 Original Article
Original Article

Affiliation:
1Greenberg Division of Cardiology, Weill Cornell Medicine, New York, NY 10021, USA
Affiliation:
2Departments of Cardiology and Nephrology, Ullevaal Hospital, University of Oslo, N-0407 Oslo, Norway
Affiliation:
1Greenberg Division of Cardiology, Weill Cornell Medicine, New York, NY 10021, USA
ORCID: https://orcid.org/0000-0001-5861-1478
Affiliation:
3Merck Research Laboratories, North Wales, PA 19454, USA
Affiliation:
2Departments of Cardiology and Nephrology, Ullevaal Hospital, University of Oslo, N-0407 Oslo, Norway
ORCID: https://orcid.org/0000-0001-7520-7645
Affiliation:
2Departments of Cardiology and Nephrology, Ullevaal Hospital, University of Oslo, N-0407 Oslo, Norway
4Division of Cardiovascular Medicine, University of Michigan, Ann Arbor, MI 48109, USA
ORCID: https://orcid.org/0000-0003-2389-0272
Affiliation:
4Division of Cardiovascular Medicine, University of Michigan, Ann Arbor, MI 48109, USA
Affiliation:
5Department of Clinical Science, University of Bergen, N-5020 Bergen, Norway
ORCID: https://orcid.org/0000-0003-4109-2311
Affiliation:
1Greenberg Division of Cardiology, Weill Cornell Medicine, New York, NY 10021, USA
Affiliation:
1Greenberg Division of Cardiology, Weill Cornell Medicine, New York, NY 10021, USA
Email: rbdevere@med.cornell.edu
Explor Med. 2022;3:128–138 DOI: https://doi.org/10.37349/emed.2022.00079
Received: November 18, 2021 Accepted: January 27, 2022 Published: March 17, 2022
Academic Editor: Carlos M. Ferrario, Wake Forest School of Medicine, USA
The article belongs to the special issue Angiotensins—A Century of Progress
Aim: The Losartan Intervention For Endpoint reduction in hypertension (LIFE) study showed less new-onset atrial fibrillation (AF) in hypertensive patients receiving losartan- vs. atenolol-based treatment. Because losartan reduces serum uric acid (SUA) levels, the aim of the present study was to investigate relations of SUA with new-onset AF in the study.
Methods: Hypertensive patients with electrocardiographic (ECG) left ventricular hypertrophy (LVH) and no prior AF (n = 8,243) were treated for 5.0 ± 0.4 years with losartan- or atenolol-based therapy. Associations of SUA with new-onset AF documented by Minnesota coding were assessed by Cox models using SUA and systolic blood pressure as time-varying covariates to take into account changes of SUA related to losartan or diuretic treatment, changes in renal function, and aging.
Results: Time-varying SUA was associated with new AF defined by Minnesota code [hazard ratio (HR) = 1.19 per 16.8 μmol/L (1 mg/dL), (95% confidence intervals (CIs), 1.12–1.26), P < 0.0001], independent of losartan treatment [HR = 0.75 (95% CIs, 0.61–0.93), P = 0.007], older age [HR = 1.95 per 7.0 years (95% CIs, 1.73–2.20), P < 0.0001], male sex [HR = 1.46 (95% CIs, 1.09–1.94), P = 0.010] and higher Cornell voltage-duration product [HR = 1.10 per 1,023 ms·mm (95% CIs, 1.01–1.21), P = 0.034]. Similar results were obtained in Cox models with SUA levels partitioned according to baseline quartiles and in which AF was defined by physician reports or by both Minnesota coding and physician reports.
Conclusions: In-treatment SUA is a strong predictor for new-onset AF in hypertensive patients, independent of effects of antihypertensive treatment, age, sex, and ECG-LVH. Further research is needed to clarify how uric acid may provoke AF (ClinicalTrials.gov identifier: NCT00338260).
Atrial fibrillation (AF) is associated with increased rates of cardiovascular morbidity and mortality in treated hypertensive patients with left ventricular hypertrophy (LVH) [1]. The incidence of AF is increased in patients with uncontrolled hypertension [2], and antihypertensive treatment reduces new-onset AF [3, 4]. In view of evidence that heart rate control is at least as beneficial as rhythm control for preventing cardiovascular events in patients with AF [5, 6], beta-blockade is an accepted mainstay of AF treatment, despite lack of evidence that it is superior to other antihypertensive treatments in preventing or managing AF. Several animal and human studies suggest that renin-angiotensin blockade reduces the occurrence of AF [7–9], and one study suggested that the beneficial effect was independent of blood pressure (BP) reduction by angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers [10].
A striking result of the Losartan Intervention For Endpoint reduction in hypertension (LIFE) study was a 42% lower rate of cardiovascular events on losartan- compared with atenolol-based treatment in patients with a history of AF [1]. This result and a 33% lower incidence of new-onset AF with losartan- compared with atenolol-based therapy occurred with similar BP lowering in both treatment arms [11]. Additional analyses suggest that this benefit of losartan is not fully accounted for by greater reduction of LVH and left atrial size in LIFE patients treated with losartan [12, 13].
Serum uric acid (SUA) has been independently related to cardiovascular events in a number of epidemiological studies [14–17], and the association seems to be stronger in women than in men and in black compared with white patients. Hyperuricemia has been shown to predict the development of hypertension [18], to be present in one-fourth of untreated hypertensive patients [18], and to be related to hypertension after adjustment for other risk factors and use of diuretics [19, 20]. SUA predicts stroke in the presence or absence of diabetes [21, 22] and is related to increased mortality in patients with congestive heart failure [23, 24] as well as to prevalent [25] and new onset AF [26–29]. The LIFE study was the first clinical trial to indicate that lowering SUA by a uricosuric drug such as losartan has a beneficial effect on cardiovascular outcome independently of changes in renal function or BP [30]. Because losartan reduces SUA levels, the present study aimed to investigate whether lower SUA levels during treatment were associated with less new-onset AF in the LIFE study.
The LIFE study was a prospective, randomized, double-masked, parallel group study (n = 9,193) that evaluated the long-term effects of losartan- compared with atenolol-based antihypertensive therapy on cardiovascular morbidity and mortality in patients with hypertension and electrocardiographic (ECG)-LVH. The LIFE study design, organization, clinical measures, endpoint definitions, basis for choice of comparative agents, statistical power calculations, recruitment details, baseline characteristics, 1-year follow-up, and primary results have been published [31–32]. The LIFE patients with AF at study baseline (n = 342) had a lower rate of cardiovascular events with losartan- compared with atenolol-based treatment [1]. The present study was done in 8,243 LIFE participants with ECG-documented sinus rhythm, no history of AF, and available SUA measurements at study baseline.
As previously described, patients aged 55–80 years with previously treated or untreated hypertension and ECG signs of LVH were randomized to initial therapy with 50 mg/day of losartan or atenolol after 1–2 weeks on placebo if they had sitting systolic BP 160–200 mmHg and/or diastolic BP of 95–115 mmHg [32]. In both groups, hydrochlorothiazide 12.5 mg was added in case of insufficient BP lowering, after which study drug was increased to 100 mg/day and supplemented with hydrochlorothiazide 25 mg or additional antihypertensive therapy to reach target BP of < 140/90 mmHg. Patients were enrolled from June 1995 to May 1997 and were followed ≥ 4 years (mean 5.0 ± 0.4 years) with regular visits. Sitting BP was recorded 24 h post-dose (range 22–26 h) and blood was obtained for SUA determination at baseline and at annual revisits during the study.
New-onset AF was documented by annual in-study ECGs by Minnesota codes including all types of AF [33] and determination of ECG-LVH by both Cornell voltage-duration product and Sokolow-Lyon voltage at a central ECG core laboratory as previously described [31, 32]. Supplemental analyses used incident AF reported by investigators during the follow-up period, either alone or in combination with new AF by Minnesota code (n = 652). Care of patients’ new onset AF was left to the discretion of local investigators. Laboratory measurements of SUA were made at central laboratories for the Nordic countries (located in Brussels, Belgium) and in the United States that assured comparability of measurements by cross-validation.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agreed to the manuscript as written. SPSS version 12.0 (SPSS, Inc., Chicago, IL, USA) was used for statistical analysis. Potential risk factors (including baseline clinical, demographic, and laboratory data) were assessed for association with new-onset AF. Cox proportional hazards models were used to compare hazard ratios (HRs) between study treatment allocation groups (losartan or atenolol) and to evaluate contributions of differences in the degree of LVH (both Cornell voltage-duration product and Sokolow-Lyon voltage as continuous variables), Framingham risk score, and other covariates. For each baseline characteristic, a univariate proportional hazards regression model was used to estimate the HR and its 95% confidence interval (CI). Variables without significant associations with new-onset AF were eliminated before developing multivariable models.
Multivariable analyses were performed using Cox regression models with inclusion of remaining baseline variables to identify those independently associated with the endpoints. Two-tailed P < 0.05 was considered significant. Multivariable Cox models were constructed using SUA and other variables re-measured at intervals during LIFE follow-up as continuous time-varying covariates to take into account variable changes of SUA from baseline among patients related to losartan treatment, changes in renal function, and aging. Supplemental Cox models using quartile ranges of SUA measurements at baseline were used to assess the predictive value of SUA levels in defined ranges for subsequent new AF during intensive antihypertensive treatment in LIFE. HRs were calculated to assess the risk of developing new-onset AF in patients whose SUA fell in the 1st through 4th quartiles at baseline using sex-specific 2nd, 3rd, and 4th SUA quartile ranges as follows: < 309, 309–353, 354–408, and ≥ 409 μmol/L for men; and < 255, 255–297, 298–345, and ≥ 346 μmol/L for women. The 1st through the 4th quartiles of baseline SUA contained n = 2,033, 2,070, 2,100, and 2,040 patients, respectively, at enrolment. These analyses were repeated using SUA within these sex-specific ranges at each visit during the LIFE study as a time-varying predictor of subsequent AF. A modified Kaplan-Meier plot [34] was constructed to illustrate the association between time-varying SUA in these ranges and the rate of new-onset AF.
The present study was performed in 8,243 LIFE participants with ECG-documented sinus rhythm, no history of AF, and available SUA measurements at study baseline. The eligible subjects had a mean age of 66.8 years and 46% were women. Patients treated with losartan had, compared with those on atenolol, similar baseline characteristics with respect to proportion of male sex (54% vs. 55%, respectively), mean age (66.8 years in each group), mean Framingham risk score (22.1 vs. 22.3, respectively), history of diabetes (13% in each group), and mean baseline systolic BP (174 mmHg in each group).
Assessment of univariate relations between baseline variables and new-onset AF (Table 1) identified significant positive associations with older age, non-black ethnicity, higher systolic BP, presence of LVH by Cornell voltage-duration product criteria, prevalent coronary heart disease and cardiovascular disease, lower serum potassium, and higher urinary creatinine/albumin ratio. New-onset AF was not associated with baseline smoking habit, total cholesterol, high-density-lipoprotein (HDL)-cholesterol, or plasma glucose levels. Similarly, baseline SUA level did not predict subsequent incident AF (P > 0.20).
New-onset AF in relation to baseline characteristics
| Variable | New-onset AF, n (%) | HR (95% CIs) | P | |
|---|---|---|---|---|
| Sex | Men | 187 (5.0) | 1.22 (0.99–1.50) | 0.070 |
| Women | 186 (4.1) | |||
| Age, years | ≥ 65 | 306 (6.0) | 3.01 (2.30–3.94) | < 0.0001 |
| < 65 | 67 (2.1) | |||
| Ethnicity | Black | 8 (1.6) | 0.34 (0.17–0.68) | 0.001 |
| Non-Black | 362 (4.7) | |||
| Systolic BP, mmHg | > 173 | 228 (5.5) | 1.62 (1.31–2.00) | < 0.0001 |
| ≤ 173 | 145 (3.5) | |||
| Cornell voltage-duration product | LVH+ | 273 (5.2) | 1.61 (1.26–2.04) | < 0.0001 |
| LVH– | 91 (3.3) | |||
| Coronary heart disease | Present | 106 (5.9) | 1.91 (1.19–3.04) | 0.006 |
| Absent | 22 (3.2) | |||
| Cardiovascular disease | Present | 116 (5.8) | 1.44 (1.15–1.80) | 0.002 |
| Absent | 257 (4.1) | |||
| Serum potassium, mmol/L | < 4.1 | 165 (5.2) | 1.30 (1.05–1.61) | 0.019 |
| ≥ 4.1 | 207 (4.1) | |||
| Urine albumin/creatinine (log) | < 1.25 | 147 (3.7) | 1.44 (1.16–1.78) | 0.001 |
| ≥ 1.25 | 208 (5.3) | |||
Because SUA and BP levels both changed substantially during the LIFE study, a further multivariable Cox model considered SUA and systolic BP as time-varying covariates. In this model, time-varying SUA strongly predicted subsequent new-onset AF by Minnesota code [HR = 1.19 per 16.8 μmol/L (1 mg/dL), (95% CIs, 1.12–1.26), P < 0.0001] independent of the positive associations of new AF with higher Cornell voltage-duration product [HR = 1.10 per 1,023 ms·mm (95% CIs, 1.01–1.21), P = 0.034], older age [HR = 1.95 per 7.0 years (95% CIs, 1.73–2.20), P < 0.0001], male sex [HR = 1.46 (95% CIs, 1.09–1.94), P = 0.010], and the protective effect of losartan treatment [HR = 0.75 (95% CIs, 0.61–0.93), P = 0.007]. Systolic BP, Framingham risk score, and previous cardiovascular disease did not contribute significantly to this model.
SUA at baseline and at each annual follow-up visit was divided into four groups based on sex-specific quartile ranges of SUA at baseline, as described above. Additional univariate and multivariable Cox models that adjusted for the same covariates were used to compare SUA level in quartiles with new-onset AF. Patients were analyzed as belonging to the quartiles they belonged to from one year to the next thus the term “time-varying”. SUA in quartile 4 was strongly predictive of new-onset AF defined by Minnesota code in the univariate as well as in multivariate analyses (Table 2).
Univariate and multivariable Cox models for the prediction of subsequent new-onset AF by SUA quartiles using first quartile as reference
| Variable | Number of events | Univariate | Multivariable | ||||
|---|---|---|---|---|---|---|---|
| 2nd Quartile HR (95% CI) | 3rd Quartile HR (95% CI) | 4th Quartile HR (95% CI) | 2nd Quartile HR (95% CI) | 3rd Quartile HR (95% CI) | 4th Quartile HR (95% CI) | ||
| Baseline | 371 | 1.24 (0.94–1.65) P = 0.130 | 1.00 (0.75–1.35) P = 0.980 | 1.09 (0.81–1.46) P = 0.567 | 1.21 (0.91–1.61) P = 0.195 | 0.95 (0.70–1.29) P = 0.736 | 1.03 (0.75–1.40) P = 0.872 |
| Year 1 | 294 | 1.95 (1.31–2.91) P = 0.001 | 1.70 (1.12–2.58) P = 0.012 | 1.92 (1.32–2.80) P = 0.001 | 1.58 (1.09–2.30) P = 0.016 | 1.67 (1.16–2.40) P = 0.006 | 1.63 (1.15–2.31) P = 0.007 |
| Year 2 | 242 | 1.40 (0.94–2.07) P = 0.095 | 1.25 (0.83–1.90) P = 0.290 | 1.70 (1.17–2.46) P = 0.005 | 1.50 (1.07–2.11) P = 0.02 | 1.11 (0.77–1.59) P = 0.579 | 1.53 (1.09–2.13) P = 0.013 |
| Year 3 | 186 | 1.81 (1.05–3.12) P = 0.033 | 1.82 (1.05–3.16) P = 0.033 | 2.56 (1.56–4.21) P < 0.0001 | 1.42 (0.95–2.10) P = 0.084 | 1.29 (0.86–1.92) P = 0.215 | 1.69 (1.17–2.43) P = 0.005 |
| Year 4 | 130 | 1.89 (0.96–3.70) P = 0.064 | 1.75 (0.88–3.46) P = 0.110 | 2.18 (1.18–4.04) P = 0.013 | 1.07 (0.70–1.66) P = 0.748 | 1.29 (0.85–1.94) P = 0.228 | 1.54 (1.06–2.24) P = 0.025 |
| Year 5 | 50 | 1.52 (0.38–6.09) P = 0.552 | 2.53 (0.70–9.19) P = 0.159 | 4.26 (1.30–13.93) P = 0.017 | 1.18 (0.67–2.07) P = 0.569 | 1.25 (0.74–2.13) P = 0.407 | 2.10 (1.31–3.38) P = 0.002 |
1st through 4th SUA quartiles at baseline using sex-specific ranges: < 309, 309–353, 354–408, and ≥ 409 μmol/L for men and < 255, 255–297, 298–345, and ≥ 346 μmol/L for women. 1st quartile n = 2,033, 2nd quartile n = 2,070, 3rd quartile n = 2,100, 4th quartile n = 2,040. Multivariable models included age, sex, time-varying Cornell voltage-duration product, time-varying BP, Framingham risk score, cerebrovascular disease, and treatment status
In a Cox model that considered sex-specific SUA level partitions and systolic BP as time-varying covariates and included time-varying Cornell voltage-duration product, Framingham risk score, and age as well as categorical variables of baseline cerebrovascular disease, gender, and treatment group, SUA in the 4th quartile strongly predicted new-onset AF by Minnesota Code [HR = 1.65 (95% CIs, 1.19–2.29), P = 0.003] independent of effects of systolic BP [HR = 0.92 per 14 mmHg (95% CIs, 0.84–1.00), P = 0.044], Cornell voltage-duration product [HR = 1.10 per 1,023 ms·mm (95% CIs, 1.01–1.21), P = 0.033], age [HR = 1.94 per 7.0 years (95% CIs, 1.72–2.19), P ≤ 0.0001], male gender [HR = 1.44 (95% CIs, 1.08–1.92), P = 0.014], and atenolol treatment [HR = 1.35 (95% CIs, 1.10–1.66), P = 0.005]. Framingham risk score and cerebrovascular disease, which were significant in the univariate analysis, did not contribute significantly to this multivariable analysis (Table 3). Of note, SUA was second only to age as a strong predictor of new-onset AF, as shown by the Wald statistic of 30, a four-fold greater effect than that of the next most potent predictor.
Univariate and multivariable Cox models for prediction of new-onset AF by sex-specific SUA quartiles and systolic BP as time-varying covariates
| Variable | Number of subsequent events | Univariate | Multivariable | Wald | ||
|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |||
| SUA 2nd Quartile | 373 | 0.59 (0.43–0.80) | 0.001 | 0.87 (0.58–1.30) | 0.499 | 0.46 |
| SUA 3rd Quartile | 373 | 0.83 (0.65–1.07) | 0.158 | 1.14 (0.79–1.64) | 0.499 | 0.46 |
| SUA 4th Quartile | 373 | 1.84 (1.50–2.25) | < 0.001 | 1.65 (1.19–2.29) | 0.003 | 8.88 |
| Systolic BP, per SD | 373 | 0.95 (0.87–1.04) | 0.273 | 0.92 (0.84–1.00) | 0.044 | 4.06 |
| Cornell voltage-duration product, per SD | 373 | 1.15 (1.05–1.25) | 0.003 | 1.10 (1.01–1.21) | 0.033 | 4.56 |
| Framingham risk score, per SD | 373 | 1.25 (1.14–1.38) | < 0.001 | 0.94 (0.82–1.08) | 0.401 | 0.71 |
| Cerebrovascular disease | 373 | 1.50 (1.20–1.86) | < 0.001 | 1.20 (0.96–1.50) | 0.114 | 2.50 |
| Age, per SD | 373 | 1.89 (1.69–2.12) | < 0.001 | 1.94 (1.72–2.20) | < 0.001 | 115.0 |
| Male gender | 373 | 1.27 (1.03–1.55) | 0.022 | 1.44 (1.08–1.92) | 0.014 | 6.01 |
| Atenolol treatment | 373 | 1.40 (1.14–1.72) | 0.001 | 1.35 (1.10–1.66) | 0.005 | 7.97 |
1st through 4th SUA quartiles at baseline using sex-specific ranges: < 309, 309–353, 354–408, and ≥ 409 μmol/L for men and < 255, 255–297, 298–345, and ≥ 346 μmol/L for women. 1st quartile n = 2,033, 2nd quartile n = 2,070, 3rd quartile n = 2,100, 4th quartile n = 2,040. SD: standard deviation
Additional time-varying covariate analyses comparing the 2nd, 3rd, and 4th sex specific quartiles of SUA with the 1st quartile separately as predictors of new-onset AF by Minnesota code, with adjustment for time-varying systolic BP, age, sex, prevalent cerebrovascular disease, Cornell voltage-duration product, and Framingham risk score at baseline in losartan-treated and atenolol-treated patients were performed. The HRs in losartan-treated patients [HR = 0.82 (95% CIs, 0.43–1.47), P = 0.50, HR = 1.12 (95% CIs, 0.65–1.92), P = 0.68, and HR = 1.88 (95% CIs, 1.17–3.01), P = 0.009] and in atenolol-treated patients [HR = 0.99 (95% CIs, 0.55–1.76), P = 0.96, HR = 1.18 (95% CIs, 0.69–2.20), P = 0.55, and HR = 1.59 (95% CIs, 0.92–2.58), P = 0.065] did not suggest a major inter-treatment difference in the association of higher SUA with AF. The use of diuretic was identical in the two treatment arms and did not influence the SUA-AF relationship.
In parallel Cox models that were adjusted for the same covariates, new-onset AF defined by physician reports (n = 531) was independently predicted by time-varying SUA level [HR = 1.10 per 16.8 μmol/L (1 mg/dL), (95% CIs, 1.04–1.16)], as was the first occurrence of AF defined by either Minnesota code or physician reports (n = 644) [HR = 1.11 per 16.8 μmol/L (1 mg/dL), (95% CIs, 1.06–1.16), both P < 0.0001].
In a parallel Cox model that adjusted for the same covariates, new-onset AF defined by physician reports (n = 535) was higher in the 4th quartile of time-varying SUA level [HR = 1.40 (95% CIs, 1.07–1.83), P = 0.015] independent of effects of history of cerebrovascular disease [HR = 1.30 (95% CIs, 1.08–1.56), P = 0.006], age [HR = 1.65 per 7.0 years (95% CIs, 1.50–1.82), P ≤ 0.0001], and male sex [HR = 1.51 (95% CIs, 1.19–1.91), P = 0.001]. Higher systolic BP, Cornell voltage-duration product, Framingham risk score, and treatment with losartan did not contribute significantly to this multivariable analysis. In another Cox model that adjusted for the same covariates (Figure 1), new-onset AF defined by either Minnesota code or physician reports (n = 652) was higher in the 4th quartile of time-varying SUA level [HR = 1.40 (95% CIs, 1.10–1.79), P = 0.007] independently of effects of greater age [HR = 1.67 per 7.0 years (95% CIs, 1.53–1.83), P ≤ 0.0001], male sex [HR = 1.45 (95% CIs, 1.17–1.80), P = 0.001], and history of cerebrovascular disease [HR = 1.32 (95% CIs, 1.11–1.56), P = 0.001]. The Cornell voltage-duration product, higher systolic BP, Framingham risk score, and losartan treatment did not contribute significantly to this multivariable analysis.
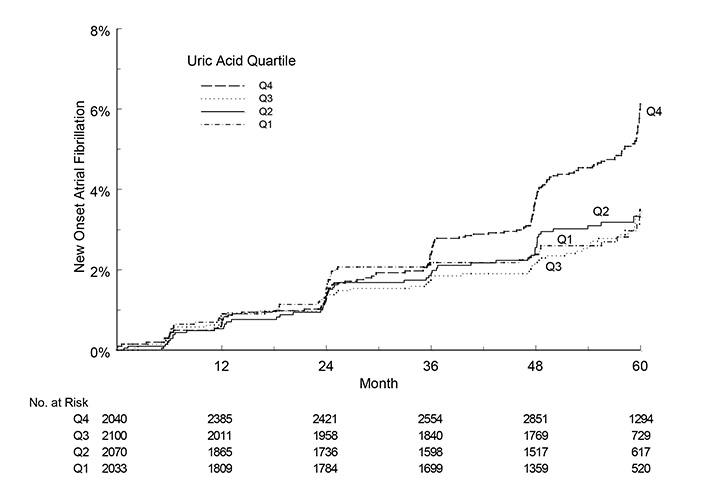
The incidence of new-onset AF (vertical axis) was higher in the 4th quartile than in lower quartiles of sex-specific SUA during five years of antihypertensive treatment (horizontal axis) as illustrated by a modified Kaplan-Meier plot. 1st through 4th SUA quartiles at baseline using sex-specific ranges: < 309, 309–353, 354–408, and ≥ 409 μmol/L for men and < 255, 255–297, 298–345, and ≥ 346 μmol/L for women. 1st quartile n = 2,033, 2nd quartile n = 2,070, 3rd quartile n = 2,100, 4th quartile n = 2,040
The present study visualized a strong relationship between time-varying SUA levels during antihypertensive treatment and new-onset AF in elderly patients with LVH. This relation between SUA and new-onset AF was present in the patients with new onset AF discovered by annual ECG and Minnesota coding, in patients reported in the adverse event data base, and when the data bases were combined. Because adverse events were not adjudicated by the endpoint committee, the two databases were not merged for all analyses. However, in as much as the results came out more or less identical compared to the incident AF taken from the ECG database, the investigators had reported new-onset AF correctly. The consistent findings irrespective of ECG detection, adverse event reports or combination of the two databases point to a strong relationship between SUA and new-onset AF.
Various mechanisms have been proposed for the effects of SUA on cardiovascular disease outcomes. SUA level is closely related to renal function and renal blood flow and may be a marker of impaired renal function. Until the beginning of this century moderately elevated SUA levels were not thought to induce renal disease. However, experimental studies suggest that induction of moderate hyperuricemia in rats causes hypertension, intra-renal arterial disease, and impaired renal function [35–38]. The hyperuricemia-associated hypertension was reversed by allopurinol, a drug that inhibits the conversion of oxalate to SUA, and by enalapril, an angiotensin-converting enzyme inhibitor. In a double blind, placebo-controlled crossover trial in 30 adolescents with hyperuricemia and hypertension, allopurinol was associated with significant reduction in office and ambulatory BP [39].
Hyperuricemia is also associated with increased platelet aggregation [40] and elevated whole blood viscosity [41]. Anker and coworkers suggested that hyperuricemia is a marker of impaired oxidative metabolism, activation of inflammatory cytokines, reduced vascular function, and even indicates a poor prognosis in heart failure [23]. Several studies have demonstrated that allopurinol improves endothelial function and peripheral blood flow in patients with type 2 diabetes, congestive heart failure [24], and in smokers [42]. Other studies suggest that SUA may affect the risk of cardiovascular disease through pro-oxidant or antioxidant effects [43, 44].
Multiple clinical studies have established a role for SUA as an independent risk factor for cardiovascular and cerebrovascular disease in various populations and for morbidity and mortality in high-risk populations with known cardiovascular and cerebrovascular disease [14, 15, 45–47] or hypertension [48]. The present study is linking higher SUA with new-onset AF in hypertensive patients who remain at high risk despite aggressive antihypertensive treatment. Previous documentation of high rates of stroke and other cardiovascular events in hypertensive patients with AF provides a hitherto unrecognized pathway via which adverse cardiovascular effects of SUA may be mediated.
Unlike prior studies of the relationship between SUA and cardiovascular outcomes that have restricted analysis to baseline measurements of SUA, the LIFE study measured SUA annually throughout the trial. This design allowed the analysis to take into account the time-varying levels of SUA and BP, variables that were affected by treatment, changes in renal function, and other factors, thereby revealing a substantially stronger relation of SUA levels to new-onset AF than could be detected by baseline measurements of SUA alone. Additionally, analyses were conducted using sex-specific partitions of SUA, based on the known sex difference in SUA levels. The predictive value of SUA in the highest quartile for new-onset AF remained strong even when sex-based partitions were used. There was a 33% reduction in the incidence of new-onset AF with losartan-based therapy compared to atenolol-based therapy with similar BP lowering in both treatment arms in the LIFE study [11]. At the same time the increase in SUA concentration during the 4.8 years was attenuated by losartan compared to atenolol, as previously described [30]. The finding was associated with the uricosuric effects of losartan and its active metabolite, therefore one might speculate whether losartan contributes indirectly, via increased uric acid excretion to reduce the risk of new onset AF.
In Figure 1 it is shown that Kaplan Meier curves split after 2–3 years into the study. This observation indicates that it takes time for SUA to influence new onset AF in patients with hypertension and LVH being aggressively treated with losartan or atenolol, two drugs that we consider preventive against new onset AF. The observation is further in line with “time-varying” SUA (and not SUA at baseline) to relate to new-onset AF.
Some few limitations of the present study need to be mentioned. The LIFE study was a prospective and randomized clinical trial set up to compare cardiovascular outcomes on the angiotensin-receptor blocker losartan vs. the beta-1 selective receptor-blocker atenolol. Incident AF and SUA were secondary endpoints and the relationship between time-varying SUA and incident AF emerged from an ad hoc analysis.
In conclusion, our data supports an association between SUA levels and increased risk of new-onset AF, and extends prior findings of the LIFE study as well as other investigations of elevated SUA levels as a possible mechanism of increased cardiovascular morbidity and mortality in hypertension. Examination of the relation between SUA levels and subsequent rates of new-onset AF in existing data sets is suggested by the present results. If the association between higher SUA and new-onset AF is further confirmed incorporation of randomized therapy to lower SUA into future studies in populations at high risk of AF will be needed for definitive determination of the role of SUA in the pathogenesis of AF.
AF: atrial fibrillation
BP: blood pressure
CI: confidence interval
ECG: electrocardiographic
HR: hazard ratio
LIFE: Losartan Intervention For Endpoint reduction in hypertension
LVH: left ventricular hypertrophy
SUA: serum uric acid
The authors thank the 945 clinical centers who participated in the LIFE study.
ESZ, KW, PMO and RBD contributed to the conception and design of this LIFE sub-study. ESZ, IMS, KW and DAH performed the statistical analyses. ESZ wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Sverre E. Kjeldsen has received ad-hoc lecture honoraria within the past 3 years from Getz, Merck Healthcare KGaA, Sanofi-Aventis and Vector-Intas. The other authors declare that they have no conflicts of interest.
The trial protocol was approved by ethical committees in all countries with participating clinical centres and done in accordance with the Declaration of Helsinki.
Written informed consent to participate in the study was obtained from all participants.
Not applicable.
The data that support the findings of this study are available from the corresponding author (RBD) upon dire need.
Merck & Co., Inc. supported the LIFE study with an unrestricted grant from 1993. Merck had a non-voting member of the Steering Committee, which designed the study and wrote the protocol. Merck did monitoring, and data was accumulated in a Merck database until study ended in 2002. Merck provided the study steering committee with free access to blinded data until 2002 and then un-blinded data after the database was cleared for queries and frozen. The steering committee including a committee statistician validated independently the main outcomes. The steering committee has always been free to analyze and interpret the data, make decisions to publish and write the papers. Merck has not had any formal role after 2002 except for sporadically provided expert statistical assistance.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Rafael Luzes ... Adalberto Vieyra
Natalia L. Rukavina Mikusic ... Mariela M. Gironacci
Gian Paolo Rossi ... Teresa Maria Seccia
Sukhwinder K. Bhullar ... Naranjan S. Dhalla
Kristin E. Reeve ... Babbette LaMarca
Casper N. Bang ... Peter M. Okin
Casper N. Bang ... Peter M. Okin
Marcello Chinali ... Richard B. Devereux
