Review
Review

Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0002-7433-9221
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0001-8061-0631
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0003-4304-1024
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0003-2027-4968
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0002-8365-0345
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0002-6859-8198
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0001-6030-4936
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
ORCID: https://orcid.org/0000-0003-1490-2577
Affiliation:
Department of Biomedical and Molecular Sciences, Queen’s University, Kingston, ON K7L 3N6, Canada
Email: tayadec@queensu.ca
ORCID: https://orcid.org/0000-0001-9062-050X
Explor Immunol. 2022;2:454–483 DOI: https://doi.org/10.37349/ei.2022.00062
Received: November 08, 2021 Accepted: March 08, 2022 Published: July 26, 2022
Academic Editor: Satish Kumar Gupta, Indian Council of Medical Research, India
The article belongs to the special issue Human Reproduction: Involvement of the Immune System
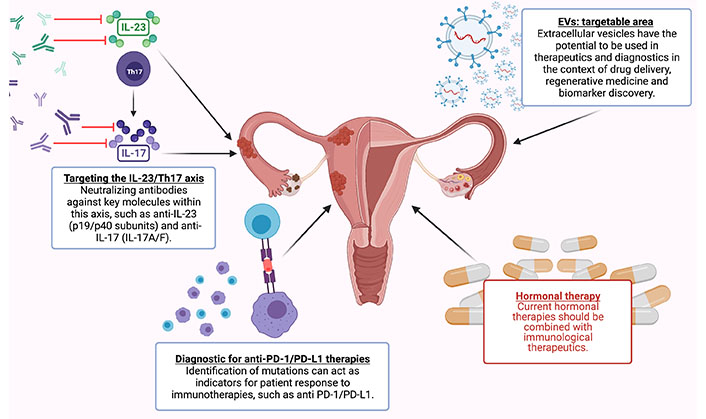
Endometriosis (EMS) is an inflammatory, gynaecologic disease characterized by the growth of endometrial tissues outside the uterus. With no satisfactory therapies or non-invasive diagnostics available, a shift in perspectives on EMS pathophysiology is overdue. The implication of immune dysregulation in EMS pathogenesis and disease progression has been an evolving area of research, with numerous immune and inflammatory pathways identified. Traditional theories regarding the establishment of endometriotic lesions have lacked mechanistic explanations for their proliferation and survival until recent research unearthed the involvement of mesenchymal stem cell (MSC) and myeloid-derived suppressor cells (MDSCs) in a complex network of immune-endocrine signaling. The unique immunology of EMS is likely owing to estrogen dominance, as endocrine imbalance reliably cultivates immune dysregulation. Many of the phenomena observed in EMS parallel immune biology seen in various cancers, including accelerated somatic mutations in endometrial epithelial cells. Here, the high mutational load leads to EMS neoantigen development which potentially contributes to the lesion immune microenvironment. As well, EMS manifests comorbidity with several chronic inflammatory diseases that share common dysregulation of the interleukin-23 (IL-23)/IL-17 pathway (as seen in inflammatory bowel disease, psoriasis, and rheumatoid arthritis). EMS is especially relevant to the study of chronic pelvic pain (CPP) as 60% of EMS patients experience this symptom and chronic inflammation is believed to be central to the process of pain sensitization. Since the onset of the disease usually occurs in adolescence, and diagnosis only occurs years later once moderate to severe symptoms have developed, it is vital to innovate non-invasive diagnostic tools for earlier detection. Several potential biomarkers are being studied, including some cytokines, gene signatures, and extracellular vesicle (EV) signatures. By incorporating the immune perspectives of EMS into our research, approaches to diagnosis, and treatment solutions, the field has more promising avenues to clearly define EMS and offer patients relief.
Endometriosis (EMS) is an inflammatory gynaecologic disease that affects about 10% of menstruating individuals worldwide. It is characterized by the growth of endometrial-like tissue at ectopic sites outside of the uterus; lesions most commonly being found on the peritoneal wall, exterior of uterus, ovary, bladder, or colon. In Canada alone, EMS is estimated to cost the healthcare system $1.8 billion annually [1]. The disease causes severe pelvic pain and infertility, and we do not have reliable non-invasive diagnostics or curative therapeutic options. To compound this difficulty, the average time between the onset of symptoms and diagnosis is approximately seven years. This is partially owing to the fact that the pathogenesis and pathophysiology of EMS are poorly understood. Aside from being under-studied, EMS is a multifaceted disease with etiologies rooted in genetics, hormonal imbalance, immune dysregulation, and phenotypic heterogeneity. Though years of research have unraveled individual contributions of these aspects, the mechanisms of their interplay remain largely elusive.
Because EMS is an estrogen-dominant disease, most first-line treatments attempt to address this imbalance through hormonal contraceptive therapies. These options do not cause regression of existing lesions, and contraception is not a satisfactory option for those who wish to conceive children. In recent years the field has acknowledged several immune cells and pathways that are dysregulated in EMS, contributing to severe inflammation, fibrosis, and angiogenesis in lesions. For this reason, immune-focused therapies with proven success in other chronic inflammatory diseases and several cancers are being explored in EMS. However, precise understanding of the complex immune-regulatory pathways is integral to the future application of immune-based therapeutics. Additionally, with high incidence of comorbidities and individual mutational variations, personalized medicine for EMS is an area needing significant research and development.
Despite these challenges, the field continually makes new discoveries in an immunological scope to understand EMS pathophysiology. The complexity of immune dysregulation in EMS is an active area of ongoing research, driving a forward deeper understanding of EMS pathophysiology and allowing us to attempt new approaches to diagnosis and treatment. In this article, we summarize current knowledge within a broader immune context and discuss areas of promising investigations with perspectives on diagnostics and treatments.
Several theories exist to explain the presence of EMS lesions, and while their applicability varies across disease phenotypes, each has its own merit. Familial associations have been documented for EMS [2–4] and have been reviewed elsewhere [5–8]. A pioneer of EMS pathogenesis research was John Sampson, and his works have been discussed extensively by Yovich et al. [9]. The widely accepted Sampson’s theory of retrograde menstruation, in our opinion, is most feasible as a part of a larger story which includes immune dysregulation and aberrant stem cell activity as discussed below.
This theory postulates that metaplasia and transdifferentiation occur in the mesothelium of the peritoneum and within the ovarian germinal epithelium, thereby transforming these cells into endometrial tissue that can cause lesion development [10]. This hypothesis may explain rare cases of EMS seen in males [11, 12]. Metaplasia could also explain rare distal EMS lesion sites such as lymph nodes [10] and lungs [13]. Some have theorized that the presence of deep infiltrating EMS (DIE) is caused by metaplasia of the coelomic epithelium, however, there is a high prevalence of rectovaginal DIE that co-occurs with other forms of EMS, where coelomic metaplasia cannot explain lesion presence [14]. Metaplasia can also arise from events unrelated to the coelomic epithelium, or from ectopic tissue implanting into peritoneal sites [15]. These theories can be applied to certain phenotypes of EMS, however, they do not account for the prevalent immune dysregulation seen in the disease pathophysiology.
The Mullerian ducts, which normally differentiate to form the female reproductive tract, are said to be involved in this etiological hypothesis of EMS. The ectopic endometrial tissue that is characteristic of EMS is theorized to originate from primitive embryonic Mullerian tissue that was misplaced during fetal development [16, 17].
Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome, caused by an anomaly in the Mullerian tract, is characterized by abnormalities or absence of the uterus and vagina, despite the normal function of ovaries and (46,XX) karyotype. MRKH patients have been shown to have a significant incidence of EMS, bolstering this hypothesis [18]. There has also been EMS documented in individuals with Mullerian anomalies, despite no obstruction in menstrual flow [19]. Additionally, cases of EMS in girls before menarche [20], as well as males with lesions along the route of the Mullerian duct, align with this hypothesis [11]. However, this theory is not sufficient for explaining the placement of lesions seen outside of Mullerian rests [12], or the immunological aspects of EMS.
Iatrogenic endometriotic growths typical of EMS can develop within incision scars from gynecologic surgery [21]. Specifically, iatrogenic scar EMS is suggested to arise from accidental auto-transplantation of endometrial cells [22–24]. Lesions may be found in incisions from cesarian, appendectomy [22], hysterotomy, tubal, trocar-site, amniocentesis, and episiotomy surgeries [25, 26]. This hypothesis explains rare cases of EMS development in those who have recently undergone pelvic surgery. Endometrial cells being transplanted into incision sites is a reality of the cesarian section, yet the occurrence of EMS in the scar is quite low, indicating other unidentified factors lead to disease development in these cases [22]. Immune tolerance during pregnancy in combination with the displacement of endometrial cells is a reasonable speculation for the etiology of lesion establishment in cases of cesarian section surgery [26]. This is evidenced by the increased risk of scar EMS for those who deliver prior to labor onset before immune tolerance has subsided [27].
Retrograde menstruation, originally proposed by Sampson [28], is currently the most widely-accepted hypothesis for the etiology of EMS. This hypothesis postulates that endometrial fragments shed during menstruation travel backward through the fallopian tubes into the peritoneal cavity, where cells can adhere and develop into endometriotic lesions.
Retrograde menstruation has been well established as a normal physiological process, occurring in 90% of menstruating individuals [29]. Some non-human primates like baboons, who also menstruate, have exhibited cases of spontaneous EMS associated with a higher prevalence of retrograde menstruation [30]. Also in baboon models, forced menstrual reflux by cervical occlusion has been shown to induce EMS [31]. Similarly, EMS and retrograde menstruation have been shown to be increased in individuals with outflow obstruction, such as those with malformation of Mullerian ducts [19]. There is also a positive association between EMS and high menstrual volume (typical of heavy periods or greater numbers of menstrual cycles) [32]. Additionally, differences in endometrial tissue adhesion capability have been identified in patients with EMS; adhesion molecules such as integrins, syndecans, cluster of differentiation 44 (CD44), intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and epithelial cadherin (E-cadherin) can be dysregulated in EMS patients, explaining the increased capacity to adhere to damaged peritoneum [12, 33].
While 90% of females exhibit retrograde menstruation, only 7–10% are estimated to have EMS [34], meaning that other factors must contribute to disease establishment. For lesion establishment to occur from displaced endometrial cells, there must be immune evasion, attachment, invasion, vascularization, and growth. Although endometrial fragments are misplaced at ectopic sites, the adaptive immune system may not mount an effective response to clear debris due to the recognition of endometrial cells as self-versus foreign antigens. Several pathways of immune dysfunction have been identified in EMS that could contribute to lesion establishment. For instance, macrophages (normally recruited for tissue repair and debris clearance) have been shown to produce increased amounts of vascular endothelial growth factor (VEGF) in EMS patients compared to controls [35]. This increase in VEGF, an angiogenic factor, could be supporting the vascularization of the lesion. Another mechanism previously described is estrogen receptor β (ERβ) interacting with apoptotic machinery to decrease tumor necrosis factor-alpha (TNF-α)-mediated apoptosis, proposed as a mechanism by which displaced cells evade immune clearance [36]. Many other mechanisms in addition to immune dysfunction can complicate the pathophysiology of the disease, such as genetic, environmental, and endocrine factors. While retrograde menstruation is a plausible theory explaining the presence of endometrial cells in the peritoneal cavity, more recent discoveries relating to stem cells and immune pathways have allowed us to theorize how immunological factors lead to EMS after endometrial cells enter the peritoneal cavity.
Somatic stem cells, also known as adult or tissue-specific stem cells, play a critical role in the regulation of adult tissue regeneration and restoration of the local microenvironment following injury [37]. The human endometrium is a dynamic tissue that undergoes cyclic regeneration through the replacement of epithelial and mesenchymal tissue following menses. A growing body of evidence suggests endometrial epithelial stem/progenitor cells are involved in the repair process, along with CD140b+CD146+ or sushi domain containing-2+ (SUSD2+) endometrial mesenchymal stem cells (MSCs), and endometrial stem cells that may be of Mullerian origin [38]. Additionally, non-resident bone marrow-derived stem cells (BMDSCs), which differentiate into populations of hematopoietic stem cells, MSCs, and progenitor cells may be involved. It is possible that these cells restore endometrial tissue. Du et al. [39] found murine uteri with reperfusion injury harbored nearly twice as many BMDSC-derived MSCs compared to uninjured uteri (42 versus 22 MSCs per 105 stromal cells). As well, BMDSCs have been shown to differentiate into non-hematopoietic lineages including kidney, intestine, lung, and muscle cells [40]. Pathogenic alteration of stem cell regulation, function, number, and location provides a basis for lesion establishment in EMS.
The MSC theory of EMS purports that endogenous endometrial stem/progenitor cells, predominantly located in the endometrium basalis, travel in the menstrual fluid to reach the peritoneum and incorporate within lesions. Several researchers have noted the presence of basalis-like tissue in the menstrual effluent and endometrium functionalis of endometriosis patients [41, 42]. As well, some have theorized a deeper plane of separation between functionalis and basalis at menstruation, theoretically including more basalis-associated progenitor cells within the menstrual effluent of endometriosis patients [43]. In the context of retrograde menstruation, this would effectively boost the progenitive capacity of menstrual effluent in endometriosis patients as proportionally more progenitor/stem cells are transported to the peritoneal cavity. One school of thought hinges on the concept that decidualization primes endometrial stromal cells to recruit MSCs and prioritize tissue remodeling as part of normal endometrium restoration after menstrual shedding [44]. It is possible that with increased or dysregulated MSCs, endometrial stromal cells herd MSCs to ectopic sites where lesion establishment occurs. In the presence of an existing lesion, local dysregulation of cytokines, such as elevated C-C motif chemokine ligand 2 (CCL2), CCL5, C-X-C motif chemokine ligand 8 (CXCL8), VEGF, and placental growth factor [45–48], contribute to an inflammatory microenvironment that may recruit stem cells to the lesion site.
The involvement of MSCs is not limited to scaffolding lesion establishment. The cells exhibit immunomodulatory properties that inhibit proliferation, activation, and differentiation of CD4+ T cells to pro-inflammatory T helper (Th) Th1 and Th17 subtypes and promote the generation of tolerogenic regulatory T (Treg) cells [49]. In addition, tissue-resident MSCs have shown the capacity to modulate the activity of natural killer (NK) cells in a manner that reduces cytotoxicity and provides feedback that positively influences the proliferative and pro-angiogenic properties of MSCs. Reduced cytotoxicity of NK cells is also well documented in EMS [50]. MSCs have been shown to polarize macrophages to the M2 phenotype through the release of prostaglandin E2 (PGE2), indoleamine 2,3-dioxygenase 1 (IDO1), CCL2, and CXCL12 [51–54]. In fact, MSCs have been found to exhibit more immunosuppressive and M2-polarizing signals in vitro when derived from EMS lesions compared to eutopic endometrium derived MSCs, which Abomaray et al. [55] theorized aids immune evasion to support lesion establishment. The secretory profile of MSCs includes chemoattractant cytokines that further MSC recruitment (CXCL12, CCL2, CXCL8) as well as antiapoptotic [insulin-like growth factor-1 (IGF-1), transforming growth factor beta (TGF-β), basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF)], proliferative [interleukin-6 (IL-6), stromal cell-derived factor-1 (SDF-1), macrophage colony-stimulating factor (M-CSF)], and angiogenic [VEGF, placental growth factor (PIGF), monocyte chemoattractant protein-1 (MCP-1), bFGF] chemokines [56]. Overall, the breadth of interactions induced by MSCs appears to promote an immunosuppressive environment, permissive to the establishment and sustainment of ectopic tissue/lesions within the peritoneal cavity.
The presence of stem/progenitor cells of BMDSC origin may be associated with disease pathogenesis, as these cells have been shown to migrate to areas of tissue damage and inflammation. A study by Sakr et al. [57] showed that CD45− donor BMDSCs favored engrafting endometriotic lesions over eutopic endometrium in EMS-induced mice following bone marrow transplantation. This evidence supports the involvement of circulating BMDSCs in endometriotic lesion establishment. Furthermore, BMDSCs could regulate the formation and maintenance of blood vessels through their supply of myeloid cells, such as macrophages and mast cells, which release angiogenic factors including VEGF, TNF-α, and angiopoietin [38, 58]. Neoangiogenesis is a key feature in EMS lesion establishment and survival.
Chen et al. [59] found endometrial stromal cells interact with BMDCs to induce them to differentiate into stromal, epithelial, or leukocyte cell types via paracrine activity. BMDCs inhibit lymphocyte immune responses through increased expression of programmed cell death 1 (PD-1) and its ligand PD-L1 expression on T cells. Indeed, elevated serum PD-1/PD-L1 expression was observed in eutopic and ectopic endometria, as well as serum-derived CD4+/CD8+ T cells in patients with versus without EMS, the occurrence of which is reported to be exacerbated by elevated estradiol [60]. The chemotactic capacity of BMDSCs is specially enhanced with elevated CXCL12 levels, as is seen in EMS lesions [61]. In a mouse model of EMS, elevated CXCL12 has been shown to support the recruitment of bone marrow cells, a source of endothelial progenitor cells, to allow for neovascularization of EMS lesions [61, 62].
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of myeloid progenitor cells with potent immunosuppressive and angiogenic activity. Several studies have noted an association between MDSC accumulation and the progression of inflammatory diseases, including EMS [63–66]. MDSCs are present in small numbers in healthy tissue, although they rapidly expand in response to inflammatory disease through interactions with molecules such as IL-1β, IL-6, IL-10, and TNF-α [65, 67]. Elevated concentrations of these inflammatory cytokines in the peritoneal fluid (PF) of EMS patients may also be implicated in the transformation of monocytes into MDSCs [68–70]. The presence of chemokines CXCL1, CXCL2, and CXCL5 at the site of the lesion promotes MDSC migration to the ectopic environment [63]. Furthermore, Guo et al. [71] observed significantly elevated concentrations of CCL3 and CCL5 in EMS patients, cytokines associated with recruitment of MDSCs. Alongside this effect, recruited MDSCs may further secrete C-X-C motif chemokine receptor 2 (CXCR2), which has been associated with interferon-γ (IFN-γ)-induced upregulation of immunosuppressive molecules (e.g., PD-1, PD-L1, CD152) that promote exhaustion of CD4+ and CD8+ T cells [66]. The presence of CXCR2+ MDSCs may further enhance the production of IL-6, which is implicated in lesion establishment through the promotion of epithelial-mesenchymal transition, a pathogenetic mechanism whereby endometrial cells transition from an epithelial phenotype to a more motive and invasive mesenchymal phenotype [72]. Chen et al. [64] associated the presence of elevated levels of monocytic-MDSCs (M-MDSCs) in peripheral blood and PF of EMS patients with suppressed pro-inflammatory Th and cytotoxic T cells in peripheral blood and enhanced anti-inflammatory Treg cells in the PF compared to healthy controls. Thus, the recruitment and accumulation of excess MDSCs in the ectopic environment may be responsible for the protection and promotion of endometriotic implants in the peritoneal cavity. In addition, Chen et al. [64] detected the presence of elevated M-MDSCs was further associated with an increase in the production of reactive oxygen species (ROS), possibly via the Notch signaling pathway [64, 73]. Findings by Ngô et al. [74] propose the involvement of ROS in increasing the proliferative rate of stromal and epithelial cells, both of which contribute a large proportion of the lesion’s composition.
MDSCs further contribute to the development of EMS through cell-to-cell interactions. Morales et al. [75] have described the capacity of MDSCs to enhance the inflammatory responses of mast cells. As well, mast cells release chemoattractants to increase MDSC recruitment and expansion, predominantly through the promotion of CCL2 secretion and elevated IL-17 production [76–78]. Within lesions, mast cells may further interact with endometrial cells to secrete CCL8, which promotes endometrial cell migration and angiogenesis at the site of release, and may further upregulate pro-inflammatory factors such as IL-6, IL-10, IL-13, TNF-α, and VEGF [79, 80].
The involvement of MSCs and MDSCs in EMS lesion establishment is a promising hypothesis with strong evidence. The findings to date are congruent with a story of EMS pathogenesis where dysregulated immune signaling bridges the gap between retrograde menstruation and lesion establishment.
EMS has long been considered an endocrinological disease. With recent research identifying significant immune dysregulation in the disease pathophysiology, we must consider the relationships between endocrine and immune systems. As EMS is an estrogen-dependent disease, hormonal therapies are the first-line treatments offered to patients, including combined estrogen and progesterone contraceptives, synthetic androgens, and gonadotrophin-releasing hormone (GnRH) agonists [81–83]. These therapies do not stop the progression of lesion growth and in many cases fail to provide patients with pain relief [84]. Estrogen has a complex role in immune functions as it can activate pro- and anti-inflammatory pathways in different cell types at different cycle points [85–87]. All immune cells express receptors for estrogen (ERα, ERβ) [86] while several express progesterone receptors (PRs), and respond to hormonal stimulation by altering inflammatory behavior, differentiating, or activating, depending on the cell and signal [88–94]. Several immune cell populations are pertinent in EMS immunopathophysiology; including but not limited to mast cells, macrophages, and various types of T cells, which are all affected by estrogen dominance.
Estrogen and progesterone can activate ERs and PRs as transcriptional factors, but also as non-genomic membrane receptors to rapidly activate cellular responses in leukocytes [95–97]. Hormonal stimulation also alters the immune signaling of the endometrium. High estrogen induces endometriotic epithelial and stromal cells to secrete more inflammatory and chemokinetic factors, including CCL5, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6, IL-8, and CXCL12 [98–100]. Since endometriotic lesion tissue overexpresses aromatase (CYP19), estradiol is biosynthesized in high amounts [101]. Combining this with impaired local metabolic enzyme activity, including poor conversion of 17β-estradiol to estrone by 17β-hydroxysteroid dehydrogenases (17β-HSD) [102], the lesion hormonally contributes to its own local inflammatory microenvironment.
Endocrine and immune systems work closely together in the menstrual cycle, and menstruation is a highly immunologically active process. It is known that most immune cells are hormonally regulated and their presence in endometrial tissue follows a cyclic fluctuation according to the menstrual cycle, with an increase in abundance of macrophages, mast cells, neutrophils, NK cells, and Th1 cells at day 26–28 [90, 103–105]. The systemic hormonal imbalance in EMS manifests as both abnormally high estrogen and also the development of progesterone resistance; a lack of response to progesterone by endometriotic tissue. In the normal endometrium, progesterone regulates appropriate apoptosis and counteracts epithelial proliferation induced by estrogen. Receptors for estrogen (ERα, ERβ) and progesterone (PR-A, PR-B) are expressed abnormally in endometriotic tissue; lesions express more ERβ and less ERα than healthy endometrium, and generally low transcript levels of PR mRNA [106, 107]. With the absence of PRs in endometrial stromal cells, normal progesterone-stimulated production of the enzyme 17β-HSD-2 is disabled, hindering estradiol metabolism and further maintaining a high estrogen level [108]. This is compounded by estradiol production resulting from aromatase activity in endometriotic tissue, as CYP19 expression is significantly increased in endometriosis lesions compared to healthy endometrium [109, 110].
Several immunological alterations are associated with supraphysiological estrogen. For example, high estrogen is associated with a systemic increase in G-CSF as granulocytes are important in the process of ovulation [111]. Normally progesterone rescues this effect, but in the progesterone-resistant state of EMS, estrogen may contribute to chronic leukocyte colony stimulation (Figure 1A). Likewise, other immune impacts of progesterone are relevant in EMS, where inflammatory events normally rescued by progesterone are left unregulated. Progesterone inhibits chemokine secretion by stromal cells [100], opposing estrogen’s agonistic effect on chemokine secretion in stromal cells (Figure 1B). Progesterone also inhibits mast cell migration [112], secretion and histamine release [113]. Mast cells fluctuate cyclically and are actively involved in the menstruation process [114, 115], and it is known that mast cells can mature and activate upon estrogenic stimulation [95]. Mast cells have been found in higher quantities in the tissue of endometriotic lesions compared to eutopic endometrium and the endometrium of healthy women [116, 117]. In addition, the vital mast SCF has been observed in higher concentrations in women with EMS [118].
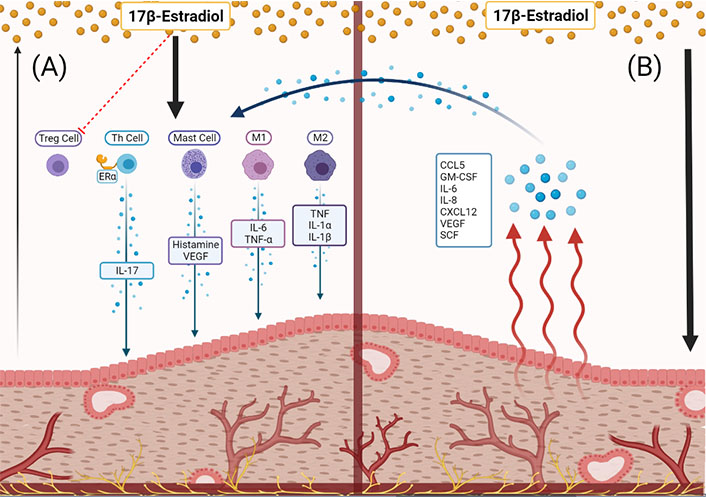
The immune-endocrine relationships of the endometriotic lesion. (A) Estradiol is sourced from systemic circulation and, to a small extent, the lesion tissue itself. Estradiol acts on immune cells to secrete inflammatory, angiogenic, chemokinetic molecules which contribute to lesion growth and disease progression; (B) estradiol causes the endometriotic lesion to produce immune signaling molecules [CCL5, GM-CSF, IL-6, IL-8, CXCL12, VEGF, stem cell factor (SCF)], subsequently acting on local immune cells to further increase inflammation, angiogenesis, and proliferation of the lesion
EMS patients have a significantly higher number of endometrial and peritoneal macrophages as compared to healthy women during the proliferative phase, and other reports found significantly higher macrophages in EMS patients regardless of menstrual phase [119, 120]. Macrophages polarize into either M1 or M2 phenotype, which have distinguished cytokine profiles and downstream actions. Generally, the M1 phenotype is pro-inflammatory, it is the classically activated macrophage that scavenges and launches immune responses to infections [121]. Meanwhile, M2 macrophages are alternatively activated and have generally anti-inflammatory downstream effects [121, 122]. Studies of macrophages under estrogenic stimulation have observed polarization to the M2 phenotype, which is considered more permissive, and this may support ectopic endometriotic lesion proliferation by allowing decidualized endometrial cells to escape clearance and survive/proliferate at ectopic sites [123–126]. Macrophages from EMS patients were found to express much more ERβ, which induces more inflammatory secretions from macrophages such as TNF, IL-1α, and IL-1β (Figure 1A) [119, 127, 128]. Since the immunomodulatory actions of estrogen are complex, other studies have found estradiol-induced polarization of macrophages to the M1 phenotype which performs inflammatory roles secreting cytokines like IL-6 and TNF-α, which may contribute to the increased inflammation of the EMS immune microenvironment [129]. Macrophages also contribute to pain symptoms in concert with estrogenic activation in EMS [87, 130]. These endocrine relationships require further investigation to improve our understanding of the roles of macrophages in EMS pathophysiology.
T cells have been identified for their role in various immunological pathways of EMS pathophysiology, and most T cell populations are influenced by estrogen [104]. Th cells exhibit an interesting relationship to estrogen and progesterone balance, where the pro-inflammatory Th1 population rises in the estrogen-dominant follicular phase while a Th2 population takes over in the luteal phase to contribute their immunoregulatory role, as observed by Faas et al. [91]. As well, estradiol has PD-1-dependent and -independent suppressive actions in the function of Treg cells (Figure 1A) [131]. Importantly, Mohammad et al. [132] elucidated the significance of ERα activation in the function, regulation, and survival of T cells by observing poor activation and survival of ERα-knockout T cells in a mouse model of colitis. A particularly interesting inflammatory pathway involving Th17 cells has been noted in EMS, and increased Th17 level in the PF of patients has been identified in association with disease severity [133]. Fuseini et al. [134] showed that ERα in Th17 cells increases the IL-23/IL-17 inflammatory pathway by upregulating IL-17A production. This inflammatory pathway has become increasingly relevant to our understanding of EMS pathophysiology, specifically relating to the recruitment of macrophages [135]. The involvement of estradiol and other endocrine factors is a significant aspect of T cell function and should be considered in researching EMS pathophysiology.
An important question to consider is whether the immune-endocrine relationship in EMS pathophysiology is bi-directional. It is known that hormones influence immune cell actions and immune signaling. However, a less observed relationship is the impact of immune events on sex hormone metabolism and receptor regulation. Some pathways in both normal and disease scenarios have been identified. In endometrial carcinoma, estrogen-stimulated IL-6 has been found to upregulate stromal cells’ expression of aromatase, driving forward metabolism of androgens to 17β-estradiol, resulting in a positive feedback loop of IL-6 and E2 production [136]. Both aromatase and IL-6 have roles identified in the pathophysiology of EMS [137], and given this feedback loop has been identified in other estrogen-dependent diseases, e.g., various breast cancers [138], this very feedback loop could exist in the disease. In several endocrine-dependent cancers, ERα can be activated by IL-1β and TNF-α via the IkappaB kinase-beta (IKK-β) pathway [139]. These cytokinic ER activation pathways are not well studied in EMS, though given the existing parallels between EMS and cancer, the capacity of immune signaling to activate sex steroid hormone receptors may be worth a deeper investigation.
Knowledge of the endometriotic microenvironment is continuously expanding with new information centered around angiogenesis, cytoarchitecture, etiology, and immune cell presence; the field is diversifying. These seemingly independent fields are all relevant to novel establishing tissues. This framework allows for parallels to be made to malignant cancerous tissues. This is not to equate EMS to cancer, but to provide an alternative perspective in analyzing endometriotic lesions through the lens of cancer research. The breadth of cancer research has made discoveries that could prove beneficial to the field of EMS research. As this school of thought has been gaining traction in the field, we hope to provide insight into the pathophysiology of EMS through this perspective. With a focus on somatic mutations and immune cell dysfunction, there may be insights into potential biomarkers and novel therapeutics for EMS.
Genomic studies in EMS patients typically aim to comment on disease etiology and heritability [140, 141]. One emerging topic is somatic mutations and their influence on EMS pathophysiology. Whole exome sequencing of EMS has revealed the presence of single nucleotide variations in ectopic and eutopic sites, with mutually exclusive mutations across these locations [142]. Alterations to genes supporting cell adhesion, apoptosis, and cell cycle pathways have all been identified and maintain a heterogeneous presentation across patients. These pathways have similarities with known cancer driver mutations, allowing for proliferation and metastasis of tissues. Cancer driver mutations such as AT-rich interaction domain-containing protein 1A gene (ARID1A), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), and Kirsten rat sarcoma virus proto-oncogene (KRAS) mutations have been found in the epithelium of EMS patients [143–145]. There is still a large debate over the role that these mutations play in the potential progression of EMS to endometrial cancers, however, even in non-cancerous lesions, such as those in DIE, these mutations are present [143]. The acquisition and interpretation of this data have been extensively reviewed elsewhere [140], however, one dimension of this discussion yet to be brought to EMS is the impact of somatic mutations on immune function. Cancer literature highlights the influence that somatic mutations can have on immune cell infiltration as well as cytotoxic capacity [146–149], thus calling us to discuss the effect of mutations in pathways beyond just the cytoarchitecture of the lesion itself.
Mutational load, the amount of somatic mutations present within a tissue, has been correlated with tumor leukocyte infiltration [146, 148, 149]. This in part could be due to the over-activation of the mutated signaling pathways having downstream targets that contribute to inflammation, such as IFN-γ and TGF-β [147, 150, 151], however, the formation of neoantigens is where the current focus lies. Neoantigens are proteins that have been modified due to mutations, and that present as novel targets for the immune system. With a higher mutational load, there is the potential for greater neoantigen formation and thus a greater capacity for mounting an immune response against these new targets (Figure 2) [146]. Across various cancers, there is an association with mutational load, neoantigen formation, tumor leukocyte infiltration, and the expansion of the T cell repertoire [148, 149]. Comparing the ratio of gene expression within T cells infiltrating these genetically different tissues, there is an increase in cytolytic capacity, as measured by granzyme A expression as well as an increase in Treg marker forkhead box p3 (Foxp3), potentially counterbalancing the immune cell clearance taking place [148]. Due to this heightened tumor leukocyte infiltration, the mutational load can be associated with better patient outcomes, however, this is very microenvironment specific, as the functional capacity of the infiltrating cells still needs to be considered. Additionally, the mutational landscape at each tumor or potentially lesion site has its own microenvironment. Within EMS, both intra- and inter-lesion heterogeneity of mutations has been identified, thus it is fair to assume that the neoantigens present will differ at all locations. This adds complexity, in that parallel immune interactions are taking place with specificity to their microenvironment. EMS literature has identified the presence of CD8+ T cells as well as an abundant immune infiltrate at the lesion site [152, 153]. Further research is needed however to elucidate these immune populations and to identify the functional status of these cells. This information would bring us one step closer to understanding the lesion’s complex network, of which somatic mutations are a part.
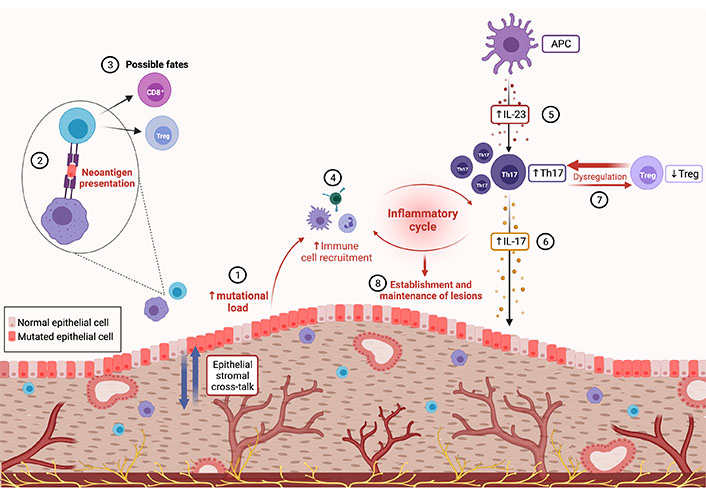
Lesion immune microenvironment and inflammatory cycle. (1) Inherent mutational predispositions differ across epithelial and stromal cells. Epi-genetic stromal cell alterations promote an environment that stimulates further replication and mutational acquisition within epithelial cells. (2) This increased presence of somatic mutations allows for the formation of neoantigens which are detected by antigen-presenting cells (APCs) and presented to adaptive immune cells. (3) The lymphocyte fate from these interactions is yet to be determined, forming a novel target of the investigation to define immune phenotype and activation status of T cells in response to lesion-specific neoantigens. (4) Phenotypic fate is not yet determined, but higher immune cell infiltration has been seen in lesions with increased somatic mutations. Elevated immune cell infiltration is a facet of another potentially dysregulated pathway within the lesion microenvironment. (5) IL-23, which is increased in EMS, is mainly secreted by APCs. (6) This cytokine is crucial for the expansion and stabilization of “pathogenic” Th17 cells resulting in high levels of IL-17, which has potent pro-inflammatory and pro-angiogenic actions in EMS. (7) IL-23 may also then promote the dysregulated ratio of Th17 and Treg cells seen in EMS derailing immune homeostasis. (8) As IL-17 receptor A (IL-17RA) is expressed on immune cells including T cells, IL-17 may promote Th17 cells to induce inflammation, recruit neutrophils, and further produce pro-inflammatory cytokines, supporting the establishment and maintenance of endometriotic lesions
As previously mentioned, it is the epithelial cells within endometriotic lesions as well as tumors, that harbor the vast majority of somatic mutations [143–145, 154]. Stromal cells have been reported to have very few or no somatic mutations, however, they do carry epigenetic modifications [154–156]. This marked difference in mutation presence is explained by the relationship between these tissues and their contribution to the lesion/tumor site; epithelial cells are under the influence of stromal cells [150]. The epigenetic modifications of stromal cells cause alterations to their receptor expression and cytokine release [157]. Both mechanisms are exploited to promote the proliferation of epithelial cells, increasing their mutation acquisition. Within EMS this can manifest as increased ERβ, supporting proliferation by heightening the sensitivity to estrogen, PR downregulation contributing to progesterone resistance, and increased paracrine cytokine production of growth factors (HGF and TGF-β) targeted to the epithelium [150, 157]. Once combined, these signals increase the rate of epithelial proliferation and mutation. Advancing our understanding of this complex relationship will allow for a greater understanding of these inherent cell properties and how mutations fit in their physiology.
The study of somatic mutations within EMS is in its infancy. New findings continue to highlight the dynamic nature of these tissues, guiding us to adjust our perception of normal tissues and the role of cancer driver mutations. Lac et al. [158] identified that 50% of healthy endometrium contains cancer driver mutations and that acquisition will increase proportionally with age. Mutations are no longer relegated to the abnormal, requiring us to adjust our perspective of “normal” tissues. We must seek to understand the pathways they exploit and the potential room for their influence at various stages of disease progression. Adding to this complexity is the presence of allelic variations in mutations, thus while the presence of one may influence tumor leukocyte infiltration and correlate with positive patient outcomes, another may not [147, 159]. The importance of individual microenvironments cannot be understated and will eventually be the focus in therapeutics, as immune-centered therapies continue to rise. Anti-PD-1/PD-L1 therapies in various cancers have looked to mutational load as an indicator of patient success with immunotherapies [146, 147, 160]. This underscores the need for understanding the status of immune cells present within the lesion and how they interact with these mutations to capitalize on those networks. Finally, more work is needed considering a place for neoantigens within EMS. The prevalence of these mutations in normal tissues may be preventing the immune system from mounting a response to those within endometriotic lesions as it does not recognize these as foreign. The selection process of T cells within the reproductive tract as well as the repertoire of these cells needs further research to understand at what point these mutations can be recognized as foreign and then cleared.
Chronic pelvic pain (CPP) is defined as non-menstrual pain originating from the pelvic region which typically persists for more than 6 months [161, 162]. CPP is estimated to affect 26% of the world’s female population, accounting for 40% of laparoscopies and 12% of hysterectomies annually in the US [162]. In regards to EMS, which is commonly characterized by debilitating CPP, more than 60% of all EMS patients report CPP [163]. In fact, of the 60% of laparoscopies performed for CPP of which an abnormality is identified, 85% of these patients reveal early-stage EMS or adhesions [162]. Existing evidence indicates that CPP can frequently persist in EMS patients following successful lesion excision, despite a lack of lesion regeneration and/or the usage of hormonal therapies [163, 164]. Furthermore, it is well known that the number or spread of lesions, as classified by the American Society for Reproductive Medicine (ASRM) staging, is not indicative of one’s degree of pain [161, 165–167]. This supports the idea that EMS-related CPP is not entirely reliant on the presence or number of lesions and is likely driven by other factors. Moreover, due to the complexity of CPP symptomology, which is reviewed extensively elsewhere [162, 168], identifying the exact mechanism(s) of CPP is extremely challenging. However, several hypotheses have been proposed to be the cause of CPP, including immune dysfunction, neuropathies, and psychosocial modulators [162, 169].
The scope of CPP is broad, however, the aspect of immune dysfunction is a very understudied area. While the mechanistic link between CPP and EMS remains undefined, one of the avenues being explored is the link between CPP, EMS, and inflammation. There are various evidence that CPP results from a localized pro-inflammatory milieu containing upregulated Th cells, mast cells, and the secretion of inflammatory mediators [162, 169, 170]. EMS is characteristically known for its chronic inflammatory milieu [171]. This link is particularly of interest due to the well-documented immune-centered comorbidities in EMS and their role in CPP, as discussed elsewhere [169, 170, 172]. The dysregulated inflammation in CPP/EMS increases the infiltration of immune cells, such as mast cells, macrophages, and lymphocytes [169, 171, 173], which further amplifies inflammation in the microenvironment. Specifically, mast cells promote the infiltration of other inflammatory cells that further activate mast cells, creating a positive feedback loop of chronic inflammation [169]. Cytokines secreted by activated mast cells (IL-6, IL-21, IL-23, and TGF-β) are also known to regulate Th17/Treg cell differentiation and plasticity, suppressing Treg control of self-tolerance which is crucial to avoid immune dysfunction [169, 170]. Mast cells may play a key role in chronic pain and hyperalgesia in EMS due to their expression of nerve growth factor (NGF) receptors, as the downstream results of binding of NGF may result in a feedback mechanism which promotes pain sensitization [169]. Furthermore, due to the shared neural pathways innervating the bladder, colon, and female reproductive tract, this can lead to cross-organ sensitization and CPP [162, 163].
It is suggested that cross-talk between the nerves and immune cells/modulators within the chronic inflammatory milieu of EMS results in the activation of central and peripheral pain pathways, magnifies pain signals, and leads to CPP [162, 169–171, 173, 174]. Particularly, PGE2, TNF-α, NGF, CCL5, IL-8, and IL-1β are key inflammatory mediators increased within the endometriotic microenvironment which directly activate sensory nerve endings and create a positive feedback loop to further exacerbate inflammation [163]. In EMS, IL-1β specifically is known to stimulate NGF promoting local neurogenesis, and is associated with severe deep dyspareunia [175, 176].
It is also crucial to consider psychosocial modulators, such as trauma, abuse, and poor mental health as possible contributing factors for CPP. Women with CPP are significantly more likely to report experiencing physical/sexual/verbal/emotional abuse during childhood [162]. Although the cause-and-effect of this pain is difficult to decipher, it is speculated that these psychosocial modulators uniquely sensitize patients due to their experience of trauma, making them more vulnerable to developing CPP. As psychosocial health breaches the scope of this article, we encourage readers to review the literature on this topic [162, 177, 178]. Overall, it is clear that CPP has an extremely complex etiology. However, as there are growing reports of these key inflammatory mediators contributing to CPP, we must improve our understanding so as to utilize this novel information that will aid in defining disease pathophysiology and the development of an effective and non-invasive therapy avenue for EMS patients suffering from CPP.
As previously mentioned, there are a few key immunological factors known to be increased in the PF of EMS patients, including IL-1β, IL-6, TGF-β1, IL-17, and VEGF [174, 179, 180]. These mediators greatly influence the microenvironment in EMS, driving neurogenesis and persistently bathing lesions in a pro-inflammatory and pro-angiogenic environment that is engineered to support lesion establishment and survival [161, 165, 171, 181]. Interestingly, IL-1β, IL-6, and TGF-β1 have been well-documented to be directly involved in the differentiation of “pathogenic” Th17 cells, when in combination with IL-23 [182–185]. Specifically, TGF-β1, IL-1β, and IL-6 are known to promote Th17 cell differentiation from naive CD4+ T cells by inducing a signal transducer and activator of transcription 3 (STAT3)-dependent expression of IL-21, IL-23R, and retinoid-related orphan receptor γt (RORγt), a lineage-specific transcription factor required for Th17 cell generation which promotes expression of genes IL23R and IL17A [182–184]. IL-17 and IL-21 produced by developing Th17 cells then mediate Th17 cell amplification, while IL-23 is particularly crucial for the expansion and stabilization of Th17 cells, as well as their ability to induce tissue inflammation [182–185]. Exposure to IL-23 also reduces the concentration of anti-inflammatory IL-10 in developing Th17 cells, which may further evoke a “pathogenic” profile in these cells (Figure 2) [182, 185]. In fact, IL-17-producing Th17 cells generated with TGF-β1 and IL-6 alone have been found to be incapable of inducing autoimmune disease or producing “pathogenic” Th17 cells without further exposure to IL-23 [182, 185]. The key role of IL-23 is further underlined by evidence that loss or inhibition of the IL-23-specific p19 subunit results in protection from various autoimmune diseases, including experimental autoimmune encephalomyelitis and autoimmune colitis, whereas loss of IL-17 or IL-17RA alone resulted in no protection or exacerbation of disease [184–186].
It is important to note that these Th17 cells are highly plastic and can be driven to become either “non-pathogenic”, producing IL-10 and protective against pathogens, or “pathogenic”, producing IL-17 and contributing to chronic inflammation [182, 185, 187]. As EMS patients have elevated levels of IL-17 compared to healthy controls [180, 188], Th17 cells are likely “pathogenic” in EMS and a potential source of IL-17. This is of interest due to the well-known pro-inflammatory and pro-angiogenic actions of IL-17 in chronic inflammatory disorders and the pivotal role of this cytokine in the pathogenesis of EMS [135, 188, 189]. Furthermore, as levels of IL-23 have been found to be elevated in EMS [179, 190, 191], IL-23 is likely a key contributor in driving Th17 cells to this pathogenic profile, producing IL-17 and exacerbating EMS [188]. However, the involvement of group 3 innate lymphoid cells (ILC3s) in this IL-23/IL-17 axis must also be examined in the pathophysiology of EM, as IL-23 can similarly act on ILC3s to produce IL-17 [192, 193].
There is a critical balance between Th17 and Treg cells necessary to maintain immune homeostasis [194]. Disruptions in this ratio of Th17 and Treg cells have been well documented in EMS [133, 195–197]. Specifically, patients with EMS have been found to have lower Treg cells (tolerant) and greater Th17 cells (inflammatory), and this Th17 increase is associated with disease severity [133, 197]. This is of interest, if IL-23 is a key contributor in driving Th17 cells to a pathogenic profile, IL-23 may then control the balance between Th17 and Treg cells which is critical to preventing chronic inflammation and autoimmunity [194, 198–200].
This evidence may call into question whether endometriosis should be investigated as an autoimmune disorder. Indeed, some studies have found an association of endometriosis with autoimmune diseases, and others have identified autoantibodies within endometriosis [201]. However, according to a systematic review and meta-analysis by Shigesi et al. [202], out of 26 investigations, only 5 had good quality evidence, of which 4 reported an association of endometriosis with one or more autoimmune diseases.
As we have discussed, the IL-23/Th17 axis is dysregulated in EMS, as well as various other chronic inflammatory diseases, including psoriasis, inflammatory bowel disease, rheumatoid arthritis, and multiple sclerosis [185, 188, 200, 203, 204]. The clinical presentation of these diseases alongside EMS has been extensively discussed [165, 202, 205–208]. Here, we will focus on the current gaps in the literature, particularly on the possible pathophysiological pathways shared between EMS and its comorbidities in order to aid in the discovery of novel therapeutic targets. The crucial role of the IL-23/Th17 axis in the pathogenesis of various chronic inflammatory diseases has been confirmed using IL-23-specific p19 subunit inhibitors [209–211]. Currently, a few p19-specific IL-23 inhibitors have been Food and Drug Administration (FDA)-approved for clinical use in the treatment of psoriasis and psoriatic arthritis, such as risankizumab, tildrakizumab, and guselkumab, which are also currently in trial for other diseases with dysregulated IL-23/Th17 axis, such as Crohn’s disease [209, 212–217].
Since the dysfunctionality of the IL-23/Th17 axis is a treatment target for the diseases comorbidly exhibited with EMS, it would also serve as a promising therapeutic target for EMS. Furthermore, as IL-23-driven Th17 cell differentiation is induced through Janus kinase (JAK)/STAT3 signaling, and this pathway is upregulated in EMS [218], this could provide an additional therapeutic target for EMS or any condition with dysregulated IL-23/Th17 axis. In fact, tofacitinib, a JAK inhibitor was tested on JAK/STAT signaling in an EMS mouse model and was found to result in EMS lesion regression and reduced adhesion burden as compared to controls [218]. This dysfunctional signaling pathway may be a potential therapeutic target for EMS comorbidities, such as psoriasis, as STAT3 has recently been shown to be a key player in the pathophysiology of psoriasis and psoriatic-like inflammatory conditions [219]. STAT3 has also been found to be positively correlated with rheumatoid arthritis and STAT3 loss was found to block joint inflammation in mouse models of arthritis [220–222]. This evidence suggests that both the IL-23/IL-17 axis and JAK/STAT3 signaling pathways may be viable therapeutic targets deserving further exploration in the treatment of EMS and related comorbidities (Figure 3).
The currently available options for EMS treatment are predominated by hormonal therapies, but many other opportunities exist to treat the immune dysregulation of EMS and target specific immune pathways. As EMS is a highly complex disease, treatment plans should move towards a personalized medicine approach, incorporating the individual’shormonal and immunological profiles that can be captured through extracellular vesicle (EV) signatures.
The current gold standard for diagnosis of EMS is diagnostic laparoscopy with inspection of the abdominal cavity and histological confirmation of suspected lesions. Considering the complex manifestations of EMS, obtaining the correct preoperative diagnosis is fundamental in defining the best treatment strategy. The World Endometriosis Research Foundation has agreed that one of the key research priorities for EMS is the development of reliable non-invasive tests [223]. However, in a systematic review, May et al. [224] evaluated over 100 biomarkers for EMS in serum, plasma, and urine, and could not identify any biomarker or diagnostic panel to recommend for clinical application. The heterogeneous nature of EMS poses a challenge in developing diagnostics capable of distinguishing different phenotypes and stages of EMS. Furthermore, the high incidence of comorbidities experienced by EMS patients create another challenge in finding diagnostic biomarkers; overlapping inflammatory and endocrine mechanisms of comorbid diseases can confound EMS signs and symptoms. Since surgery is invasive and expensive, other tests including imaging and biomarkers [glycoproteins, growth factors, microRNAs (miRNAs), and long noncoding RNAs (lncRNAs)] have been evaluated for their non-invasive diagnostic potential [225, 226].
Immunological markers and inflammatory cytokines have been extensively studied in EMS patients, and despite an abundance of findings, no diagnostic value has been established. The most representative are IL-1, IL-6, IL-8, IFN-γ, MCP-1, and TNF-α [224]. Higher serum levels of IL-6 in EMS patients have been reported [227], though two systematic reviews could not verify a link between elevated serum levels of IL-6 and EMS due to testing parameters varying between studies [224, 228]. Several studies have shown higher levels of TNF-α in the serum of women with EMS [229–231], while others reported no difference [232, 233]. The concentration of IL-8 is reported to be increased in the PF of women with EMS [234] in correlation with disease stage severity [235]. As well, Carmona et al. [236] have reported that IL-8 is significantly higher in the serum of patients with ovarian endometrioma compared to healthy controls, but not in the serum of DIE patients. Several cytokines have been reported with higher serum values in earlier stages [232]. If ranges can be clearly defined, this could potentially help differentiate the EMS stage during diagnosis.
As discussed earlier, IL-17A and IL-23 are heavily involved in the pathogenesis of EMS and are found at significantly higher concentrations in the PF of EMS patients than in controls [188, 237, 238]. Furthermore, IL-17 along with TNF-α are involved in increasing IL-8 secretion [239]. As these cytokines have all been observed in higher levels in EMS patients’ serum or PF, and are all relevant to Th17-mediated inflammation, this axis is worth investigating further in terms of diagnostic panel design.
Other ILs have been studied in EMS, such as IL-4 which has been found to have higher serum values in adolescents with EMS [240]. As well, elevated serum IL-32 has been observed in EMS patients compared to controls [241]. Specifically, in cases of DIE, IL-33 has been found elevated in the serum and PF of patients versus healthy controls, with levels correlating to disease severity [242]. In peripheral blood, IL-8, MCP-1, and CCL5 showed potential as biomarkers, being significantly increased in EMS patients versus controls [48]. NK cells (CD57 + CD16) as potentially viable biomarkers have been reported to be lower in EMS and elevated one month after surgery to resect EMS lesions [243]. Interestingly, the expression of a killer inhibitory receptor known as killer inhibitory receptor 2 DL1 (KIR2DL1), on NK cells is reported to be higher in women with EMS [244]. This may explain why NK cells are less active in the disease, but more importantly, this discrepancy may be useful in identifying EMS. Copeptin, a molecule increased in inflammatory conditions, has been found significantly higher in EMS patient serum compared to controls and in correlation with disease stage severity [245]. In a study by Tuten et al. [246] the inflammatory biomarker glycoprotein YKL-40 was significantly elevated in patients with EMS compared to healthy controls, and positively correlated with disease severity. While several immunological biomarkers have been studied in EMS, many have failed repeatability tests. As well, most of these biomarkers are not EMS-specific enough to reliably diagnose EMS, given comorbid inflammatory diseases. For immunological biomarkers to be applicable in diagnostics, more research is required to define a unique panel of analytes.
In recent years, exosomes have emerged as a novel source of biomarkers in liquid biopsy has emerged with the potential to be used for early diagnostics and drug delivery-based therapeutics. Exosomes are a unique subset of EVs [247], at 30 nm to 150 nm in size they carry proteins, DNA, RNA, miRNA, and lncRNA [248]. These vesicles play an important role in the exchange of biological information between different cells; the incorporated genetic molecules can be taken up by neighboring or distant cells, where their contents enact downstream functions in recipient cells [248]. A study by Sun et al. [249] showed that exosomes isolated from the eutopic endometrium of a mouse model of EMS polarized macrophages into an M2-like phenotype and caused a reduction in their phagocytic ability, thereby enhancing the development of lesions. Furthermore, primary human endometrial stromal cell cultures from EMS patients were found to secrete exosomes with a unique proangiogenic miRNA profile, and these exosomes conferred proangiogenic (tubulogenic) properties to human umbilical vein endothelial cells (HUVECs) in culture [250]. The link between exosomes, immune function, and angiogenesis in EMS is what makes exosomes an exciting route to explore in noninvasive biomarker research.
It has been reported that exosomes from ovarian endometrioma patients showed high levels of ecto-nucleotidases compared to simple ovarian cysts [251]. Ecto-nucleotidases regulate extracellular ATP and rising extracellular adenosine levels, contributing to the local immune suppression necessary for EMS progression [251]. Detecting ecto-nucleotidases in more accessible biological fluids would make them promising non-invasive EMS biomarkers [251]. Our group has demonstrated that EVs from EMS patient tissues and plasma samples carry unique signatures of miRNAs and lncRNAs [252]. Endometrial (epithelial and endothelial) cell cultures treated with patient and control plasma-derived EVs exhibited signatures of inflammatory and angiogenic cytokines involved in EMS progression [252]. miRNA-lncRNA proteomic signatures and networks inherent in EMS patient EVs have the potential to be used as biomarkers [252]. Further, Zhang et al. [253] showed that two exosomal miRNAs, miR-22-3p and miR-320a are significantly upregulated in the serum of EMS patients. The two miRNAs may play an important role in EMS occurrence and development and have the potential to be biomarkers for the diagnosis of EMS [253]. Recently it was reported that exosomes isolated from PF samples of 28 women at different stages of the disease showed specific proteins in the exosomes that were absent in the controls [254]. Five proteins were found exclusively in the EMS groups: peroxiredoxin 1 (PRDX1), histone H2A type 2-C, annexin A2 (ANXA2), inter-alpha-trypsin inhibitor heavy chain 4 (ITIH4), and the tubulin α-chain [254]. These proteins specifically found in EMS patient exosomes open up new avenues as biomarkers. It was illustrated in a study by Qiu et al. [255] that the exosomal antisense hypoxia-inducible factor (aHIF) is highly expressed in serum and endometriotic lesion tissue of EMS patients. This study also demonstrated that upon HUVECs internalizing exosomes from EMS cystic stromal cells, exosomal aHIF regulated angiogenesis-related genes, facilitating angiogenesis [255]. This highlights the potential clinical value of circulating exosomal aHIF as a biomarker. Overall, despite extensive efforts to find EMS biomarkers, no previous candidate has achieved the sensitivity and specificity required for a viable diagnostic tool. Exosomes show great promise as biomarkers and as novel drug delivery vehicles in cellular therapy (Figure 3). We must standardize the protocols and identification methods such that exosome biomarkers can be used in clinical settings.
Various phenotypes of EMS have distinct mechanisms, and as a result, a single distinctive biomarker is not feasible for all EMS cases. This is a central challenge to developing diagnostics, and further research is needed to identify a panel of biomarkers that accounts for the heterogeneous manifestations of EMS.
EMS is a highly complex, heterogeneous disorder that has multi-factorial etiology intertwined in genetic, hormonal, immune, and environmental predispositions. Recent evidences have uncovered the dynamic nature of the EMS lesion microenvironment with particular emphasis on immune-inflammatory changes and the evolving relationship between endocrine, immune, and pain aspects. While the general consensus in the literature is that EMS is a systemic disorder, further evidence is required to validate this notion. The puzzling question yet to be answered is whether the lesion microenvironment contributes to the inflammatory changes or arises as a response to the growing lesion. Analysis of inflammatory markers in PF and within the lesion itself can only provide a snapshot of the disease captured at the time of sample collection. However, we need to consider how long a patient has been harboring these lesions and associated chronic inflammation, hormonal imbalance, somatic mutations, or co-morbidities that might shape EMS lesion architecture. Another knowledge gap is the molecular/cellular underpinning of endometriotic lesion initiation and subsequent progression. Does immune dysfunction in an EMS patient facilitate lesion initiation? Studies focused on single-cell RNA sequencing within the lesion microenvironment will provide new context associated with the immune and non-immune basis of disease-specific pathways. It is important to keep in mind that the chronic nature of the disease combined with concurrent therapies to relieve lesion growth and pain further complicates the precise understanding of the pathophysiology of the disease.
There is growing consensus in the EMS research community that perhaps each EMS phenotype such as DIE, ovarian endometrioma, and peritoneal lesions have unique pathogenetic mechanisms, yet most studies lump EMS as one entity. Furthermore, associated comorbidities pose a significant challenge to understanding the pathophysiology of EMS. Another major consideration is the lack of suitable animal models that would mimic the spontaneous human disease scenario. Despite these challenges, research for the last decade in the EMS field has revealed new insights into disease pathogenesis via cell-cell communication through EVs, miRNAs, genome-wide association studies, etc. Going forward, the integration of new advances in genomics, proteomics, and lipidomics, combined with the generation of new experimental animal models including humanized mice will help pinpoint specific mechanisms contributing to EMS lesion growth and identify new diagnostic and therapeutic targets.
aHIF: antisense hypoxia-inducible factor
BMDSCs: bone marrow-derived stem cells
CCL: C-C motif chemokine ligand
CD44: cluster of differentiation 44
CPP: chronic pelvic pain
CXCL: C-X-C motif chemokine ligand
DIE: deep infiltrating endometriosis
EMS: endometriosis
ERβ: estrogen receptor β
EV: extracellular vesicle
GM-CSF: granulocyte-macrophage colony-stimulating factor
IFN-γ: interferon-γ
IL: interleukin
JAK: Janus kinase
lncRNAs: long noncoding RNAs
MCP-1: monocyte chemoattractant protein-1
MDSCs: myeloid-derived suppressor cells
miRNAs: microRNAs
MSCs: mesenchymal stem cells
NGF: nerve growth factor
NK: natural killer
PD-1: programmed cell death 1
PD-L1: programmed cell death-ligand 1
PF: peritoneal fluid
PRs: progesterone receptors
STAT3: signal transducer and activator of transcription 3
TGF-β: transforming growth factor beta
Th: T helper
TNF-α: tumor necrosis factor-alpha
Treg: regulatory T
VEGF: vascular endothelial growth factor
AM conceived the main themes and subsections of the article. AM, DS, KZ, DH, KS, and SA reviewed and interpreted literature and wrote body of paper. AM, DS, KZ devised schematic figures. DS created illustrations. HL reviewed literature, advised on article design, and revised body of text. CT and MK advised on article design and direction and offered expertise on the article subject matter. CT oversaw the writing process and critically revised body of text.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Gustaaf Albert Dekker, Pierre Yves Robillard
Kushaan Khambata ... Satish K. Gupta
Gursaran P. Talwar ... Krishna M. Ella
Julia Szekeres-Bartho
Shigeru Saito ... Sayaka Tsuda
Noémie Abisror ... Arsene Mekinian
Ruchi Sachdeva, Rahul Pal
Shibin Cheng ... Surendra Sharma
Betcy Susan Johnson, Malini Laloraya
Pier Luigi Meroni ... Francesco Tedesco
Marijke M. Faas
Alaa Kazhalawi ... Nathalie Lédée
Chiara Agostinis ... Roberta Bulla
Mickey V. Patel ... Charles R. Wira
Thanh Luu ... Joanne Kwak-Kim