an inflammatory response [49]. It has been reported that microglia phagocytose tau [50], Aβ [51], α-syn [52], and TDP-43 [53] aggregates. The toxic protein aggregates may thus increase neurodegeneration.
I propose a hypothetical viewpoint that generic symptoms of impaired cognitive abilities and reduced memory, aggressive and even psychotic behaviors, as well as neurodegeneration, are largely due to protein aggregates and downstream effects. This view seems to agree with the observation that the cases where protein aggregates are abundant show more severe pathology, including dementia. However, Aroor et al. [13] could not confirm a strong correlation between the amount of the intracellular aggregates and extracellular deposits of tau and Aβ, respectively, and cognitive decline in refractive forms of epilepsy. This could be due to the different toxicity of the sequestered forms of irreversible and solid aggregates in comparison to more toxic soluble oligomers. Therefore, more research in this direction is needed in the future. For example, more structural characterization of inclusion bodies would be possible with cryo-electron microscopy and atomic force microscopy (AFM). Soluble oligomers could also be detected by AFM. Pore formation in cellular membranes might lie at the limit of super-resolution optical microscopy—STED (stimulated emission depletion).
It can be agreed that protein aggregation impedes neuronal function in two ways. It reduces the amount of the active protein (thus reducing its function) and it produces generic toxicity by protein aggregates, among them oxidative stress, mitochondrial dysfunction, neuroinflammation and finally neurodegeneration.
The most likely mechanism implies that the toxic prefibrillar oligomers interact with cell membranes, destabilising them and modifying ion balance, possibly by formation of membrane pores [54]. The rise of the intracellular free Ca2+ and ROS is one of the earliest modifications in the path to cell death following cell exposure to early amyloid (pre-fibrillar) aggregates [54, 55] (see scheme in Figure 1).
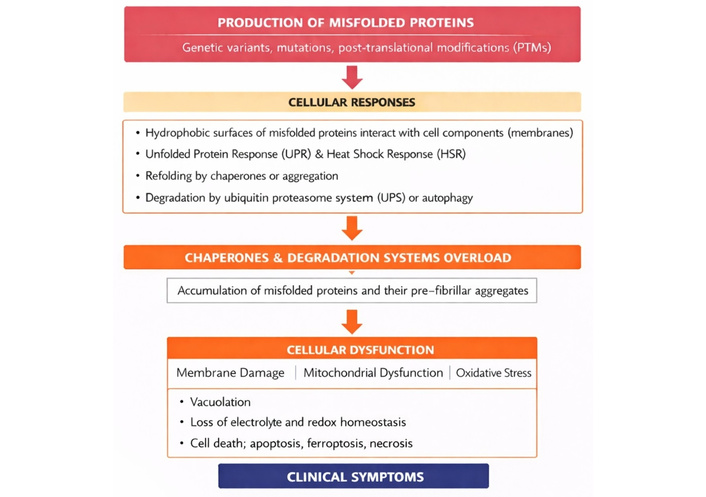
Schematic overview of the cellular consequences of misfolded and aggregated proteins leading to neurodegeneration.
This scheme illustrates the sequential cellular events triggered by the production of misfolded proteins due to genetic variants (mutations), or post-translational modifications. The cascade of cellular responses begins with hydrophobic exposure and membrane interaction by the misfolded protein oligomers, followed by failure of chaperone systems and activation of stress responses such as the UPR and heat shock response (HSR). Misfolded proteins may get refolded by the help of chaperones, degraded, or aggregated. When these proteostasis mechanisms are impaired or overwhelmed, aggregated proteins accumulate. The fully formed, mature amyloid fibrils are considered to be protective (when sequestered), whereas the pre-fibrillar oligomers might be more toxic. These latter interact with membranes, destabilising them and modifying ion balance possibly by formation of non-specific membrane pores [54]. The rise of the intracellular free Ca2+ and ROS is one of the earliest events, following cell exposure to early pre-fibrillar aggregates. Finally, this culminates in mitochondrial dysfunction, cell death (neurodegeneration), and the emergence of clinical symptoms [54, 55].
Strengthening proteostasis might be a way forward to fight protein aggregation. This can occur through three complementary mechanisms. Namely: (1) Chaperone stimulation enhances protein folding capacity by increasing chaperone activation and assisting in the proper folding of nascent or misfolded proteins. (2) Autophagy enhancement promotes the degradation of protein aggregates and facilitates the autophagic removal of damaged cellular components via the lysosomal pathway. (3) Strengthening the UPS improves the targeted degradation of misfolded and defective proteins through ubiquitin tagging and proteasomal processing. Together, these mechanisms maintain protein homeostasis and mitigate proteotoxic stress, which is critical for cellular health and the prevention of aggregation-related diseases.
Some of these therapeutic strategies have been explored by Scannevin [56]. There are diverse strategies proposed: 1st, diminishing protein synthesis of an aggregating protein, 2nd, preventing its aggregation by either small molecules or by stimulating chaperone proteins, 3rd, disaggregating the aggregates and clearing them from the cell. Point 1 is possible with the new RNA and DNA editing techniques. Point 3 is being explored by mTOR-dependent or mTOR-independent modulators of autophagy, such as rapamycin and analogues. Autophagy can also be increased by life-style changes. While, point 2 inhibiting protein aggregation by small molecules and disaggregating the aggregates is also part of the new therapeutic adjustments. Often surprisingly, substances which inhibit protein aggregation also act as anti-oxidants. Such is the case with curcumin, for example. In the case of supplementation, one also has to take care when to add a substance, because increasing autophagy, or regulating excitotoxicity [57] might be beneficial only in initial stages of neurodegenerative diseases (before progressive neurodegeneration takes place).
Till now, treatment of most neurodegenerative diseases stays symptomatic. More promising are multi-target and synergistic approaches, where natural substances such as resveratrol or quercetin are supplemented. In the field of AD, PD, ALS, HD and lysosomal disorders as well as healthy aging, some small-molecule modulators of proteostasis have been implemented (Table 1).
| Proteostasis process | Compound (class) | Mechanism | Neurodegenerative relevance | References |
|---|---|---|---|---|
| Chaperone stimulation | Arimoclomol | HSF1 co-inducer, ↑ Hsp70/Hsp40 | ALS, IBM, lysosomal disorders | [58, 59] |
| Celastrol | HSF1 activator | Reduces polyQ and α-syn aggregation | [60] | |
| 17-AAG (geldanamycin derivative) | Hsp90 inhibitor → HSF1 activation | Broad proteotoxicity models | [61] | |
| Novel HSF1 activators | Direct HSF1 agonism | Emerging neurodegeneration candidates | [62] | |
| Autophagy enhancement | Rapamycin/Rapalogs | mTOR inhibition | AD, PD, HD, ALS | [63] |
| Trehalose | mTOR-independent autophagy | Clearance of mutant huntingtin, α-syn | [64] | |
| Spermidine | Polyamine, autophagy inducer | Neuroprotection, healthy aging | [65] | |
| Resveratrol/Curcumin | AMPK/SIRT1 activation | Broad autophagy-mediated protection | [66, 67] | |
| UPS strengthening | IU1 (USP14 inhibitor) | Enhances proteasomal degradation | Misfolded protein clearance | [68] |
| Proteasome activators | Allosteric 20S/26S activation | Counters age-related proteasome decline | [69] | |
| UPS-modulating small molecules | Ubiquitination/Assembly modulators | AD, PD, ALS models | [70] |
The table presents some small molecule modulators, drugs already used for other neurodegenerative conditions, such as AD, ALS, HD or in reversing ageing. AAG: alkyladenine glycosylase; AD: Alzheimer’s disease; ALS: amylotrophic lateral sclerosis; AMPK/SIRT1: AMP-activated protein kinase/Sirtuin 1; HD: Huntington’s disease; HSF1: heat shock factor 1; IBM: inclusion body myositis; IU1: small-molecule inhibitor of the USP14 proteasome-associated deubiquitinating enzyme; PD: Parkinson’s disease; UPS: ubiquitin proteasome system.
Several studies have also reported on the therapeutic effects of celastrol on epilepsy [71]. But it was concluded that available research on the therapeutic mechanisms of triptolide—a potent bioactive diterpenoid triepoxide derived from the Chinese herb Tripterygium wilfordii and used in TCM for its strong anti-inflammatory, immunosuppressive, and antitumor properties—and celastrol in epilepsy is still not deep enough, and more in-depth studies are needed to further clarify their mechanisms [71]. Rapamycin was also probed as a possible substance to treat epilepsy and showed some anti-seizures effect in acquired epilepsy model [72, 73].
The evidence reviewed here demonstrates that protein misfolding and aggregation are observed across several monogenic epileptic syndromes, the PMEs, such as LD, ULD, and other disorders with epilepsy as a feature, such as FENIB, and Dravet syndrome.
When we refer to the “vicious cycle” between excitotoxicity and protein aggregation; excitotoxic insults promote misfolding and aggregation, while aggregates themselves exacerbate excitotoxicity, neuroinflammation, and microglial activation. As the manuscript states, protein aggregation is one of the initiating factors for microglia activation.
Mutations in key proteins either directly promote aggregation or disrupt proteostasis pathways, leading to intracellular inclusions, which may be a protective form of the aggregates. More dangerous are soluble protein oligomers prone to interact with membranes (ER, mitochondria, lysosomes) and also interfere with synaptic trafficking.
Downstream effects of protein aggregation include ER stress, Ca2+ imbalance, oxidative damage, mitochondrial impairment, and ultimately neurodegeneration (see Figure 1).
The clinical observation shows in most cases that syndromes with more abundant protein aggregates show more severe cognitive decline and behavioral disturbances, including dementia.
At the same time, the lack of a strict correlation between aggregate load and cognitive impairment in some refractory epilepsies suggests that the form of the aggregates (irreversible and solid or soluble and oligomeric) and cellular context (to which organelles they attach) may be more relevant for the clinical deterioration.
From a therapeutic standpoint, the review underscores that targeting proteostasis represents a promising strategy also for the rare epileptic syndromes. Enhancing chaperone activity, stimulating degradation by UPS and autophagy, might help restore cellular balance and reduce proteotoxic stress. Nevertheless, one should pay attention to the role of mTOR hyperactivation in some epileptic conditions.
Small-molecule modulators (see Table 1), lifestyle interventions, and emerging genetic approaches offer complementary avenues for intervention. However, timing is critical: interventions that modulate autophagy or excitotoxicity may be beneficial primarily in the early stages of disease, before irreversible neurodegeneration occurs.
AD: Alzheimer’s disease
AFM: atomic force microscopy
AMPA: amino-3-hydroxy-5-methylisoxazolepropionic acid
CNS: central nervous system
ER: endoplasmic reticulum
FCD: focal cortical dysplasia
FENIB: familial encephalopathy with neuroserpin inclusion bodies
LBs: Lafora bodies
LD: Lafora disease
NMDA: N-methyl-D-aspartate
NS: neuroserpin
PMEs: progressive myoclonus epilepsies
ROS: reactive oxygen species
tPA: tissue-type plasminogen activator
ULD: Unverricht-Lundborg disease
UPR: unfolded protein response
UPS: ubiquitin proteasome system
EŽ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. The author read and approved the submitted version.
The author declares that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The author acknowledges support by the Slovenian Research and Innovation Agency—ARIS program P1-0140 (led by B. Turk). The funders had no direct role in the review paper design, data collection and analysis and decision to publish.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 611
Download: 24
Times Cited: 0
Hoong Wei Gan ... William P. Whitehouse
Jerónimo Auzmendi, Alberto Lazarowski
Toshimitsu Suzuki ... Kazuhiro Yamakawa
Rodolfo Cesar Callejas-Rojas ... Ildefonso Rodriguez-Leyva
Anwaar M. Bennour ... Heba A. El-Zawawi
