 Commentary
Commentary

Affiliation:
MPS Brazil Network, Hospital de Clínicas de Porto Alegre, Porto Alegre 90410-000, Brazil
Email: yorran_montenegro@hotmail.com
ORCID: https://orcid.org/0000-0002-0684-1818
Explor Neurosci. 2025;4:1006103 DOI: https://doi.org/10.37349/en.2025.1006103
Received: February 24, 2025 Accepted: July 11, 2025 Published: July 28, 2025
Academic Editor: Ameneh Rezayof, University of Tehran, Iran
Mucopolysaccharidosis type IIIB (MPS IIIB), or Sanfilippo Syndrome type B, is a lysosomal storage disorder caused by mutations in the NAGLU gene, which encodes the enzyme alpha-N-acetylglucosaminidase, responsible for the degradation of heparan sulfate. Progressive accumulation of undegraded glycosaminoglycans primarily affects the central nervous system, resulting in severe neurodegeneration. Cellular findings reveal impaired intracellular trafficking, especially within the Golgi apparatus, linked to GM130 depletion and accumulation of GM2 and GM3 gangliosides. Endocytic vesicles fail to properly fuse with lysosomes due to genetic defects, disrupting lysosomal degradation. This contributes to oxidative stress, mitochondrial dysfunction, and mitophagy failure, which collectively drive neuronal apoptosis. MPS IIIB shares pathways with Alzheimer’s and Parkinson’s, suggesting cellular aging processes. Given the lack of specific treatment, modulation of inflammatory pathways such as TLR4 emerges as a potential therapeutic strategy.
Mucopolysaccharidoses correspond to a group of seven pathologies, subdivided into 13 subgroups, classified according to the mutation involving glycosaminoglycan degradation enzymes [1–3]. Mucopolysaccharidosis type III (MPS III) or Sanfilippo Syndrome is caused by mutations in enzymes that degrade heparan sulfate, a glycosaminoglycan. Four enzymes are characterized as subtypes of MPS III: N-sulfoglucosamine sulfohydrolase gene (SGSH EC 3.10.1.1) (MPS IIIA), N-alpha-acetylglucosaminidase gene (NAGLU EC 3.2.1.50) (MPS IIIB), heparan acetyl-CoA: alpha-glucosaminide N-acetyltransferase gene (HGSNAT EC 2.3.1.78) (MPS IIIC), N-acetylglucosamine-6-sulfatase gene (GNS EC 3.1.6.14) (MPS IIID) [2]. The main clinical characteristic related to Sanfilippo Syndrome is the involvement of the nervous system.
Clinical findings can be divided into three phases. First phase corresponds to the age of 1–3 years, it is possible to observe the first signs of delayed neuropsychomotor development; second phase corresponds to the age of 3–4 years, it is possible to observe a regression in the developmental milestones achieved; the progression is gradual, often leading these individuals to death in the third decade of life, characterizing third phase [4]. No therapies are available. Clinicians operated with symptomatic management [5, 6]. Is it possible that the heterogeneity of neurological symptoms in MPS III is associated with the cellular aging process, especially neuronal aging?
Among the main symptoms associated with Sanfilippo Syndrome IIIB, especially after the first decade of life, there is an increasing neuronal death characterized by ventricular enlargement and well-defined turns [6]. It is believed that part of this neuronal death can be explained by the molecular aspects of cellular cytotoxicity in these patients. Considering the general characteristics of neuronal morphology in these patients, it is observed demyelinated neurons [7], impaired neuronal dendritic arborization [8], and axonal dystrophy [9]. These findings are consistent with the characteristics found in imaging exams of the brain structure in these patients.
In the cellular and functional context, especially with regard to synaptic connections and their repercussions on cognitive activity in patients with Sanfilippo Syndrome IIIB, a progressive and evident neuropsychomotor delay is observed [4]. Hocquemiller and colleagues [10], studying the main features in synaptic connections in animal models with MPS IIIB, observed a lower percentage of neurite regression in neurons from Mucopolysaccharidosis IIIB mice in culture, as well as a significant increase in the elongation of these neuronal bodies. The findings reflect a significant reduction in synaptic plasticity in this disease, which may explain the progressive cognitive deficits. The researchers’ findings present another curious characteristic: higher immunoreactivity of anti-βIII-tubulin, anti-MAP2 in dendrites, and anti-MAP2 expression in axons. These findings reflect an accumulation of dense proteins in the axonal stream, possibly related to considerable defects in axonal transport, especially in membranous intracellular traffic. Other findings also included anti-βIII-tubulin, mRNA GAP43, an important indicator of dysfunction in the Golgi complex, which corroborates the hypothesis of defects in intracellular membranous trafficking [11]. The effects of the central nervous system extend to the periphery in a similar way [12].
In a complex way, neuronal molecular findings in MPS IIIB also demonstrate important gene expression alterations that contribute to the hypothesis of a defect in intracellular membranous trafficking. iPSCs cells of patients with MPS IIIB show a morphological disruption of the Golgi related to GM130 depletion (Golgi cisterns organization) [13] associated with accumulation of secondary products (GM2 and GM3). These resulting byproducts significantly increase cellular cytotoxicity signaling, contributing to oxidative stress that can activate the cellular apoptotic program [11, 12, 14]. Thus, the association of defects in intracellular membranous trafficking, accumulated secondary products, and failure in the primary process of lysosomal degradation of glycosaminoglycans significantly increases markers of cellular stress, causing cell death in these patients [15, 16].
Although research has shown the importance of neurons in the neuropathology of MPS IIIB, glial cells have a direct role in modulating metabolic pathways important for neuronal survival or death. Glial cells contribute to the establishment of neuronal changes through their homeostatic function, especially the TLR4 activation pathway (Figure 1) [17, 18]. This molecular pathway is known for its pro-inflammatory characteristics and directly assists in the processes of neuronal apoptosis. These findings are consistent in both human patients and animal models [11]. The molecular findings indicate important therapeutic possibilities, especially for a pathology without an available therapy such as MPS IIIB. Exogenous molecular modulators may be useful to preserve homeostasis in patients. Myers and colleagues [19] indicated that inhibition of p38 in Schwann cell and neuron culture led to axonal regeneration, confirming its potential as a therapeutic target to downregulate TNFα signaling and attenuating neuroinflammatory symptoms.
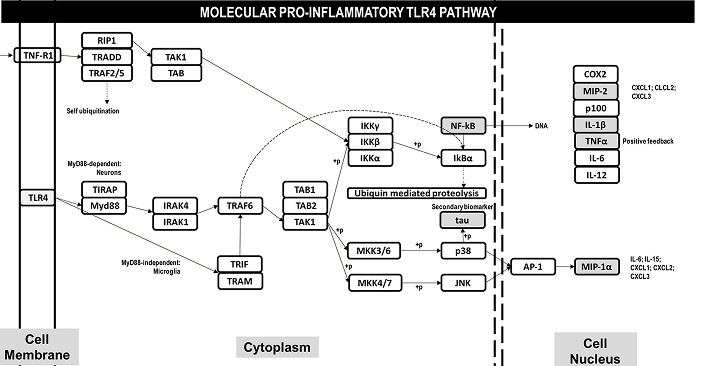
Molecular pro-inflammatory pathway activated in the central nervous system in patients with Mucopolysaccharidosis type III. The activation in glial cells occurs in a MyD88-independent pathway, with translocation of the TRIF-TRAM complex and subsequent activation of TRAF6. Released cytokines and pro-inflammatory products activate, via TLR4 receptors, a pro-inflammatory pathway MyD88-dependent in neurons, with translocation of IRAK4-IRAK1 and subsequent activation of TRAF6. The events of TRAF6 activation resulting in two directions: i) release of NF-kB to the nucleus and expression of interleukines, as well TNF-alpha resulting in a positive feedback for pro-inflammatory activation via TNRR1 receptors; ii) MAPK signaling pathway, resulting in secondary tau-phosphorylation and release AP-1 to the translocation of AP-1 to the nucleus, resulting in expression of MIP-1α inducing pro-inflammatory citokines
The best way to understand the importance of the TLR4 molecular pathway and the processes of cytotoxicity, cell death, and aging is to understand how this pathway contributes to other neurological conditions. TLR4 molecular pathway in Alzheimer’s and Parkinson’s diseases generates two of the main byproducts of these diseases: phosphorylated tau and amyloid-B proteins, respectively [20–23]. Interestingly, these proteins play important roles in axonal trafficking and intramembranous transport, similar to what occurs with the accumulated products in MPS IIIB. Furthermore, in Sanfilippo Syndrome IIIB itself, phosphorylated tau and amyloid-beta proteins also have a significant increase in concentration [17, 18].
Alzheimer’s and Parkinson’s diseases are also important to provide important insights into how byproducts related to intracellular membranous trafficking can directly affect cell viability. The harmful effects of these products are responsible for mitochondrial dysregulation [23]. Mitochondrial dysregulation in these diseases, as well as in Mucopolysaccharidosis itself, has a similar profile. Complex II + III (SCCR) and Mitochondrial ATP Synthase C Subunit (SCAMAS) protein accumulation in MPS IIIB are important markers that reveal a failure of mitochondrial fission-fusion homeostasis, revealing a possible imbalance [23]. Similarly, other MPS III subtypes also demonstrate findings like these [24]. The chain of events discussed so far, starting from increased oxidative stress due to degradation products in MPS IIIB and other neurological diseases results in: i) increased O2 concentrations; ii) dysregulation of Ca2+ homeostasis; iii) excitoxicity; iv) increased membrane permeability; and, v) activation of the mitochondrial defense system related to the progression of neurodegenerative diseases [24, 25] (Figure 2). Specifically for Mucopolysaccharidosis IIIB, events that reduce cell viability can be described as follows (Figure 3): 1) intracellular trafficking appears to function properly when compared to normal neuronal activity; 2) however, there is a mismatch involving vesicles originating from the Golgi complex, which is associated with low GM130 expression and the consequent accumulation of side products, specifically GM2-positive vesicles; 3) endocytic activity occurs as expected, with the formation of primary endosomal vesicles within the intracellular space; however, the fusion of these vesicles with LAMP1-positive lysosomes—required for the formation of mature degradative vesicles—is inefficient due to genetic defects; 4) this results in impaired protein degradation and defective maturation of endovesicles; 5) neuroinflammatory activation in neurons occurs via glial pathways, involving nitric oxide production (particularly iNOS), cytokine release, and the activation of TLR4 receptors—key components of pro-inflammatory pathways—which contribute to oxidative stress in mitochondria; 6) this mitochondrial stress induces mitophagy, but the GM2-positive vesicles are not properly degraded, leading to the accumulation of inactive mitochondria and secondary products such as GM2, GM3, and mitochondrial vesicles.
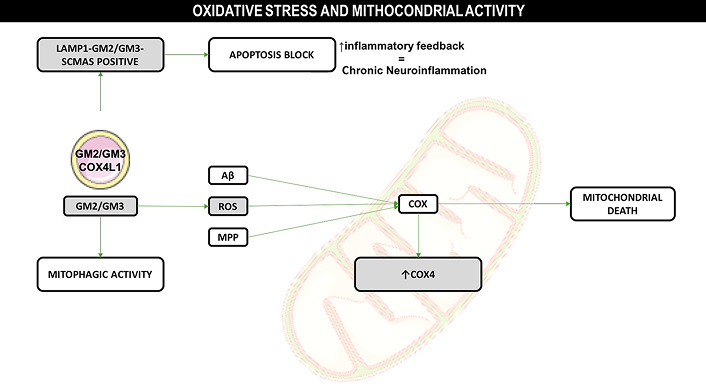
The release of pro-inflammatory cytokines and nitric oxide from glial cells induced pro-inflammatory responses in the neurons, and caused mitochondrial oxidative stress. Mitophagic activity was observed for GM2 and GM3 vesicles (Golgi complex origin), and a posteriori overexpression of Cox4, resulting in mitochondrial death. Another observation was the encompassing of mitochondrial GM2/GM3 positive vesicles for the lisosomal activity, indicating survival mechanism activation, resulting in apoptosis block, positive inflammatory feedback, and chronic neuroinflammatory profile. Image provided by Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/)
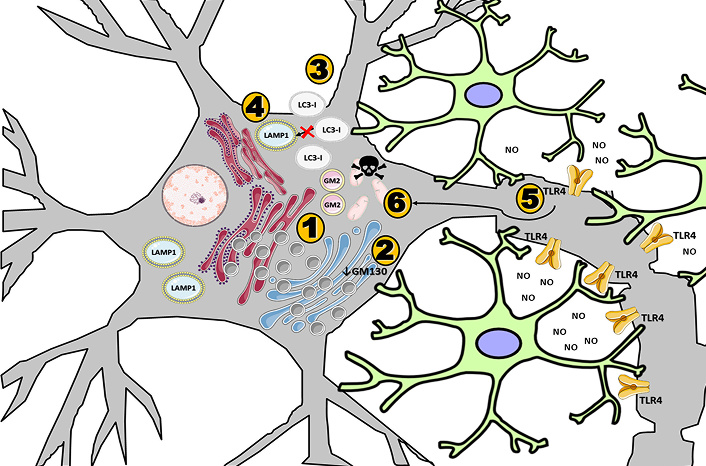
Schematic representation of neuroinflammatory processes in Mucopolysaccharidosis type IIIB. Image provided by Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/)
Data suggest mitochondria as a central element in the process of senescence and cell viability. More specifically, the findings suggest that there is a depletion in ATP production [25] and an increase in oxidative stress [26, 27]. Apparently, these findings extend to other lysosomal storage diseases such as MPS IIIB [28]. These findings suggest new ways of thinking about therapeutic interventions, aiding in the development of technologies that help these patients [29, 30]; however, it is necessary to think about short and medium-term solutions while the technologies are not developed.
Considering molecular modulators already approved and in use as therapeutic tools implies a therapeutic possibility for MPS IIIB. Some studies have already demonstrated possible useful drugs to reach this molecular target efficiently [31], including an application in other neurodegenerative diseases [32, 33]. Several studies have indicated possible molecules that can assist in the TLR4 modulation process [33–35]. I believe that this modulation can provide an important strategy in MPS IIIB, and bearing in mind the clinical trials already carried out with modulators [36]. I believe that the benefit could be studied in the population of these patients, helping to improve their quality of life, as well as the progression of neurological symptoms.
MPS III: Mucopolysaccharidosis Type III
YHAM: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
The author declares that there is no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2341
Download: 22
Times Cited: 0
