Review
Review

Affiliation:
1Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
ORCID: https://orcid.org/0000-0003-3587-9767
Affiliation:
1Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
2Department of Public Health and Maritime Transport, Faculty of Medicine, University of Thessaly, 41222 Volos, Greece
ORCID: https://orcid.org/0000-0002-3809-4271
Affiliation:
1Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
3Department of Medical Laboratory Science, College of Basic Health Sciences, Achievers University, Owo, Ondo State 341101, Nigeria
Email: uthmanadebayo85@gmail.com
ORCID: https://orcid.org/0009-0000-2000-7451
Affiliation:
1Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
ORCID: https://orcid.org/0009-0008-2544-7606
Affiliation:
4School of Global Health, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
ORCID: https://orcid.org/0000-0003-0138-3261
Affiliation:
5Faculty of Medicine and Health Sciences, SIMAD University, Mogadishu, Somalia
6SIMAD Institute for Global Health, SIMAD University, Mogadishu, Somalia
ORCID: https://orcid.org/0009-0006-5991-4052
Affiliation:
7Department of Clinical Services, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
ORCID: https://orcid.org/0000-0003-2945-3153
Affiliation:
1Department of Medical Laboratory Science, Neuropsychiatric Hospital, Aro, Abeokuta 110101, Nigeria
ORCID: https://orcid.org/0009-0005-0757-4810
Affiliation:
8Department of Nursing Science, College of Health Sciences, Bowen University, Iwo, Osun State 231004, Nigeria
ORCID: https://orcid.org/0009-0007-4698-1333
Affiliation:
9Seventh Day Adventist Hospital, Asamang, Ghana
10The Royal (Dick) School of Veterinary Studies and the Roslin Institute, University of Edinburgh, EH25 9RG Midlothian, UK
ORCID: https://orcid.org/0000-0002-9836-0834
Affiliation:
11Department of Global Health and Development, London School of Hygiene and Tropical Medicine, WC1E 7HT London, UK
12Research and Innovation Office, Southern Leyte State University, Sogod 6606, Philippines
13Center for Research and Development, Cebu Normal University, Cebu 6000, Philippines
ORCID: https://orcid.org/0000-0002-2179-6365
Explor Neurosci. 2025;4:1006112 DOI: https://doi.org/10.37349/en.2025.1006112
Received: July 30, 2025 Accepted: September 22, 2025 Published: October 12, 2025
Academic Editor: Thomas Heinbockel, Howard University College of Medicine, USA
Schizophrenia (SZ) is a complex psychiatric disorder characterized by disruptions in cognition, perception, and behavior, contributing significantly to the global burden of psychiatric disorders and necessitating ongoing research into its pathophysiology, diagnosis, and treatment. This narrative review explores recent insights into SZ research, highlighting the genetic, neurochemical, and neurodevelopmental factors that contribute to the disorder. Emerging evidence underscores the dynamic interplay between neurotransmitter imbalances, particularly involving dopamine, glutamate, and gamma-aminobutyric acid (GABA), and neuroinflammation, oxidative stress, and immune dysregulation in the pathophysiology of SZ. Neuroimaging, clinical staging models, and multi-omics technologies have deepened our understanding of structural and functional brain abnormalities, identifying potential biomarkers for early detection and subtyping. This has refined diagnostic frameworks and informed precision psychiatry approaches. Advances in pharmacological treatments, including trace amine-associated receptor 1 agonists, glutamatergic modulators, psychedelics, and anti-inflammatory agents, offer new therapeutic possibilities beyond conventional dopamine antagonists. Novel targets, such as N-methyl-D-aspartate (NMDA) receptor modulation and neuroprotective strategies, are also being explored to address negative and cognitive symptoms. Additionally, non-pharmacological interventions, such as neuromodulation techniques, digital therapeutics, and psychosocial interventions, are promising complementary strategies. Digital phenotyping, machine learning (ML), and artificial intelligence (AI)-driven tools enable real-time symptom tracking, early risk prediction, and personalized care delivery. Despite these advancements, challenges remain in early diagnosis, treatment adherence, and equitable access to mental health care, particularly in low-resource settings. Therefore, addressing these barriers requires interdisciplinary collaboration, public health education, and the integration of scalable, culturally sensitive, and AI-based mental health innovations. Future research should prioritize multi-omics integration, longitudinal and transdiagnostic studies, biomarker validation, and the real-world implementation of personalized interventions to improve outcomes and quality of life for individuals living with SZ.
Schizophrenia (SZ) ranks among the most serious psychiatric illnesses, marked by significant disruptions in thought, perception, and emotions. It generally appears in early adulthood and is linked to a shorter lifespan, frequently resulting from suicide [1]. It requires long-term management that integrates medical, psychological, and psychosocial interventions [2]. Globally, SZ affects approximately 0.3% to 1% of the population, with no significant difference in prevalence between men and women [3, 4]. Despite its relatively low prevalence, the disease contributes significantly to the global burden of disease, accounting for millions of years lived with disabilities (YLDs) [3, 5]. The prevalence of SZ is higher among migrants than among native-born individuals and is more prevalent in urban areas and among ethnic minorities [2, 4]. The economic burden of SZ is substantial, with indirect costs, such as lost productivity, contributing to 50–85% of the total costs [5, 6]. This disorder affects not only patients but also their families, caregivers, and society, imposing a considerable humanistic burden [7]. A key challenge is the treatment gap, as numerous patients, especially in low- and middle-income countries (LMICs), do not receive sufficient care [8]. Hence, addressing this gap is essential for reducing the socioeconomic impact of SZ and improving healthcare outcomes [8, 9].
The need for updated insights into SZ is driven by the complex and multifactorial nature of the disorder, which involves a combination of genetic, neurobiological, and environmental factors. Recent research has highlighted the importance of revising traditional constructs and diagnostic criteria to incorporate emerging findings and develop more personalized and patient-centered treatment strategies [10–12]. Emerging research on the pathophysiology of SZ has identified key molecular and neural circuit alterations that contribute to the symptoms of the disorder [13, 14]. Additionally, the identification of distinct dimensions of negative symptoms, such as apathy and diminished expression, has provided new insights into their behavioral, cognitive, and neural correlates, which are crucial for developing targeted treatments [15]. Hence, integrating new findings into clinical practice is crucial for enhancing the accuracy of diagnosis and effectiveness of treatment. The use of objective diagnostic and therapeutic tests, informed by recent research, can increase the rigor of clinical assessments and guide the development of more effective treatment plans [16, 17]. By bridging the gaps in our knowledge of the causes, mechanisms, and therapies of SZ, we can advance towards a more accurate and efficient strategy for handling this intricate disorder [10, 18]. Thus, this study aims to provide a comprehensive review of SZ, focusing on its pathophysiology, genetic and neurobiological underpinnings, diagnostic advancements, and emerging therapeutic strategies for its treatment.
We conducted an extensive literature search in PubMed, Scopus, and Google Scholar using Medical Subject Headings (MeSH) in PubMed and carefully selected keywords in the other databases. Search strings combined terms such as “schizophrenia”, “psychotic disorders”, and “antipsychotics” with Boolean operators (AND/OR). We limited the inclusion to peer-reviewed, English-language publications that offered contemporary insights into SZ, spanning neurobiological mechanisms, diagnostic advances, pharmacological and non-pharmacological treatments, and persistent management challenges. Although we did not impose a publication year restriction, we prioritized more recent articles to ensure currency and comprehensiveness. Eligible records included original research, systematic reviews, meta-analyses, narrative reviews, perspectives, and commentaries that addressed the pathophysiology, diagnosis, or treatment of SZ, provided they were available in their full text. Studies that did not align with the review focus were excluded after the screening. We synthesized the evidence narratively using thematic analysis to organize the key findings into coherent themes. The material was organized under headings that collectively provided a structured overview of pathophysiology, diagnosis, treatment, and future prospects. For clarity, the results were grouped into five interrelated domains—advances in understanding the pathophysiology of SZ, advances in diagnosis and early detection, emerging therapeutic approaches, challenges in SZ management, and future directions and research priorities—while adopting an integrative, stratified approach that preserves the interconnected nature of SZ’s biology, clinical presentation, and care pathways, rather than treating these domains as isolated silos.
SZ is a complex psychiatric disorder characterized by genetic and epigenetic factors. Understanding the contributions of these factors is crucial for unraveling the etiology and pathophysiology of this disease.
The genetic basis of SZ has been extensively explored using genome-wide association studies (GWASs), linkage studies, and candidate gene approaches. GWASs are large-scale studies that compare deoxyribonucleic acid (DNA) variants across thousands of individuals to identify risk-associated regions. Specific single-nucleotide polymorphisms (SNPs), which are small changes in a single DNA base associated with SZ, have been identified in diverse populations (Figure 1). For instance, a study in the Arab-Israeli population by Alkelai and others [19] in 2011 identified associations between SZ and SNPs within six genes, including leucine rich repeat flightless interacting protein 1 (LRRFIP1), neuromedin B receptor (LOC645434/NMBR), acyl-CoA synthetase long chain family member 3 (ACSL3), twist family BHLH transcription factor 2 (TWIST2), UDP glucuronosyltransferase family 1 member A1 (UGT1A1), and EF-hand domain family member D1 (EFHD1) (Table 1) [19]. Notably, UGT1A1 and EFHD1 were also associated with SZ in a German case-control sample, although with an opposite allelic effect for EFHD1, suggesting ethnic differences or a potential flip-flop phenomenon [20]. LRRFIP1 is a transcription factor involved in neurogenesis [19], while epidermal growth factor receptor (EGFR), previously linked to SZ, is a growth factor receptor important for cellular signaling and epidermal growth. UGT1A1 regulates drug metabolism, neurotransmitter levels, and toxin excretion, whereas EFHD1 contributes to neuronal differentiation. NMBR influences fear, anxiety, and stress susceptibility in animal models, linking it to psychiatric phenotypes. To illustrate the broader relevance of these associations, Alkelai et al. [19] also found an intronic SNP in the dedicator of cytokinesis 4 (DOCK4) in a Jewish-Israeli cohort. DOCK4 is crucial for neurodevelopment and has been implicated in dyslexia and autism, conditions that share genetic overlap with SZ (Table 1). This finding emphasizes the interconnections between neurodevelopmental disorders.
In European-American populations, studies by Wang et al. [21, 22] linked zinc finger and AT-hook domain containing (ZFAT), a gene involved in immune cell survival and autoimmune diseases, to SZ onset. This supports the idea that immune dysregulation contributes to the risk of SZ. Further evidence comes from autoimmune conditions, such as thyroid disease and multiple sclerosis, which share risk pathways with SZ. Similarly, Bergen et al. [23] examined bipolar disorder (BD) and SZ in a Swedish cohort and identified significant SNPs in the major histocompatibility complex (MHC), emphasizing its role in neurodevelopment and brain connectivity. The Irish SZ Genomics Consortium replicated these findings, identifying calcium voltage-gated channel subunit alpha1 I (CACNA1I), a calcium channel gene. Other calcium channel genes, such as calcium voltage-gated channel subunit alpha1 C (CACNA1C), calcium voltage-gated channel auxiliary subunit beta 2 (CACNB2), and CACNB3, have also been implicated in both SZ and BD, suggesting calcium signaling dysregulation as a shared neurobiological mechanism [24]. Other European studies have highlighted genes with diverse biological roles. For example, Rietschel et al. [25] identified SNPs in autophagy and Beclin 1 regulator 1 (AMBRA1), cut like homeobox 1 (CUX1), vaccinia related kinase 2 (VRK2), and coiled-coil domain containing 68 (CCDC68)/transcription factor 4 (TCF4), with VRK2 later replicated in a Han Chinese sample, strengthening its global relevance and link to SZ (Table 1).
Despite these advances, GWAS findings face limitations due to small effect sizes, the polygenic nature of SZ, and the unclear biological mechanisms. Replication remains a challenge, especially across ethnically diverse cohorts, because the results can be confounded by population stratification and inconsistent study designs [26]. Despite decades of intensive research and billions of dollars invested, genetics has not provided transformative insights into the etiology or treatment of SZ. A recent commentary even argued that “genetics has failed schizophrenia”, as the field remains largely descriptive without producing clinically actionable biomarkers or therapeutic breakthroughs [27]. This critical perspective underscores the need to balance enthusiasm for genomic findings with the recognition of their current limitations.
Genetic and epigenetic contributions to SZ.
| Gene | Function | Findings/Association with SZ |
|---|---|---|
| LRRFIP1 | Transcription factor | Involved in neurogenesis, identified in the Arab-Israeli population |
| NMBR | Neuromodulation | Linked to fear, anxiety, and stress responses |
| UGT1A1 | Drug metabolism & neurotransmitter regulation | Associated with SZ in Arab-Israeli and German populations |
| DOCK4 | Neurodevelopment | Related to dyslexia, autism, and SZ |
| ZFAT | Immune system regulation | Associated with SZ and autoimmune disorders |
| CACNA1C, CACNB230, CACNB327 | Calcium channel genes | Implicated in both SZ and bipolar disorder |
| AMBRA1 | Autophagy & neurotransmission | Impacts memory, anxiety, and prepulse inhibition |
| NDST3 | Synaptogenesis & neurodevelopment | Identified as a SZ risk locus in Ashkenazi Jewish and Han Chinese populations |
| 22q11.2 deletion region (TBX1, GLN1, COMT) | Neurodevelopment & neurotransmitter metabolism | Strongly linked to SZ risk |
| Erbb4, SLC1A3 | Synaptic plasticity | Disruptions linked to cognitive impairments in SZ |
AMBRA1: autophagy and Beclin 1 regulator 1; CACNA1C: calcium voltage-gated channel subunit alpha1 C; CACNB230: calcium voltage-gated channel auxiliary subunit beta 230; COMT: catechol-O-methyltransferase; DOCK4: dedicator of cytokinesis 4; GLN1: glutamine synthetase 1; LRRFIP1: leucine rich repeat flightless interacting protein 1; NDST3: N-deacetylase and N-sulfotransferase 3; NMBR: neuromedin B receptor; SZ: schizophrenia; TBX1: T-box transcription factor 1; UGT1A1: UDP glucuronosyltransferase family 1 member A1; ZFAT: zinc finger and AT-hook domain containing; Erbb4: Erb-b2 receptor tyrosine kinase 4; SLC1A3: solute carrier family 1 member 3.
Beyond broad GWAS findings, several specific genes have been linked to neurodevelopmental and synaptic plasticity pathways relevant to SZ. For example, AMBRA1 is involved in autophagy, neurotransmission, and signal transduction. Mouse studies have demonstrated its influence on memory, anxiety, and prepulse inhibition, which are relevant to the pathophysiology of SZ. Betcheva et al. [28] identified an SZ-associated SNP in Hedgehog acyltransferase (HHAT), which plays a role in carcinogenesis and neurodevelopment. In Ashkenazi Jewish populations, Lencz et al. [29] identified N-deacetylase and N-sulfotransferase 3 (NDST3) as a novel SZ susceptibility locus. This gene, which is highly expressed in the hippocampus and cerebellum, plays a role in axon guidance, synaptogenesis, and neurodevelopment [29]. The association between NDST3 and SZ was later replicated in a Han Chinese sample. Similarly, Goes et al. [30] identified markers in the 22q11.2 deletion syndrome region in Ashkenazi Jewish cohorts, highlighting its association with SZ. This region contains genes such as T-box transcription factor 1 (TBX1), glutamine synthetase 1 (GLN1), and catechol-O-methyltransferase (COMT), which are considered strong SZ-related copy number variations (CNVs) (Table 1) [30].
East Asian studies have also contributed to the understanding of SZ genetics. For example, while Japanese studies by Ikeda et al. [31] (2011), Shi et al. [32] (2011), and Yamada et al. [33] (2011) yielded mixed results, they provided evidence supporting neurogenic locus notch homolog protein 4 (NOTCH4), ornithine aminotransferase (OAT), and sulfotransferase family 6B member 1 (SULT6B1), which are previously linked to SZ risk. Shi et al. [32] (2011) also identified SNPs in Wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1)/fibroblast growth factor receptor 1 (FGFR1), LSM1 homolog, U6 small nuclear ribonucleic acid (RNA) associated (LSM1), BRP44 (also known as SLC25A32, solute carrier family 25 member 32), and DDB1- and CUL4-associated factor 6 (DCAF6), with FGFR1 playing a critical role in neurodevelopment and SZ-like phenotypes in animal models [30]. Microsatellite studies have also provided unique insights. Shibata et al. [34], using pooled Japanese DNA, identified SZ susceptibility regions in solute carrier family 23 member 3 (SLC23A3), cyclic nucleotide phosphodiesterase domain containing 1 (CNPPD1), and family with sequence similarity 134 member A (FAM134A), which exhibit brain-specific expression despite unclear functions. Wong and others [35] identified an SZ-associated SNP near renin binding protein (RENBP), a gene involved in blood pressure and sodium homeostasis. This supports a potential neurobiological connection between SZ and BD, as angiotensin II regulation affects dopaminergic activity [35]. Despite the breadth of findings across populations and gene targets, a critical challenge remains in the inconsistency and lack of replication of certain SNP associations. Differences in sample size, population structure, environmental exposures, and statistical methodologies contribute to these conflicting findings, necessitating greater harmonization of study designs. Additionally, many studies remain associative rather than mechanistic, limiting our understanding of the functional consequences of the identified loci. Integrating multi-omics approaches (e.g., transcriptomics, epigenomics, and proteomics) with imaging and behavioral phenotypes could provide deeper mechanistic insights [36]. However, it is increasingly clear that genetic studies alone will not provide a unifying model for SZ. The field may need to pivot from a purely gene-discovery framework toward integrative models that account for gene-environment interactions, epigenetic regulation, and developmental trajectories to generate clinically meaningful advances.
Although many implicated genes are involved in neurodevelopment, immune regulation, and synaptic plasticity, there is still no unified model that explains how these mechanisms converge to produce the clinical spectrum of SZ. Current evidence supports multiple overlapping hypotheses: the dopamine hypothesis (positive symptoms), glutamate hypothesis (cognitive and negative symptoms), and the neurodevelopmental model. Each is partly supported by genetic evidence, but none is solely sufficient [37, 38]. These findings collectively underscore the intricate genetic and epigenetic basis of SZ. The heterogeneity of SZ genetics is evident in the discovery of risk-associated SNPs across various populations. Moreover, the role of epigenetic mechanisms in SZ susceptibility is highlighted by genes involved in immunological responses, cellular signaling, and neurotransmission. However, the interpretation of epigenetic data remains complicated by methodological limitations, such as tissue specificity, postmortem sample variability, and a lack of longitudinal data. The interplay between genetic and epigenetic mechanisms in the presence of environmental risk factors, such as prenatal infections, urban living, or psychosocial stressors, remains poorly understood, marking a major gap in translational research [39, 40].
Furthermore, GWAS has identified numerous genetic loci associated with SZ, highlighting its polygenic nature. Notably, 108 chromosomal regions have been linked to the risk of SZ [41]. Key genetic markers identified include regions associated with genes such as B-cell CLL/lymphoma 11B (BCL11B), chromodomain helicase DNA binding protein 7 (CHD7), and E1A binding protein P300 (EP300), which are implicated in cognitive functions and may contribute to the cognitive deficits observed in SZ [41]. Additionally, genes such as ankyrin 3 (ANK3) and NDST3 have been implicated in both SZ and BD, suggesting a shared genetic basis for these psychiatric conditions [42]. Epigenetic mechanisms, including DNA methylation, histone modifications, and non-coding RNAs, play a significant role in SZ by regulating gene expression without altering DNA sequences. These modifications can be influenced by both genetic predisposition and environmental exposure [43–45]. For example, DNA methylation and histone modification patterns have been observed in the prefrontal cortex of individuals with SZ, affecting genes involved in neurotransmission and neurodevelopment [46, 47], whereas miRNAs show potential as biomarkers for SZ, with differential expression patterns linked to the disorder [45, 48].
Environmental factors, such as maternal immune infection, childhood trauma, and substance exposure, can lead to epigenetic changes that influence the risk of SZ. These factors interact with an individual’s genetic makeup, potentially leading to the development of the disorder [49, 50]. However, the temporal sequence and causal relationships among these factors remain unclear. Most epigenetic findings are correlative, lacking mechanistic validation in human models, and the reversibility or permanence of these marks in the human brain is still speculative. Future studies integrating longitudinal epigenomic profiling with neuroimaging and behavioral assessments may clarify the dynamic interactions underlying disease progression [51]. Epigenetic modifications serve as molecular “scars” from environmental exposure, which can affect brain function throughout life and may even be inherited across generations [45, 49]. This perspective not only highlights the risks but also opens potential therapeutic avenues. Drugs targeting epigenetic mechanisms or lifestyle interventions, such as stress reduction and nutrition, may eventually be used to modify the disease trajectory, although this remains an underexplored frontier.
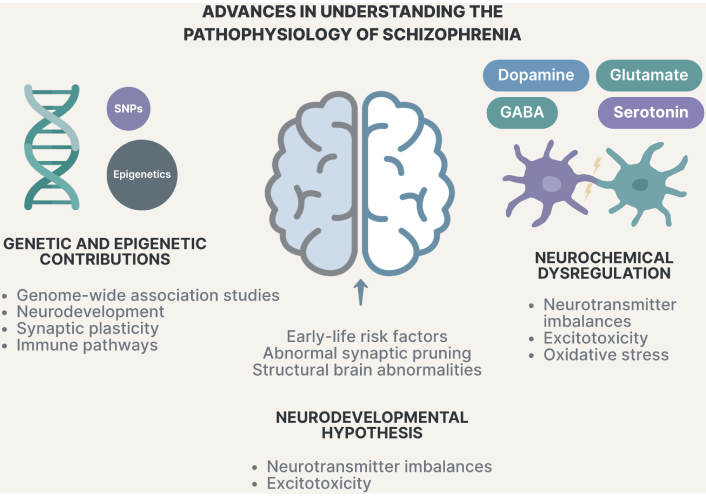
Advances in understanding the pathophysiology of schizophrenia. SNPs: single-nucleotide polymorphisms; GABA: gamma-aminobutyric acid.
The neurodevelopmental hypothesis proposes that disruptions in brain development during early life contribute to the onset of SZ. These disruptions can include structural and functional abnormalities, as well as prenatal risk factors [52]. The widely cited “two-hit” model explains that perinatal insults may create a latent vulnerability, which is later unmasked during adolescent synaptic pruning. This process is often preceded by prodromal symptoms, such as cognitive deficits or social withdrawal [53]. Several clinical frameworks have been developed to improve early detection. For example, McGorry’s clinical staging model and the “basic symptoms” paradigm provide structured ways to identify individuals at high risk of psychosis and to guide preventive interventions [54, 55]. Prodromal signs, including mild cognitive or motor impairment, suggest gene-environment interactions during critical developmental stages [56, 57]. Twin studies estimate the heritability of SZ at 60–80%, but they also emphasize the importance of gene-environment interaction [58]. GWAS have identified 287 loci implicating cortical interneurons and synaptic plasticity genes in SZ, supporting abnormal synapse formation/pruning during development [59–61]. However, most loci confer a modest risk and often lack replication across different ethnic groups [62]. In addition, polygenic risk scores (PRSs), although promising, currently have limited predictive value for clinical use [63, 64].
Structural brain changes also provide evidence supporting the neurodevelopmental hypothesis. These include ventricular enlargement and alterations in the medial temporal and frontal lobes [65]. Prenatal immune challenges, such as maternal infections, can disrupt dopamine system development, which may lead to psychosis in later life [66]. The complement component 4A (C4A) locus has been linked to excessive synaptic pruning, while CNVs in neurexin 1 (NRXN1) and 22q11.2 deletion syndrome highlight overlapping developmental pathways between SZ and autism [67–70]. Induced pluripotent stem cell (iPSC) models have further shown that genes such as NRXN1 and zinc finger protein 804A (ZNF804A) can affect neuronal excitability and dendritic structure, although reproducibility remains a challenge [71–76]. By combining genomic, transcriptomic, and cellular models, researchers may gain a clearer insight into how genes and the environment interact across the lifespan [77, 78]. Epigenetic influences include prenatal complications and hypoxia, with genes such as AKT serine/threonine kinase 1 (AKT1), brain-derived neurotrophic factor (BDNF), dystrobrevin-binding protein 1 (DTNBP1), and glutamate metabotropic receptor 3 (GRM3) interacting with obstetric problems [79, 80]. Up to 50% of SZ candidate genes are hypoxia-regulated [81]. CNVs affecting axonal guidance and synaptic genes are more common in SZ, although penetrance is variable, suggesting environmental modifiers [82–85]. Placental genomic risk scores have recently been used to link prenatal pathology to SZ vulnerability, especially in male offspring [86, 87]. Obstetric complications, including hypoxia, preeclampsia, and low birth weight, approximately double the risk of SZ [88–91].
Moreover, prenatal viral infections and maternal immune activation (MIA) disrupt neuronal differentiation and white matter, leading to long-term deficits [92–98]. The COVID-19 pandemic [severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)] has raised new concerns regarding possible future neurodevelopmental outcomes in exposed populations [52, 99]. Prenatal stress and nutritional deficiencies (e.g., vitamin D or folate deficiency) also increase the risk of SZ [94–96]. Early life adversity, including neglect and abuse, can interact with genetic risk pathways [100, 101]. Genes such as Erb-b2 receptor tyrosine kinase 4 (Erbb4) and solute carrier family 1 member 3 (SLC1A3) provide mechanistic links between MIA and impaired synaptic plasticity [102]. Taken together, these findings suggest that reconciling classical neurochemical models with developmental perspectives is essential. Dopamine dysregulation may help explain positive symptoms, whereas N-methyl-D-aspartate (NMDA) receptor hypofunction appears to underlie cognitive and negative symptoms of SZ. Prenatal infections and other environmental triggers likely act in combination with genetic predisposition, driving immune activation and abnormal neurodevelopment [103, 104].
Comparative table of competing hypotheses in SZ.
| Hypothesis | Key neurobiological mechanism | Supporting evidence | Limitations |
|---|---|---|---|
| Dopamine hypothesis | Dysregulated dopamine signaling (↑ mesolimbic; ↓ mesocortical) | Antipsychotic efficacy targeting D2 receptors; PET imaging; genetic modulation (e.g., COMT) | Does not fully explain cognitive or negative symptoms; not all patients respond to D2 antagonists |
| Glutamate hypothesis | NMDA receptor hypofunction leading to excitotoxicity | NMDA antagonists (PCP, ketamine) mimic SZ symptoms; synaptic pruning disruptions | Clinical translation is inconsistent; poor response to glutamatergic modulators in human trials |
| Inflammatory hypothesis | Elevated cytokines (e.g., IL-6, IFN-γ) affect neurotransmitter systems | Elevated inflammatory markers in FEP and relapsed patients; cytokine-genetic interactions | Biomarker variability; confounding by treatment/phase; few validated causal mechanisms |
| Neurodevelopmental model | Early disruptions in brain development lead to latent vulnerability | Structural brain abnormalities; “two-hit” model; placental gene expression; CNVs | Low specificity; many early-life insults do not lead to SZ without other risk factors |
| Kynurenine hypothesis | Excess KYNA impairs NMDA signaling and alters dopamine balance | Elevated KYNA in CSF/brain; linked to cognitive symptoms; cytokine-driven pathway shifts | Poor spatial resolution; limited in vivo tracking; unclear therapeutic windows |
| Gut-brain axis | Dysbiosis affects glutamate/GABA/tryptophan metabolism and neuroinflammation | Correlation with symptom severity; links to immune response, barrier integrity, and neurotransmitter levels | Limited replication; confounded by diet, medication, and environment |
CNVs: copy number variations; COMT: catechol-O-methyltransferase; CSF: cerebrospinal fluid; FEP: first-episode psychosis; GABA: gamma-aminobutyric acid; IFN-γ: interferon-gamma; IL-6: interleukin-6; KYNA: kynurenic acid; NMDA: N-methyl-D-aspartate; PET: positron emission tomography; SZ: schizophrenia.
Neurochemical dysregulation in the brain involves complex interactions among various neurotransmitters and pathways. Beyond the well-known role of dopamine, emerging research highlights the significance of glutamate, gamma-aminobutyric acid (GABA), serotonin, and the kynurenine pathway in neuroinflammation and neurodegenerative diseases (Tables 2 and 3) [105]. An imbalance between glutamate and GABA can lead to neurodegeneration by disrupting the synaptic strength. This imbalance is not only a neuronal issue but also involves glial cells, which modulate neuroinflammation and contribute to neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases (Figure 1) [106, 107]. Glutamate, the primary excitatory neurotransmitter in the brain, plays a crucial role in synaptic plasticity, learning, and memory by acting on ionotropic and metabotropic glutamate receptors and is implicated in various neuropsychiatric disorders. Dysregulation of glutamate neurotransmission can lead to neuro-excitotoxicity, affecting neuronal plasticity and growth, and is linked to conditions such as depression and cognitive impairments [107]. The glutamate hypothesis of SZ emerged from observations that NMDA receptor antagonists, such as ketamine and phencyclidine (PCP), can induce both positive and negative symptoms, as well as cognitive deficits similar to those seen in patients with SZ. This suggests that NMDA receptor hypofunction may contribute to the disorder by disrupting excitatory neurotransmission and synaptic plasticity in the brain. Despite its experimental support, the glutamate hypothesis faces limitations in clinical translation due to heterogeneity in patient populations and dosage sensitivity in clinical trials (Table 2) [108, 109]. Astrocytes play a critical role in glutamate regulation by controlling the synaptic uptake and recycling of glutamate. In SZ, astrocytic dysfunction leads to impaired glutamate uptake and excessive extracellular glutamate accumulation, contributing to excitotoxicity and neuronal damage. Protoplasmic astrocytes, which form astrocytic cradles around synapses, are particularly affected by this process. Disrupted glutamate transporter function in astrocytes leads to prolonged glutamate signaling, exacerbating NMDA receptor dysfunction and synaptic instability [110, 111]. Inflammatory mediators drive A1 astrocyte activation in SZ, leading to reduced glutamate clearance, oxidative stress, and excitotoxic neuronal injury. This imbalance between glutamate clearance and excessive glutamate release in SZ contributes to excitotoxicity, which damages neuronal circuits involved in cognition, emotion regulation, and executive function. Oxidative stress in SZ is linked to the abnormal activation of the complement cascade by A1 astrocytes, amplifying neuroinflammation and synaptic damage [111, 112]. This results in disrupted neuroplasticity and synapse loss in the prefrontal cortex and hippocampus, impaired working memory, executive function, and learning abilities, and increased vulnerability to environmental stressors and neurodevelopmental abnormalities [105].
Neurodevelopmental, neurochemical, and environmental contributions to SZ pathogenesis.
| Variable | Impacts on SZ |
|---|---|
| Developmental factor | |
| Prenatal insults (hypoxia, maternal infections) | Increases risk by altering neurodevelopmental pathways |
| Synaptic pruning abnormalities | Leads to disrupted neuronal connectivity and increased vulnerability to psychosis |
| Structural brain abnormalities | Enlarged ventricles, changes in the frontal and temporal lobes |
| C4A locus in the MHC region | Implicated in excessive synaptic pruning |
| Copy number variations (CNVs) | Linked to SZ and developmental disorders |
| Induced pluripotent stem cell (iPSC) models | Demonstrate how NRXN1 deletions disrupt synaptic function |
| Neurochemical dysregulation factor | |
| Glutamate (NMDA receptor dysfunction) | Leads to excitotoxicity and cognitive impairments |
| GABAergic dysfunction | Disrupts the excitatory-inhibitory balance |
| Dopamine (mesolimbic and mesocortical pathways) | Excess in the mesolimbic system (positive symptoms), deficit in the mesocortical system (negative symptoms) |
| Kynurenine pathway | Increased kynurenic acid disrupts glutamate and dopamine neurotransmission |
| Cytokine-mediated neuroinflammation | Elevated IL-6, TNF-α, and IFN-γ contribute to neurotransmitter dysregulation |
| Environmental risk factors | |
| Prenatal maternal infection (influenza, CMV, rubella, Toxoplasma gondii) | Triggers immune activation leading to neurodevelopmental disruption |
| Obstetric complications (hypoxia, preeclampsia, emergency C-section) | Increases SZ risk by impairing brain development |
| Maternal stress & malnutrition | Affects fetal brain development and neurotransmitter synthesis |
| Early-life adversity (childhood trauma, abuse, neglect) | Linked to increased risk of SZ and substance use disorder |
| Gut dysbiosis & microbiota alterations | Affects neurotransmitter balance and neuroinflammation |
C4A: component 4A; GABA: gamma-aminobutyric acid; IFN-γ: interferon-gamma; IL-6: interleukin-6; MHC: major histocompatibility complex; NMDA: N-methyl-D-aspartate; NRXN1: neurexin 1; SZ: schizophrenia; TNF-α: tumor necrosis factor-alpha.
In contrast to the glutamatergic model, dopaminergic dysregulation contributes to the development of positive, negative, and cognitive symptoms of SZ. The classical model suggests that hyperactive dopaminergic transmission, particularly due to increased dopamine D2 receptor sensitivity, underlies the symptoms of SZ (Tables 3 and 4) [113]. However, the modern dopamine hypothesis proposes a more nuanced model in which low dopamine levels in the mesocortical pathway contribute to negative and cognitive symptoms, whereas hyperactive dopamine transmission in the mesolimbic pathway drives positive symptoms (Table 4). A unifying hypothesis further integrates genetic, environmental, and immunological factors that collectively alter striatal dopamine signaling, leading to the development of psychosis [114, 115]. The dopamine hypothesis is strongly supported by antipsychotic drug efficacy, which predominantly targets D2 receptors but fails to fully explain the spectrum of cognitive and negative symptoms of SZ. Inflammatory cytokines, such as interferon-gamma (IFN-γ) and interleukin-6 (IL-6), alter dopamine availability by affecting its synthesis, transport, and receptor binding. Clinical studies have shown that chronic inflammation in SZ may drive dopamine dysregulation by depleting tetrahydrobiopterin (BH4), altering the neurotransmitter balance, and increasing oxidative damage [116]. Pharmacological targeting of dopamine dysregulation involves D2 receptor blockade, vesicular monoamine transporter 2 (VMAT2) inhibition, and tyrosine hydroxylase (TH) inhibition. However, dopamine-targeted treatments alone do not fully address cognitive and negative symptoms, suggesting a need for multi-targeted interventions that also address neuroinflammation and oxidative stress [105]. Although both glutamate and dopamine models offer valuable insights, few studies have systematically compared their predictive validity across clinical subtypes of SZ. Future integrative models must reconcile these hypotheses by considering neurotransmitter interactions, differential circuit vulnerabilities, and individual patient profiles [117]. The cytokine hypothesis of SZ suggests that elevated levels of pro-inflammatory markers, such as IL-6, tumor necrosis factor-alpha (TNF-α), IFN-γ, IL-1β, and IL-8, contribute to altered neurotransmitter function, oxidative stress, and structural brain changes, ultimately leading to the development of SZ symptoms [7, 118]. Cytokines, which regulate immune responses and brain development, are significantly altered in patients with SZ. Studies have shown persistently elevated inflammatory markers in patients experiencing first-episode psychosis (FEP) and acute relapses, particularly in medication-naïve patients [119, 120]. Cerebrospinal fluid (CSF) analyses further support this link, revealing elevated concentrations of IL-6, IL-1β, TNF-α, and kynurenic acid (KYNA) in both relapsed and first-episode patients with epilepsy [121, 122]. The specificity and causality of inflammatory biomarkers in SZ remain inconsistent, with some studies confirming elevated cytokine levels, while others report variability based on medication status, illness phase, and methodological differences, necessitating the development of standardized biomarker panels and longitudinal studies [123, 124].
The kynurenine pathway, a major route of tryptophan metabolism, is closely associated with neuroinflammation. Dysregulation of this pathway can lead to an imbalance in neuroactive metabolites, contributing to neurodegenerative diseases and psychiatric disorders (Table 2) [125, 126]. Alterations in the kynurenine pathway can affect serotonin levels and influence mood and anxiety disorders. The metabolites of this pathway can act as endogenous anxiogens or anxiolytics, affecting anxiety and depression [127, 128]. The kynurenine hypothesis of SZ suggests that elevated levels of KYNA, a glutamate receptor antagonist, contribute to the positive symptoms of SZ. Increased KYNA concentrations in the CSF and brain tissue of patients with SZ suggest that dysregulation of the kynurenine pathway of tryptophan metabolism plays a crucial role in the disorder [129]. While evidence from postmortem and CSF studies supports KYNA elevation, limitations in spatial resolution and potential postmortem artifacts weaken causal inference. Human in vivo imaging of kynurenine metabolites remains challenging and is underdeveloped. Tryptophan metabolism is predominantly directed towards the kynurenine pathway, with 90% of tryptophan being converted into kynurenine-derived metabolites. Inflammatory cytokines such as IFN-γ, IL-6, and TNF-α induce enzymes that shift tryptophan metabolism towards KYNA production (Table 3) [105, 130]. The knowledge gap in SZ pathophysiology is identifying neuroprotective metabolites within the kynurenine pathway and understanding the regional specificity of KYNA accumulation and its interaction with glutamatergic and dopaminergic systems in different brain areas [131]. Targeting the kynurenine pathway and its metabolites offers potential therapeutic strategies for managing neuroinflammation and related disorders such as AD. Modulating this pathway could help reduce neurotoxic metabolites and enhance neuroprotective metabolites [126, 132]. However, the translation of these findings to clinical settings is still in its infancy, with limited trials on kynurenine modulators and insufficient data on their long-term safety and efficacy [133]. Gut dysbiosis, an imbalance in the gut microbiota, is linked to neurochemical dysregulation in SZ by affecting glutamate, GABA, and tryptophan metabolism, leading to dopaminergic overactivity and psychosis [134, 135]. The gut-brain axis, a bidirectional communication network, is critical for neurotransmitter regulation and immune responses. Disruptions in this axis, as seen in SZ, lead to increased gut permeability, systemic inflammation, and neuroinflammation, exacerbating neurotransmitter imbalance. This leads to an impaired excitatory-inhibitory balance in the brain and excitotoxicity (Table 3) [105, 136]. Microglia, the primary immune cells in the brain, play a crucial role in neuroinflammation and are implicated in SZ pathogenesis. Microglial activation can lead to excessive synaptic pruning and neuronal damage, contributing to the cognitive and structural brain changes observed in patients with SZ [137–139]. Elevated levels of pro-inflammatory cytokines and reduced levels of anti-inflammatory cytokines have been reported in SZ, suggesting an imbalance that may exacerbate neuroinflammation and contribute to the symptoms of the disorder [140–142]. Genetic factors, such as variations in the complement system, may also influence microglial activity and cytokine production, further linking immune dysregulation to SZ [137, 143]. Patients with SZ often exhibit altered gut microbiota composition, which may correlate with symptom severity and treatment response [144, 145]. The microbiome influences the adaptive immune system, including T-cell and B-cell function, which can impact neuroimmune regulation and contribute to the pathophysiology of the disorder [145]. Current SZ models often oversimplify neurotransmitter systems, neuroinflammation, and gut-brain axis dysregulation, failing to account for patient heterogeneity. This necessitates future studies using integrative multi-omics approaches and longitudinal designs to untangle the causality and develop precision-targeted therapies [146].
Mapping core symptoms to implicated neural circuits and neurotransmitters.
| Symptom category | Key symptoms | Implicated neural circuits | Associated neurotransmitters/mechanisms |
|---|---|---|---|
| Positive symptoms | Hallucinations, delusions | Mesolimbic pathway (e.g., nucleus accumbens, amygdala) | ↑ Dopamine (D2 receptor hyperactivity), NMDA receptor hypofunction |
| Negative symptoms | Avolition, anhedonia, affective flattening | Mesocortical pathway (e.g., prefrontal cortex) | ↓ Dopamine, ↓ NMDA receptor function, ↑ KYNA |
| Cognitive symptoms | Working memory deficits, executive dysfunction | Prefrontal cortex, hippocampus | ↓ Glutamate, ↑ KYNA, ↓ GABA, neuroinflammation |
| Affective symptoms | Anxiety, depression, mood instability | Amygdala, hypothalamus, limbic system | Serotonin, dopamine imbalance, and inflammatory cytokines |
| Motor symptoms | Catatonia, abnormal movements | Basal ganglia, cerebellum | Dopamine dysregulation, oxidative stress, and synaptic dysfunction |
↑: Increase, hyperactivity, or elevated function/levels of the specified neurotransmitter or mechanism; ↓: decrease, hypoactivity, or reduced function/levels of the specified neurotransmitter or mechanism. GABA: gamma-aminobutyric acid; KYNA: kynurenic acid; NMDA: N-methyl-D-aspartate.
Recent advances in neuroimaging have improved early SZ detection, with dopamine hyperactivity, NMDA receptor hypofunction, and hippocampal hyperactivity identified as potential biomarkers [147]. Combining structural magnetic resonance imaging (MRI) with neuropsychological testing may enhance diagnostic accuracy [142], whereas diffusion tensor imaging (DTI) has revealed increased mean diffusivity in early stage SZ, indicating structural abnormalities [148]. Positron emission tomography (PET) and magnetic resonance spectroscopy (MRS), although limited diagnostically, contribute to understanding the pathophysiology [149]. Machine learning (ML) applied to neuroimaging enables single-subject biomarker detection [147–150]. Neuroimaging consistently shows reduced hippocampal and prefrontal volumes in patients with SZ [151]. Potential neurochemical biomarkers include BDNF, dopamine, GABA, IL-1β, and IL-6, which inform drug development aimed at restoring synaptic plasticity [152]. Early phase drug development is exploring agents targeting BDNF signaling and glutamatergic balance, with default network anomalies and gamma band deficits proposed as translational biomarkers [153]. Additional candidates include proteomics, miRNAs, antibodies, the gut microbiome, and blood-based neuroinflammatory markers for diagnosis and patient stratification [154, 155].
Multiple molecular and metabolic biomarkers, such as D-serine, S100 calcium-binding protein B (S100B), dopamine receptor D3 messenger RNA (DRD3 mRNA), AKT1, G72 (also known as D-amino acid oxidase activator, DAOA), oxidative stress enzymes, and total antioxidant status, are under investigation [156–158]. These may help identify high-risk individuals and track treatment responses, especially in prodromal or FEP [157]. Artificial intelligence (AI) and deep learning applied to MRI, electroencephalography (EEG), and clinical data achieved > 80% prediction accuracy [159, 160]. ML models have been used to analyze neuroimaging, speech, behavior, and actigraphy, and forecast SZ symptoms and outcomes, such as suicidality and depression [161, 162]. Linguistic markers, semantic incoherence, and natural language processing (NLP) approaches show promise for automated SZ classification [163, 164], although model interpretability and dataset limitations remain [4]. Digital technologies, such as smartphone-based algorithms, can predict symptom trajectories with ± 1.45 Brief Psychiatric Rating Scale (BPRS) accuracy and facilitate cognitive assessment [153, 164]. Wearable and mobile tools enable remote monitoring, relapse prediction, and real-world functional evaluation [165–167]. These scalable approaches can enhance early detection, personalized treatment, and access to mental health care, especially in underserved populations [168, 169].
Trace amine-associated receptor 1 (TAAR1) agonists offer potential advantages over conventional dopamine D2 receptor (D2) antagonists as new therapies for SZ [170, 171]. Ulotaront, a TAAR1 full agonist with 5-hydroxytryptamine 1A (5-HT1A) receptor activity, has shown efficacy for both positive and negative symptoms without D2 receptor inhibition, instead modulating the glutamatergic, serotonergic, and dopaminergic systems [170, 172]. It is in phase 3 trials and is designated as an FDA breakthrough therapy, while ralmitaront, another TAAR1 agonist, is in phase 2 studies [171, 172]. Novel pharmacological strategies are being developed to address the limitations of current antipsychotics. Although psychedelics such as lysergic acid diethylamide (LSD) and psilocybin, through serotonin-glutamate interactions, have been studied for their potential effects on neural plasticity and mood regulation, recent critiques caution against the notion of a “psychedelic renaissance”, emphasizing that current evidence is preliminary, inconsistent, and potentially overstated [173–175]. Glutamatergic drugs, such as memantine and D-cycloserine, target negative symptoms and cognitive deficits [176]. Other emerging treatments include pimavanserin (negative symptoms), lumateperone and cannabidiol (total symptoms), and BI 425809 (cognition), in addition to long-acting injectable formulations for improved adherence [177]. There is a growing focus on neuroprotective and anti-inflammatory strategies due to the evidence of microglial hyperactivation and cytokine imbalances in SZ [178, 179]. Investigated adjuncts include biologics targeting immune mediators, antioxidants, non-steroidal anti-inflammatory drugs (NSAIDs), and agents such as aspirin, minocycline, COX-2 inhibitors, and omega-3 fatty acids, all of which have shown promising clinical results [179, 180]. Antidepressants, mood stabilizers, and certain antipsychotics have also demonstrated neuroprotective effects [181]. Nonetheless, while preliminary evidence is promising, many of these findings, particularly in relation to neuroinflammatory and immune-modulatory approaches, remain associative, and larger confirmatory trials are needed before they can be translated into routine clinical practice in patients with SZ. Additional approaches under study include muscarinic agonists, glycine modulators, and neurosteroid-based interventions to address cognitive and negative symptoms resistant to D2-based therapies [182].
The goal of personalized medicine in the treatment of SZ is to modify antipsychotic medications according to each patient’s unique genetic profile, which may increase effectiveness and decrease side effects (Figure 2) [183]. Particularly for clozapine in treatment-resistant SZ, pharmacogenomic methods can assist in identifying genetic markers linked to the medication response and tolerability [184]. Genes related to pharmacological processes, metabolism, transport, pleiotropic effects, and disease pathogenesis are important areas of investigation [185]. The metabolism and effectiveness of antipsychotics are greatly affected by genetic differences in cytochrome P450 enzymes, particularly cytochrome P450 family 2 subfamily D member 6 (CYP2D6), cytochrome P450 family 1 subfamily A member 2 (CYP1A2), and cytochrome P450 family 3 subfamily A member 4 (CYP3A4) [185]. Although pharmacogenomics offers intriguing options for customized therapy, larger multicenter trials are required to produce clinically useful genetic tests [183]. Incorporating genetic data with clinical information may result in more accurate treatment plans and better results for managing SZ [186].
By tailoring medicine selection and dosage according to a patient’s genetic profile, pharmacogenomics in psychiatry seeks to enhance treatment results and minimize side effects [187, 188]. Considering both pharmacokinetic and pharmacodynamic parameters, this method integrates pharmacology and genetics to inform precision mental health treatments [189, 190]. Despite initial hope, obstacles, including inconsistent definitions of treatment results and a dearth of extensive replication studies, have impeded progress [189]. Nonetheless, psychiatric pharmacogenomics has the potential to embark on a new stage of investigation and application due to developments in genomics and health informatics [189]. Importantly, although pharmacoepigenomics, pharmacomicrobiomics, and pharmacometabolomics are often highlighted as future directions, the evidence supporting their clinical utility remains preliminary and largely correlational. Accordingly, these approaches should be regarded as promising, yet experimental, avenues rather than established practices. To further customize treatment plans for mental illnesses, precision psychiatry may include pharmacogenomics using cutting-edge technologies such as pharmacoepigenomics, pharmacomicrobiomics, and pharmacometabolomics [190]. Emerging approaches in precision psychiatry also involve biotyping, which categorizes patients based on neurobiological markers, symptom clusters, or genetic profiles to match them with the most appropriate treatment. The integration of ML into this framework facilitates the discovery of clinically meaningful subtypes of SZ [155].
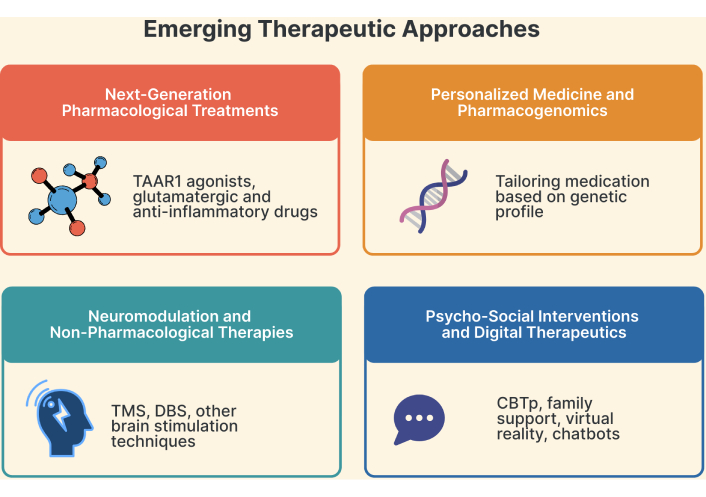
Emerging therapeutic approaches. TAAR1: trace amine-associated receptor 1; TMS: transcranial magnetic stimulation; DBS: deep brain stimulation; CBTp: cognitive behavioral therapy for psychosis.
Transcranial magnetic stimulation (TMS) and transcranial direct current stimulation (tDCS) are two neuromodulation methods that have shown promise as non-pharmacological treatments for a range of neurological and psychiatric disorders (Figure 2) [191]. This non-invasive brain stimulation technique may be probed and influenced by the central nervous system, which may have therapeutic uses in neurology, neurophysiology, cognitive neuroscience, and psychiatry [192]. Cognitive, social, behavioral, and emotional symptoms of autism spectrum disorder may be improved by TMS and tDCS, which target certain brain areas [193]. Compared to monotherapies, TMS or tDCS has shown improved efficacy in reducing symptoms of major depressive illness when combined with psychotherapy, especially mindfulness techniques, and specific drugs such as citalopram and sertraline [194]. However, in the context of SZ, current evidence indicates that TMS and deep brain stimulation (DBS) have limited or no established therapeutic benefits. Instead, electroconvulsive therapy (ECT) remains the only neuromodulation technique with proven clinical efficacy in SZ, particularly in treatment-resistant cases [195].
DBS is a new therapeutic option for SZ that is resistant to existing treatments. Numerous brain targets have been investigated, including the habenula, substantia nigra pars reticulata, subgenual anterior cingulate cortex, and nucleus accumbens [196, 197]. Some patients’ symptoms improved after stopping stimulation, according to a preliminary randomized crossover experiment [196]. According to a systematic review, nine out of ten stimulated patients saw a decrease in symptoms, and the adverse effects were similar to those of other DBS applications [197]. Evidence suggesting that SZ is a circuit illness responsive to striatal manipulation supports the justification for DBS in this condition [198]. Nonetheless, the overall body of evidence remains inconclusive, and DBS should be regarded as highly experimental and not a recommended treatment option for SZ at this stage of research. To completely understand the processes, predictors of improvement, and best ways to combine neuromodulation and non-pharmacological approaches with other treatments, further research is required using larger samples, randomized controlled designs, and standardized protocols. Moreover, neuromodulation can be refined through real-time neurofeedback, closed-loop stimulation, and integration with AI-driven systems that personalize stimulation parameters based on symptom fluctuations or brain state biomarkers [199]. Finally, caution is warranted when integrating AI into therapeutic strategies. AI systems may inadvertently induce psychotic-like experiences in otherwise healthy individuals, and there is also the risk of AI itself generating psychotic content when the data are incomplete. These safety considerations must be carefully addressed before widespread clinical adoption of AI-assisted interventions [200, 201].
To address enduring symptoms and enhance functioning, psychosocial interventions, such as cognitive behavioral therapy for psychosis (CBTp), are essential supplements to pharmaceutical treatment for SZ [202, 203]. By improving reality testing and belief assessment, CBTp and metacognitive training assist patients in comprehending and managing positive symptoms [204]. Family participation, psychoeducation, supported employment, and lifestyle modification are additional successful treatments [203]. To overcome resource constraints, digital interventions, such as serious video games and smartphone applications, present promising ways to expand access to psychosocial therapies [203, 205]. For the best results, these psychosocial strategies must be combined with early medication and all-encompassing care to encourage recovery and general well-being in people with SZ [203].
Virtual reality (VR) is a potential method for psychosocial therapy in patients with SZ. Research has demonstrated that VR-based treatments can improve social skills, emotional perception, and job interview ability [206, 207]. VR therapies have been shown to improve social cognition, negative symptoms, and social functioning [207, 208]. VR-based social cognition and interaction training (VR-SCIT) has demonstrated benefits in enhancing emotion perception and metacognition, with greater treatment compliance than traditional therapies [208]. Additionally, VR applications have been demonstrated to be safe and effective for assessing and treating social and cognitive deficiencies in people with SZ and for streamlining conventional rehabilitation programs, despite obstacles, including high prices and maintenance requirements [209].
Recent studies have highlighted the promise of digital and AI-driven therapies for managing SZ. Maximizing treatment outcomes can be achieved by combining antipsychotic medications with psychosocial therapies [203]. Particularly in times of provider shortage, digital therapies, such as chatbots and smartphone applications, offer potential solutions for better access to mental health treatment [210]. These tools can assist with psychoeducation, symptom monitoring, and diagnosis [210]. Although a thorough evaluation of the therapeutic alliance and ethical implications is required, AI-driven psychotherapy has the potential to improve the treatment of several mental health conditions, including SZ [211]. SchizoBot, an AI-powered chatbot that administers cognitive behavioral therapy for the management of SZ, is an example of this, with excellent accuracy in preliminary testing [212]. These digital treatments provide accessible and affordable substitutes for conventional therapy, which may enhance patient outcomes and assist medical professionals in therapy delivery [212]. An emerging frontier is digital phenotyping, which uses smartphone sensor data (e.g., location, speech, sleep, and typing speed) to passively monitor behavioral patterns and predict relapse or treatment response. Combined with wearable technologies and remote assessments, this approach enables continuous and ecologically valid tracking of patient states, which is critical for timely intervention [213].
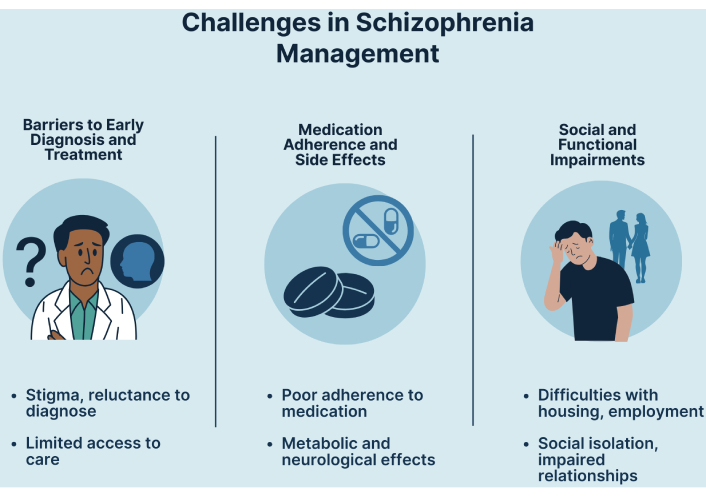
A major obstacle to early identification is the lack of clarity surrounding the diagnosis of SZ. Clinicians are frequently cautious when diagnosing SZ, particularly in the absence of certain risk indicators. The stigma attached to SZ contributes to this reluctance, as individuals may initially be misdiagnosed or have their diagnosis delayed because they are initially labeled as having mood or anxiety problems (Figure 3) [214, 215]. This diagnostic shift can result in significant delays in appropriate treatment, which is crucial for improving long-term outcomes [216, 217]. Treatment-related barriers play a significant role in managing early-onset SZ. Additionally, the side effects associated with antipsychotic medications can be particularly severe in younger populations, contributing to premature mortality and complicating treatment adherence [218]. Societal factors also contribute to the barriers faced in early diagnosis and treatment of the disease. The stigma surrounding mental health disorders, particularly SZ, can lead to a lack of understanding and support from healthcare providers and the public [219]. This stigma not only affects patients’ willingness to seek help but also influences clinicians’ attitudes towards diagnosis and treatment decisions [219].
In many low-resource settings, mental health services are underfunded, resulting in shortages of trained personnel and necessary resources. In Sierra Leone, district services are dependent on discretionary funding from District Health Management Teams, while psychiatric hospitals receive support only from the Freetown City Council [220]. The current dearth of trained individuals is exacerbated by the lack of funding, which restricts the availability of services and affects the hiring and retention of qualified mental health professionals [221, 222]. Furthermore, the integration of mental health services into primary healthcare systems is frequently inadequately executed, resulting in fragmented care. For instance, a study conducted in Liberia found that the lack of qualified mental health professionals significantly restricts the country’s ability to address the population’s mental health needs [221]. Effective clinical care is further hampered by this fragmentation, which is exacerbated by undertrained medical personnel who do not know enough about mental illnesses [223]. Moreover, the general lack of knowledge about mental health concerns leads to the belief that mental health treatment is not important, which influences government funding for such services [224]. Additionally, the general public’s ignorance of mental health concerns feeds the belief that mental health care is not important, which influences government funding for these services [225].
Long-term use of antipsychotic medications has been linked to significant benefits in relapse prevention and symptom control [226]. However, long-term use of antipsychotic medications is not without drawbacks, as many patients experience negative side effects, including metabolic syndrome and extrapyramidal symptoms, which can result in a lower quality of life and increased health risks (Figure 3) [227]. Furthermore, medication adherence is crucial in the context of long-term treatment. Patients’ prognoses may worsen because of nonadherence to antipsychotic treatment, which might increase the likelihood of relapse, rehospitalization, and suicide attempts [228]. Research has demonstrated that treatments, including psychoeducation and long-acting injectable formulations intended to improve adherence, can improve treatment outcomes [229, 230]. Antipsychotic drugs span three main generations: first-generation (typical), second-generation (atypical), and third-generation (recently described). First-generation antipsychotics, such as haloperidol and chlorpromazine, have historically been effective in reducing positive symptoms but are strongly associated with extrapyramidal side effects and tardive dyskinesia [231]. Second-generation (atypical) antipsychotics, including risperidone, olanzapine, and clozapine, are widely prescribed because of their relatively lower risk of movement disorders, although they frequently cause metabolic, cardiovascular, and endocrine complications [231, 232]. Third-generation antipsychotics, such as aripiprazole, brexpiprazole, and cariprazine, act as dopamine system stabilizers and are considered to have a more favorable safety and tolerability profile, particularly with respect to metabolic and neurological adverse effects, although long-term evidence is still evolving [233]. One of the most adverse effects of antipsychotic medications is metabolic side effects; therefore, patients treated with these medications have increased rates of morbidity and mortality [234, 235]. Olanzapine and clozapine, two atypical antipsychotics, are especially well-known for causing weight gain and metabolic syndrome, which includes elevated blood pressure, insulin resistance, and dyslipidemia [236, 237]. Managing patients using antipsychotic drugs can be extremely difficult when neurological side effects, including akathisia, extrapyramidal symptoms, and neuroleptic malignant syndrome, are present. Although atypical and third-generation antipsychotics are generally associated with fewer extrapyramidal symptoms than first-generation agents, none are risk-free, and careful monitoring remains essential in clinical practice [238, 239].
People with SZ face significant barriers to employment, housing, and social integration, which negatively impact their overall functioning and quality of life. These challenges are compounded by symptom-related impairments, social stigma, and a lack of strong support networks (Figure 3) [240, 241]. Cognitive impairments, negative symptoms, and social skill deficits contribute to lower employment rates among individuals with SZ than in the general population [242, 243]. Difficulties in maintaining focus, engaging in social interactions, and managing workplace demands make securing and retaining employment particularly challenging for individuals with ADHD. Finding stable housing is another critical issue, as individuals with SZ often experience financial constraints, health-related challenges, and discrimination [244, 245]. Research indicates that inadequate housing exacerbates social isolation and worsens mental health, whereas stable housing is linked to better psychological well-being and increased community engagement [244]. Supportive housing programs tailored to their needs, along with community-based services, play a vital role in ensuring long-term housing stability [245]. Deficits in social cognition, including difficulties in interpreting and responding to social cues, further hinder social integration [246, 247]. These challenges contribute to feelings of loneliness and social withdrawal [248]. However, interventions such as social skills training have been shown to improve social functioning and reduce loneliness [249]. Supportive community environments that foster social connections can enhance a sense of belonging and well-being [248, 249].
SZ has a strong polygenic architecture, which means that its pathophysiology is influenced by a wide range of genetic variants [241, 250]. Neuroimaging research has shed important light on the anatomical alterations in the brains of individuals with SZ. For example, early-onset SZ has been linked to gray matter decreases [251], highlighting the necessity of early intervention to prevent cognitive and functional losses. According to Adamu et al. [252], structural alterations can influence the development of diagnostic and therapeutic techniques, and tracking brain morphology may be a powerful method for assessing the effectiveness of treatments in clinical settings. People with SZ have been reported to benefit from lifestyle changes, such as exercise, in addition to pharmaceutical therapies. Regular exercise may result in significant cognitive gains, as demonstrated by Takahashi et al. [253], who showed that physical activity can increase hippocampal volume and improve short-term memory. This aligns with findings regarding cognitive deficits in SZ; improved neuroplasticity is associated with both physical activity and cognitive rehabilitation programs, although the precise neurobiological underpinnings remain unclear [254]. Additionally, the relationship between SZ and circadian rhythms is becoming a crucial area for treatment. The possibility that circadian-based therapy could enhance symptom management is highlighted by Delorme et al.’s [255] hypothesis that circadian disruption may constitute a fundamental pathogenic component of SZ. By adjusting these therapies to the patients’ sleep-wake cycles, symptoms can be lessened, and general health can be enhanced. Emerging clinical trials are now testing melatonin agonists, light therapy protocols, and sleep-wake scheduling interventions to evaluate their ability to reduce cognitive impairment, stabilize mood, and improve treatment adherence in patients with SZ. Such chronotherapeutic approaches represent a low-cost, non-invasive adjunct to pharmacotherapy, with promising early phase results that warrant larger randomized trials [256]. Understanding how endophenotypes, such as cognitive abilities and sensory gating, contribute to SZ will help improve intervention techniques. Practitioners can customize their treatments more precisely to each patient’s unique neurobiological profile by identifying these biological markers and assessing how they manifest in patients. This could ultimately improve therapeutic outcomes [257].
Peer support plays a crucial role in enhancing the recovery and well-being of individuals with SZ. Online peer networks provide valuable social connections, empowerment, and access to information, helping individuals navigate their conditions more effectively [258]. Structured community-based interventions further enhance recovery by connecting individuals with mental health services and peer support networks, thereby improving access to care and social support [259]. Research highlights that strong social support systems help mitigate the impact of SZ on personality and social interactions, promoting greater stability and functional outcomes [260]. Engaging peer support specialists in mental health services benefits those receiving assistance and enhances the mental well-being of peer supporters themselves [261]. This dual impact reinforces peer support as a sustainable and effective model for long-term mental health care, fostering community resilience and empowerment [262].
Equally important is examining how the structural brain patterns of people with SZ develop over time. Lieslehto et al. [263] discovered that certain gray matter changes occurred over nine years, especially in the hippocampus and medial prefrontal cortex. This suggests that long-term continuous observations are necessary to capture the dynamic characteristics of these changes [263]. The significance of antipsychotic medication adherence and its correlation with long-term consequences necessitates a longitudinal approach. Glick et al. [264] stressed the need for consistent, long-term care; however, they also acknowledged that results should be interpreted cautiously because of possible confounding variables. Research on the length of untreated psychosis offers crucial information about the significance of early intervention in a longitudinal setting, highlighting the importance of long-term research to track the effects and timing of medication on symptoms and changes in the brain over time. Moreover, Shrivastava et al. [265] found a strong correlation between the length of untreated psychosis and worse long-term results, indicating that early and consistent intervention may improve prognosis and recovery potential. Longitudinal studies can also shed light on how genetic and environmental factors interact to cause diseases. Martin et al.’s [266] work demonstrates how genetic predispositions might influence study participation in longitudinal research, potentially affecting the validity of long-term treatment efficacy and outcome data. Recognizing the various paths that SZ can take, including the potential for recovery and long-term functional gains, highlights a growing body of knowledge based on longitudinal studies.
Genomic, proteomic, and metabolomic data are combined in multi-omics integration to unravel the intricate pathophysiology of SZ. By translating research findings into therapeutic applications, this method can provide a more comprehensive view of the interactions involved in the pathophysiology of the condition [267, 268]. Recent developments in proteomics, such as mass spectrometry and liquid chromatography, have facilitated the combination of proteome data with other omics. A thorough multi-omics approach to SZ requires an understanding of protein interactions and functions, which can only be achieved through integration [267]. Tools such as the integrated risk gene selector (iRIGS) predict high-confidence risk genes from SZ GWAS data using multi-omics data. Potential therapeutic uses are suggested by the fact that these genes are often expressed in brain tissues and could be targets for already available medications [268]. Multi-omics may classify genes within significant loci, but without large data volumes, it cannot consistently boost the discovery of new genes. Therefore, expanding sample sizes is crucial for uncovering novel genes and loci in SZ research [269]. Developments in data mining and statistical techniques are essential for the successful integration of multi-omics data. These techniques can improve our understanding of the biological mechanisms underlying SZ by assisting in the identification of biomarkers, functional modules, and causal pathways [270]. Importantly, multi-omics approaches are beginning to inform drug repurposing pipelines, where compounds originally developed for oncology, immunology, or metabolic disorders are systematically tested against SZ-related molecular signatures. This translational step could accelerate the availability of novel pharmacological interventions [271].
AI can improve SZ treatment by analyzing large-scale genetic, clinical, and behavioral data to personalize the interventions. Explainable AI models, such as the Personalized Advantage Index and Bayesian Rule Lists, help identify patient subgroups that may respond better to specific treatments, improving clinical outcomes [258]. Digital health technologies and mobile applications can facilitate real-time monitoring, patient-clinician communication, and tailored interventions. However, challenges such as small datasets and model evaluation limitations require methodological improvements to ensure accurate and reliable AI applications [272]. In parallel, neuromodulation therapies, such as repetitive TMS (rTMS), tDCS, and DBS, are being explored as adjunctive treatments, with preliminary evidence suggesting benefits for treatment-resistant hallucinations, negative symptoms, and cognitive dysfunction. Combining AI-driven patient stratification with neuromodulation trials may help identify individuals most likely to benefit from these approaches [273].
Mental health disorders place a significant burden on public health systems, necessitating increased global investments. Several countries have committed to expanding 24-hour mental health services, crisis hotlines, and community-based interventions [274]. However, international funding remains insufficient, with mental health comprising only 0.3% of total health development assistance, emphasizing the need for greater financial support, especially in LMICs [275]. Expanding funding and governance in countries such as Brazil and integrating mental health services into general healthcare systems in China offer promising models for improving accessibility and service delivery [276]. Schools play a crucial role in early mental health interventions. Establishing comprehensive school mental health systems (CSMHS) through cross-sector partnerships, medicaid coverage, and professional training ensures that students receive timely support [277, 278]. However, sustainable funding mechanisms must be developed, as many current programs rely on fee-for-service structures that do not adequately support preventive and ongoing care [279]. As mental health funding expands, it is essential to avoid repeating the past mistakes of overemphasizing biological paradigms, which can lead to coercion and unnecessary pathologization. Mental health initiatives must uphold human rights and be culturally appropriate, particularly in post-pandemic recovery efforts [280]. Public education programs are crucial for enhancing mental health literacy and reducing the stigma surrounding mental illness. However, their success relies on cultural adaptation and integration into the host country. Direct interactions between the public and individuals with mental illnesses foster mutual understanding and reduce stigma. Advocacy can shape public attitudes and drive policy changes. Comprehensive mental health policies and anti-discrimination laws address stigma and improve service accessibility [281–284].
In summary, future directions for SZ research and care must integrate cutting-edge biological discoveries (e.g., multi-omics, biomarkers, and neuroimaging) with innovative therapies (chronotherapy, neuromodulation, and AI-driven precision psychiatry), while simultaneously strengthening community-based recovery models, digital tools, and sustainable health system investments. This multidimensional strategy can advance both scientific understanding and practical therapeutic outcomes in people living with SZ.
SZ is a multifactorial psychiatric disorder influenced by genetic, neurodevelopmental, neurochemical, and environmental factors. Research has highlighted the roles of glutamatergic, dopaminergic, and immune system dysfunction in its pathophysiology. Emerging tools, such as digital phenotyping, multi-omics, and ML, are improving early diagnosis and personalized care. New therapies, including TAAR1 agonists, glutamatergic modulators, psychedelics, and anti-inflammatory agents, complement traditional dopamine-based treatments, whereas neuromodulation techniques (TMS and DBS) support precision psychiatry. Key challenges include early diagnosis, treatment adherence, integration of mental health into primary care, and stigma reduction, especially in LMICs. Future efforts should focus on longitudinal studies, validated biomarkers, culturally sensitive interventions, and strengthening the global mental health infrastructure through interdisciplinary collaboration and public education.
AI: artificial intelligence
AKT1: AKT serine/threonine kinase 1
AMBRA1: autophagy and Beclin 1 regulator 1
BD: bipolar disorder
BDNF: brain-derived neurotrophic factor
CACNB2: calcium voltage-gated channel auxiliary subunit beta 2
CBTp: cognitive behavioral therapy for psychosis
CNVs: copy number variations
COMT: catechol-O-methyltransferase
CSF: cerebrospinal fluid
DBS: deep brain stimulation
DNA: deoxyribonucleic acid
DOCK4: dedicator of cytokinesis 4
EFHD1: EF-hand domain family member D1
FEP: first-episode psychosis
FGFR1: fibroblast growth factor receptor 1
GABA: gamma-aminobutyric acid
GWASs: genome-wide association studies
IFN-γ: interferon-gamma
IL-6: interleukin-6
KYNA: kynurenic acid
LMICs: low- and middle-income countries
LRRFIP1: leucine rich repeat flightless interacting protein 1
MIA: maternal immune activation
ML: machine learning
MRI: magnetic resonance imaging
NDST3: N-deacetylase and N-sulfotransferase 3
NMBR: neuromedin B receptor
NMDA: N-methyl-D-aspartate
NRXN1: neurexin 1
RNA: ribonucleic acid
SNPs: single-nucleotide polymorphisms
SZ: schizophrenia
TAAR1: trace amine-associated receptor 1
tDCS: transcranial direct current stimulation
TMS: transcranial magnetic stimulation
TNF-α: tumor necrosis factor-alpha
UGT1A1: UDP glucuronosyltransferase family 1 member A1
VR: virtual reality
VRK2: vaccinia related kinase 2
TAO: Conceptualization, Investigation. OJO: Conceptualization, Data curation, Investigation, Writing—original draft, Writing—review & editing. UOA: Conceptualization, Data curation, Investigation, Writing—original draft, Writing—review & editing. KBO: Conceptualization, Data curation, Investigation, Writing—original draft. SSM: Conceptualization, Data curation, Investigation, Writing—original draft, Writing—review & editing. MMA: Conceptualization, Data curation, Investigation, Writing—original draft, Writing—review & editing. AKA: Conceptualization, Data curation, Investigation, Writing—original draft, Writing—review & editing. ABO: Writing—original draft. JOO: Writing—original draft, Writing—review & editing. GE: Writing—review & editing. DELP: Writing—review & editing, Supervision. All authors have read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.