 Mini Review
Mini Review

Affiliation:
1National Council for Scientific and Technical Research (CONICET), Buenos Aires C1425FQD, Argentina
2Institute of Physiopathology and Clinical Biochemistry (INFIBIOC), School of Pharmacy and Biochemistry (FFyB), University of Buenos Aires (UBA), Buenos Aires C1113AAD, Argentina
ORCID: http://orcid.org/0000-0002-4819-2713
Affiliation:
2Institute of Physiopathology and Clinical Biochemistry (INFIBIOC), School of Pharmacy and Biochemistry (FFyB), University of Buenos Aires (UBA), Buenos Aires C1113AAD, Argentina
Email: alazarowski@gmail.com; naiatom@ffyb.uba.ar
ORCID: http://orcid.org/0000-0001-8979-7631
Explor Neurosci. 2025;4:100692 DOI: https://doi.org/10.37349/en.2025.100692
Received: January 22, 2025 Accepted: April 27, 2025 Published: June 04, 2025
Academic Editor: Jinwei Zhang, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, China
The article belongs to the special issue Advances in Epilepsy Research
All living beings, from microorganisms to plants, animals, and humans, require iron as an essential micronutrient for their lives. However, iron overload can constitute a scenario prone to damage to the organism, including oxidative stress, deterioration of cellular and subcellular membranes, and thus leading to cell death. This process involves unrestricted lipid peroxidation caused by the generation of reactive oxygen species (ROS) because of an abrupt increase in free Fe2+ in the cytoplasm, all of which leads to subsequent membrane damage and iron-dependent cell death, now known as “ferroptosis”. This process can be induced by convulsive stress, and conversely, inducing seizures, and in both situations under a context of neuroinflammation. In this critical review, we will highlight the most relevant aspects of this recently described mechanism, which has been studied little in epilepsy, its impact on the prognosis of the disease, and its effects on the development of central and/or peripheral comorbidities, including SUDEP (sudden unexpected death in epilepsy).
In the 1850s, the French physician Armand Trousseau warned his colleagues “Do not prescribe iron preparations to patients with tuberculosis”. Trousseau was able to document that iron supplementation in the diet of his tuberculous patients with some degree of anemia worsened their clinical picture compared to those who did not receive additional iron [1]. It is known that the production of ROS (reactive oxygen species) plays a role in protection against pathogenic microorganisms, but they can also cause tissue damage during this protection and trigger inflammation, where excess iron favors this process leading to cell death [2]. According to these observations, at the beginning of the 21st century, it was indicated that iron overload increases the virulence of iron-dependent pathogens, which could play a devastating role in their progression and exacerbate overall mortality in patients infected with such microorganisms [3, 4]. More recently it was reported that ferroptosis is the main mechanism of cell death induced by Mycobacterium tuberculosis [4].
In 2012, Sheftel et al. [5] described a long history of millions of years of evolution in iron homeostasis. In the same year, Dixon et al. [6] coined the concept of “iron-dependent programmed cell death” which they called ferroptosis that is a condition directly related to oxidative stress as previously described by Alvira et al. [2].
Iron overload increases the severity of inflammatory processes, in which there is not only a consistent relationship between seizures and inflammatory cytokines (IL-6, TNF-α, and IFN, etc.) but also the inflammation itself generates increased iron deposits [7]. Since its description in 2012, there have been more than 10,000 scientific articles dedicated to ferroptosis. Although ferroptosis can lead to neurodegenerative processes associated with mitochondrial dysfunction, mitophagy [8], autophagy, or ferritinophagy [9], and are implicated in hypoxic brain injury [10, 11], less than 100 articles link ferroptosis with epilepsy.
Hypoxia, oxidative stress, and inflammation are always present in neurodegenerative processes where seizures can be both the cause and the consequence of them [12, 13]. Several brain insults such as hypoxia, trauma, micro-hemorrhages, inflammation, infections, tumors, etc., can induce oxidative stress and neurodegeneration [13] and some of them are related to ferroptosis [14].
Refractory epilepsy (RE) is characterized by the multidrug-resistance (MDR) phenotype where seizures are not controlled despite patients receiving combinations of two or more recommended anti-seizure medications (ASMs) [15]. Since the use of bromide (in 1860) more than 20 ASMs have been developed, but a constant ~30% of cases remain as RE with mentioned MDR phenotype [16].
Several hypotheses have been proposed to explain this phenomenon [17], of which the so-called “Transporter hypothesis” and “Pharmacokinetics hypothesis” have gained great relevance. These hypotheses link the expression of some members of the ABC (ATP binding cassette) transporter family (ABC-t) to the MDR phenotype. ABC-t, such as P-glycoprotein (P-gp), multidrug resistance-associated proteins (MRP1–4), and breast cancer-resistance protein (BCRP), are highly expressed in excretory organs and play a critical role in the pharmacokinetics of almost all drugs, including ASMs. Stimuli such as hypoxia, oxidative stress, excitotoxicity, and/or inflammation, in addition to triggering ferroptosis, may also promote increased expression of these ABC-t.
As indicated by the Transporter hypothesis, overexpression of ABC-t at the BBB (blood-brain barrier) level may affect the access of ASMs to the brain parenchyma [18, 19]. A very comprehensive review of the role of ABC-t expression in BBB linked to the MDR phenotype has recently been published [20].
The recurrence of uncontrolled epileptic seizures, particularly generalized tonic-clonic seizures (GTCS), can lead to hypoxic conditions not only at the central level but also at the peripheral level, promoting the overexpression of ABC-t [12]. In this context, the overexpression of ABC-t may reduce gastrointestinal absorption and increase hepatobiliary and renal excretion of drugs [16], reducing drug biodistribution (Pharmacokinetics hypothesis) [17].
Stressful conditions may affect peripheral organs that do not normally express ABC-t, such as the heart. In this sense, we have been able to document a high expression of P-gp in cardiomyocytes associated with bradycardia, an increase in the QT interval during the postictal period, and a high rate of spontaneous death in an experimental model of status epilepticus (SE) induced by pilocarpine [21]. One of the main risk factors associated with sudden unexpected death in epilepsy (SUDEP) is the high frequency of GTCS, and under these conditions, a progressive cardiac P-gp overexpression has been described, which has been considered a potential biomarker for SUDEP risk [22, 23]. In these same conditions, it has recently been described that seizures produce hemosiderin deposition in the myocardium [24], providing a link between the recurrence of GTCS and ferroptosis.
Stressful conditions produced in the brain parenchyma by uncontrolled repetitive seizures can induce neuronal expression of ABC-t, as has been demonstrated both in surgical resections of patients with drug-resistant epilepsies [25–27] and in different experimental animal models [26, 28–30]. It has been postulated that neuronal expression of P-gp in the epileptic focus, leads to a decrease in the excitatory threshold facilitating seizure recurrence [27] and increasing the stressful conditions that trigger ferroptosis.
Ferroptosis has been described as a consequence of epileptic seizures, as certain biomarkers of ferroptosis such as 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), glutathione peroxidase 4 (GPX4), cyclooxygenase-2 (COX-2), and glutathione (GSH) have been found to be altered in plasma and in brains of epileptic patients and animal models of experimental epilepsy [31–33]. In the same way, treatment with ferrostatin-1 and other ferroptosis inhibitors tends to restore the basal level of ferroptosis markers after seizures [34–37].
Under normal conditions, cells have almost all iron as Fe+3 stored in ferritin depots. A tiny portion of the total iron is found to be Fe+2 as part of the labile iron pool (LIP), which can be used for biological purposes. As is well known, Fe+2 can catalyze the formation of ROS, although cells possess the ability to counteract the deleterious effects of ROS produced by their basal metabolism through GSH. Under conditions of seizure-induced stress, this system can be thrown out of balance in several ways leading to ferroptosis.
Firstly, epileptic seizures can increase the secretion of proinflammatory interleukins [38]. In vitro experiments have shown that IL-6 treatment led microglia and astrocytes to increase hepcidin secretion, which blocks the iron-exporting action of ferroportin (FPN). At the same time, neurons and microglia increased the expression of divalent metal transporter 1 (DMT1) which incorporates iron into cells, and decreased that of FPN. In this context, microglia and neurons developed an iron-accumulating phenotype [39]. At the cellular level, iron homeostasis is regulated between its accumulation within the ‘ferritin cage’ and its release on demand by ferritinophagy (ferritin degradation). In this equilibrium, iron incorporated by cells is stored in ferritin as Fe+3, which in turn is incorporated into pre-autophagosome structures dependent on anchorage by NCOA4 (nuclear receptor coactivator 4) [40, 41]. In situations of iron requirement, the assembled autophagosome binds to lysosomes, and iron is reduced to Fe+2 and released to LIP [40]. Recent results show that inhibition of ferritinophagy can decrease ferroptosis in rat hippocampus and spinal cord [42, 43]. On the other hand, under conditions of iron overload, NCOA4 bound to an iron atom is degraded by proteasome facilitating the accumulation of charged ferritin molecules in the cytoplasm and the formation of insoluble precipitates so-called haemosiderin [40]. Furthermore, haemosiderin has been found in surgical resections of drug-resistant epileptic patients, accompanied by a marked increase in intracellular ferritin [33]. An experimental study of SE demonstrated a significant increase in ferritin in reactive hippocampal microglia, which decreased over time but remained elevated in the chronic epileptic phase post-SE and was associated with extensive cell loss. The upregulation was most intense in rats that developed a progressive form of epilepsy with five to ten seizures per day. These results suggest that increased ferritin in specific limbic regions is an indicator of brain iron accumulation (BIA) that plays a role in the progression of epilepsy, subsequent cell loss, and perhaps in epileptogenesis itself [44].
In addition to inflammation, oxidative stress plays an important role in ferroptosis caused by seizures. Several reports have shown that seizures lead to a decrease in GPX4 promoting oxidative stress [35, 45, 46]. GPX4 expression is controlled by the transcription factor NRF-2, whose activation is repressed by Keap1. A recent work has shown that epileptic seizures lead to overexpression of Keap1 promoting proteasomal degradation of NRF-2 in neurons, while inhibition of Keap1 expression stabilizes NRF-2, and increases the level of GSH, GPX4, superoxide dismutase (SOD), and the cystine/glutamate exchanger subunit SLC7A11 [47]. Neuroprotective assays designed to stimulate the NRF-2/Keap1 axis showed a decrease in oxidative stress markers and an increase in antioxidant enzymes in different models of experimental epilepsy [48–51]. In the same line of evidence, drug-resistant epileptic patients who underwent surgery of the epileptic focus and remained with seizures showed reduced NRF-2 expression and increased Keap1 expression compared to seizure-free patients after surgery [52]. Some polymorphisms in this axis have been linked to drug-resistant epilepsy. The single nucleotide polymorphism (SNP) rs2706110 G>A in NRF-2 was associated with drug resistance while the SNP rs1048290 C>G in the Keap1 gene was shown to prevent drug resistance [53].
Although GSH has an enzymatic recycling system via GSH reductase, excessive ROS production requires increased GSH synthesis. The cystine/glutamate exchanger (xCT subunit from xC-system) couples cystine entry into the cytoplasm with glutamate exit to the cell exterior [54]. Cystine is crucial for GSH synthesis, and the cystine/glutamate exchanger’s activity directly influences ferroptosis development due to seizures. While xCT expression is abundant in all cell types of the body, in the central nervous system (CNS) xCT expression is limited to astrocytes [55]. Recent reports have shown that epileptic seizures increase xCT expression, both in patients with drug-resistant epilepsies [33] and in experimental models [56]. This may translate as an increase in extracellular glutamate concentration that promotes neuronal hyperexcitability. This effect may be even greater as seizures have been reported to lead to a rapid decrease in extracellular volume promoted by overexpression of astrocytic aquaporins [57, 58]. In tumor-associated epilepsies such as glioblastoma, xCT overexpression and elevated peritumoral glutamate concentration were associated with seizure generation and complications in patient survival [59–61] while treatment with xCT inhibitors such as sulfasalazine decreased epileptiform activity in vivo and in vitro [62]. In addition, knock-out mice for xCT–/– showed reduced hippocampal glutamate levels [63] and reduced epileptiform activity in a pilocarpine-induced SE model [64].
Normal iron metabolism is a finely regulated homeostatic process. It is well known that iron absorption at the enteric level is controlled by the hepatic hormone hepcidin. Under normal conditions, hepcidin blocks FPN from enterocytes preventing iron incorporation into the bloodstream. Under certain conditions of iron demand, such as anemia, hypoxia, or hemorrhage, hepatic hepcidin synthesis decreases because the Hamp gene that encodes it, is under the control of the hypoxia-inducible factor (HIF). Under these conditions, the FPN of the enterocytes is unblocked and iron can access the bloodstream and be transported by transferrin to the target organ or even be stored in the liver and macrophages into the ferritin as a reserve deposit. Under pathological conditions, excessive accumulation of iron in tissues can lead to hemochromatosis. In this context, excessive accumulation in the liver can lead to the development of cirrhosis and thus lead to a marked decrease in hepcidin synthesis, supporting higher iron accumulation. A similar condition with BIA was recently suggested in Lafora disease which is associated with muscle spasms and epilepsy [65].
Some findings in patients link hemochromatosis to brain iron overload and the development of epilepsy [66]. Even some studies in animal models indicate that gene alterations linked to hemochromatosis increase iron accumulation in the brain [67, 68]. A very particular study designed to assess whether iron overload predisposes to epilepsy showed that the transferrin saturation of epileptic patients increased 33% above the level of controls. Eight percent of the epileptic patients showed abnormally high levels of transferrin saturation. Of these only 20% were heterozygous for the H63D mutation in the hemochromatosis gene, while the C282Y mutation was not detected, suggesting that iron overload rather than typical hemochromatosis mutations was associated with epilepsy development [69].
Consistent with these findings, several patients with hyperferritinemia have presented with epileptic seizures [70, 71]. Ferritin is made up of two chains of different molecular weights, the ferritin light chain (FTL) and the ferritin heavy chain (FTH). This hetero polymer is not only found in circulation but also in tissues as an iron reservoir due to its large storage capacity (approximately 4,500 iron atoms per molecule). Genetic variants that impact the carboxyl-terminal domain of FTL and modify its iron-binding capacity have been linked to the disease hereditary neuroferritinopathy, it is characterized by progressive neurologic symptoms and iron accumulation, particularly in the basal ganglia. The most prominent symptoms include chorea, focal dystonia, and, less often, parkinsonism [72]. Recent work has described heterozygous nonsense de novo variants in the last FTH1 exon of several pediatric patients, leading to ferritin accumulation, resonance imaging typical of neurodegeneration with cerebral iron accumulation, cellular markers of oxidative stress, and epilepsy [73].
Lipoperoxidation is the main biochemical event during ferroptosis, which requires high amounts of free Fe2+ in the cytosol to generate ROS by Fenton reaction [74]. ROS will induce lipoperoxidation of the membranes’ lipids, with the accumulation of free radicals (phospholipid-hydroperoxides, 4-HO-nonenal, malonaldehyde), that changes in the membrane’s configuration leading to cell death [75]. Under diverse types of aggressive stimuli that stress cells, such as inflammation, altered metabolism, hypoxia, seizures themselves, etc., the BIA is the feature that distinguishes ferroptosis from other forms of cell death. Iron overload is characterized by an increase in intracellular ferritin with large amounts of Fe3+ within its protein structure due to a positive dietary balance of this metal, hemochromatosis, or hemolytic anemia, bone marrow aplasia, high transfusion requirements, and even chronic inflammatory diseases. Certain clinical conditions can stimulate ferritinophagy (as specific iron-storage-protein autophagy), resulting in high cytoplasmic levels of free Fe2+ and intra- and extracellular deposits of hemosiderin. BIA with elevated ferritins can facilitate ferritinophagy that favors brain-ferroptosis. On the other hand, the generation of microbleeds due to cerebral hypoxia, brain trauma, aneurysms, hemorrhagic stroke, or due to the seizures themselves, will produce the intra-tissue accumulation of hemoglobin, activation of hemoxygenase 1 (HO-1) and release of high amounts of iron and hemosiderin accumulation, that acts itself as factor inducing seizures as observed in cavernomas [36, 76] or brain tumors [77] (Figure 1).
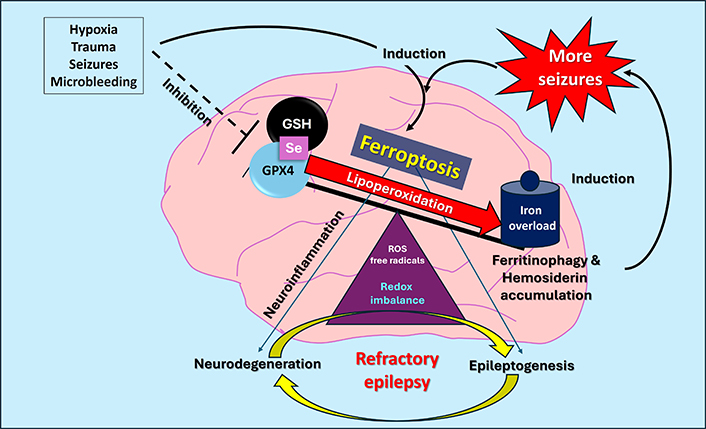
Schematic representation of the relationship between ferroptosis and epilepsy, highlighting the iron storage process by iron storage inside ferritin, oxidative stress with ferritinophagy, and hemosiderin formation as a source of neurodegeneration and new seizures. Redox imbalance with high brain iron accumulation inducing neurodegeneration and/or epileptogenesis. GPX4: glutathione peroxidase 4; GSH: glutathione; ROS: reactive oxygen species; Se: selenium
Finally, a direct link between ferroptosis and drug-resistant epilepsy can be postulated since activation of the NRF-2 pathway, the main transcription factor linked to iron metabolism and the response to oxidative stress, promoted the expression of efflux transporters such as P-gp in the mouse BBB [78]. In line with these results experiments performed with an in vitro human BBB model showed that under hypoxic conditions activation of NRF-2/HIF pathways led to overexpression of P-gp although a distinct expression profile of BCRP was observed [79], while inhibition of HIF-1α can prevent ferroptosis [80].
The evidence described in this review indicates a feedback process between seizures and ferroptosis because seizures can trigger oxidative stress processes that, via ferroptosis, result in neurodegeneration, neuronal death, and hemosiderin accumulation, which triggers the onset of new seizures. In turn, various CNS damage processes such as hypoxia, trauma, inflammation, and microbleeds can also induce ferroptosis, with the same convulsive consequences, generating a vicious cycle of perpetuating the seizure-prone or epileptogenic state.
ABC: ATP binding cassette
ABC-t: ATP binding cassette transporter family
ASMs: anti-seizure medications
BBB: blood-brain barrier
BCRP: breast cancer-resistance protein
BIA: brain iron accumulation
CNS: central nervous system
FPN: ferroportin
FTH: ferritin heavy chain
FTL: ferritin light chain
GPX4: glutathione peroxidase 4
GSH: glutathione
GTCS: generalized tonic-clonic seizures
HIF: hypoxia-inducible factor
LIP: labile iron pool
MDR: multidrug-resistance
NCOA4: nuclear receptor coactivator 4
P-gp: P-glycoprotein
RE: refractory epilepsy
ROS: reactive oxygen species
SE: status epilepticus
SNP: single nucleotide polymorphism
SUDEP: sudden unexpected death in epilepsy
JA and AL: Writing—original draft, Writing—review & editing, Resources.
Both authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by “Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación [PICT2019-01282]”. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Hoong Wei Gan ... William P. Whitehouse
Toshimitsu Suzuki ... Kazuhiro Yamakawa
Rodolfo Cesar Callejas-Rojas ... Ildefonso Rodriguez-Leyva
Anwaar M. Bennour ... Heba A. El-Zawawi
