 Review
Review

Affiliation:
1Department of Clinical-Surgical Diagnostic and Pediatric Sciences, University of Pavia, 27100 Pavia, Italy
2Unit of Pain Therapy, IRCCS Fondazione Policlinico San Matteo, 27100 Pavia, Italy
Email: silvia.natoli@unipv.it
ORCID: https://orcid.org/0000-0003-3758-5375
Affiliation:
3Department of Medical Biotechnology and Translational Medicine, University of Milan, 20133 Milan, Italy
ORCID: https://orcid.org/0000-0002-9668-3103
Affiliation:
4Division of Anesthesia and Pain Medicine, IRCCS Fondazione G. Pascale, National Cancer Institute of Naples, 80131 Naples, Italy
ORCID: https://orcid.org/0000-0002-1165-6713
Affiliation:
1Department of Clinical-Surgical Diagnostic and Pediatric Sciences, University of Pavia, 27100 Pavia, Italy
ORCID: https://orcid.org/0009-0000-5839-5461
Affiliation:
2Unit of Pain Therapy, IRCCS Fondazione Policlinico San Matteo, 27100 Pavia, Italy
Affiliation:
5Anesthesiology and Pain Department, Fondazione Istituto G. Giglio of Cefalù, 90015 Cefalù, Italy
ORCID: https://orcid.org/0000-0003-2705-1548
Affiliation:
2Unit of Pain Therapy, IRCCS Fondazione Policlinico San Matteo, 27100 Pavia, Italy
Affiliation:
2Unit of Pain Therapy, IRCCS Fondazione Policlinico San Matteo, 27100 Pavia, Italy
Affiliation:
2Unit of Pain Therapy, IRCCS Fondazione Policlinico San Matteo, 27100 Pavia, Italy
ORCID: https://orcid.org/0009-0003-4019-9456
Affiliation:
4Division of Anesthesia and Pain Medicine, IRCCS Fondazione G. Pascale, National Cancer Institute of Naples, 80131 Naples, Italy
ORCID: https://orcid.org/0000-0002-3286-9531
Explor Immunol. 2026;6:1003254 DOI: https://doi.org/10.37349/ei.2026.1003254
Received: September 23, 2025 Accepted: April 08, 2026 Published: May 27, 2026
Academic Editor: Apostolos P. Georgopoulos, University of Minnesota, USA; Bernhard Ryffel, University of Orleans, France
The article belongs to the special issue Immunology and Pain
The intricate involvement of glial cells in chronic pain mechanisms represents a paradigm shift in our understanding of pain processing. From microglial-mediated neuroinflammation to astrocytic modulation of synaptic function, glial cells emerge as critical players in the complex neurobiology of chronic pain. As research continues to unravel the multifaceted roles of these cells, novel therapeutic targets and strategies are likely to emerge, potentially revolutionizing the management of chronic pain conditions. Chronic pain is complicated by frequent comorbidities such as fatigue, sleep disturbances, cognitive impairment, and mood disorders. Here, we hypothesize that neuroinflammatory processes are at the root of pain chronification and serve as the common thread linking characteristic hypersensitivity with the cognitive and emotional comorbidities typically associated with chronic pain.
Chronic pain represents a significant global health burden, affecting millions of individuals worldwide and posing substantial challenges to healthcare systems. It is defined as pain persisting or recurring for more than three months, often becoming a predominant clinical issue for affected individuals. It is associated with significant disability, mood disorders, and reduced quality of life [1]. Chronic pain can arise from sustained nociceptive activity subsequent to a peripheral tissue lesion or from aberrant nociceptive activity following damage or disease of the somatosensory nervous system, extending beyond the expected period of healing. In addition, chronic pain may lack a clear cause, occurring in conditions such as fibromyalgia (FM) or other forms of pain in which there is no clear activation of peripheral nociceptors nor evidence of damage to the somatosensory nervous system [2, 3]. For this latter pain phenotype, the term nociplastic pain has been coined [4]. When nociplastic mechanisms are suspected, pain is disproportionate to any identifiable tissue damage, and the picture is complicated by frequent comorbidities such as fatigue, sleep disturbances, cognitive impairment, and mood disorders. Nociplastic pain may also be secondary to sustained nociceptive activity or to pain amplification after nerve injury, thus reflecting aberrant processing of nociceptive signals at the central level, characterized by gain amplification and dysregulation of endogenous pain modulatory systems. Therefore, chronic pain is often referred to as a state of increased pain sensitivity maintained by mechanisms not yet fully elucidated, which we generically call “central sensitization” [3, 5]. Previous research has focused on mechanisms of enhanced synaptic responses in the spinal cord initiated and maintained by nociceptive activity (either orthotopic, as in the case of tissue damage, or heterotopic, as in the case of neuropathic pain) and by subsequent release of excitatory neurotransmitters and neuropeptides in the synaptic cleft, activating NMDA-mediated synaptic plasticity [6].
Recent research has highlighted the pivotal role of neuroinflammation—a complex response involving glial and immune cells—in the pathogenesis and maintenance of chronic pain syndromes [7]. Microglia, the intrinsic immune cells of the spinal dorsal horn, are highly responsive to modifications in neuronal activity. Together with other glial cells, such as astrocytes, microglia can undergo changes in both gene expression and morphology and subsequently secrete pro-inflammatory mediators [7, 8]. Disruptions in microglial homeostasis directly regulate neuronal output and underpin key mechanisms of chronic pain. Evidence suggests that microglial activation in chronic pain is not limited to spinal sensitization and hyperexcitability but also involves brain regions that participate in pain processing [9]. Given the overlap between these regions and those implicated in mood disturbances, it is reasonable to hypothesize that neuroinflammatory events may contribute to centralized stages of pain chronification.
In this narrative review, the focus is on neuroinflammation as a unifying mechanism that can be secondarily engaged in nociceptive and neuropathic pain but appears to be a primary driver of nociplastic pain and may also provide a common pathophysiological substrate for frequent chronic pain–related comorbidities such as depression, anxiety, cognitive dysfunction, and fatigue.
The term “neuroinflammation” refers to inflammatory responses within the central nervous system (CNS), characterized primarily by the activation of resident immune cells, particularly microglia and astrocytes, and the subsequent release of pro-inflammatory mediators, including cytokines, chemokines, and reactive oxygen species. Indeed, specialized glial cells continuously monitor the CNS microenvironment and respond to perturbations in neuronal activity, tissue damage, or pathogen invasion.
Neuroinflammation can be triggered by diverse insults, including infection, traumatic injury, neurodegenerative processes, autoimmune reactions, metabolic dysfunction, and persistent nociceptive signaling [10]. The inflammatory cascade involves complex cellular and molecular interactions: damage-associated molecular patterns (DAMPs) released from injured cells activate pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and NOD-like receptors (NLRs), on glial cells, thereby initiating intracellular signaling cascades involving nuclear factor kappa B (NF-κB), mitogen-activated protein kinases (MAPKs), and interferon regulatory factors (IRFs) [11]. Interestingly, TLR4 can be activated by other signals, including exogenous opioids such as morphine-3-glucuronide [12].
Intracellular signaling cascades result in transcriptional upregulation of inflammatory genes and the release of mediators that amplify the response through autocrine and paracrine signaling loops [7]. These mechanisms have been associated with a variety of neurological conditions, including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and chronic pain [13].
Neuroinflammation can profoundly modulate neuronal excitability and synaptic transmission [14]. In the context of chronic pain, microglia and astrocytes shift from a homeostatic to a reactive state, disrupting neuron–glia communication and producing long‑lasting alterations in nociceptive pathways that persist beyond the initial peripheral insult [8, 14]. While these concepts were initially established in preclinical models, converging human evidence from brain imaging and cerebrospinal fluid studies now indicates that similar neuroimmune mechanisms operate in patients with chronic pain, supporting the view that neuroinflammation is a core pathophysiological component rather than a mere epiphenomenon [15–18].
A substantial body of research demonstrates that spinal dorsal horn microglia are not only necessary but also sufficient to enhance nociceptive processing [8, 19].
Upon peripheral nerve injury (PNI), microglial cells adopt a hypertrophic phenotype, retracting their processes, increasing cell body volume, and altering gene expression profiles [20]. DAMPs released from injured neurons and glia initiate pro-inflammatory cascades by activating TLRs (especially TLR2, TLR4) [21, 22]. Other studies have delineated specific signaling pathways involved in microglial activation and in the initiation of downstream processes, including adenosine 5’-triphosphate (ATP) [23], the ATP-gated P2X4 and P2X7 receptor channels [8, 20], and the fractalkine receptor (CX3CR1) [8, 24]. Via P2X7–p38 MAPK signaling, microglia release cathepsin S, which cleaves neuronal fractalkine, further activating microglia through CX3CR1 [8]. Other primary signals include colony-stimulating factor 1 (CSF-1), which is rapidly induced in dorsal root ganglion (DRG) neurons following injury and, acting through the CSF-1 receptor (CSF-1R) expressed on microglia, promotes proliferation and activation [25]. Several intracellular cascades contribute to microglial activation. These include the p38 MAPK pathway that plays a pivotal role in upregulating inflammatory genes, and IRF8, which orchestrates the expression of reactive genes, including P2rx4, IL-1β, and BDNF. Of relevance, by releasing brain-derived neurotrophic factor (BDNF), microglia induce tropomyosin receptor kinase B (TrkB)-mediated downregulation of KCC2 (potassium-chloride cotransporter 2) in dorsal horn neurons, resulting in disinhibition and hyperexcitability by inverting GABA currents [26]. IRF8 further induces IRF5, which promotes the expression of P2X4 receptors (P2X4Rs), essential for neuropathic pain development [27, 28]. A key role in activating local neuroinflammation after nerve injury is also played by NLRP3 inflammasome, a complex that mediates the maturation and release of interleukin-1β (IL-1β) by integrating signals from TLRs and P2X7 [29].
Activated microglial cells further amplify their own activation and influence neighbouring cells to propagate inflammation. Microglia release IL-1β, tumor necrosis factor-alpha (TNF-α), and C-C motif chemokine ligand 2 (CCL2), and crosstalk with neurons, astrocytes, and endothelium, amplifying nociceptive transmission and acting on other immune cells [10, 28].
Spinal microglia can also be activated by nociceptive activity associated with inflammatory pain through mechanisms involving glutamatergic transmission [8]. Sustained nociceptive signaling following inflammation and peripheral sensitization leads to increased glutamate release in the dorsal horn of the spinal cord. This glutamate not only excites postsynaptic neurons but also contributes to microglial activation through direct and indirect mechanisms involving glutamate spillover.
First, glutamate binding to neuronal NMDA and AMPA receptors triggers ATP release from neurons, which, similarly to what has been described in neuropathic pain, acts on microglial purinergic receptors P2X4 and P2X7, thereby initiating microglial activation and downstream pro-inflammatory signaling [30, 31].
Second, glutamate directly activates metabotropic glutamate receptors (mGluRs) expressed on microglia. Although microglia express multiple mGluR subtypes, chronic pain models predominantly implicate group I receptors (mGluR1 and mGluR5). Group I mGluR activation (via mGluR1 and mGluR5) induces microglial intracellular Ca2+ mobilization and modulates MAPK and NF-κB signaling pathways, thereby altering the profile and magnitude of pro-inflammatory cytokine release [32, 33]. However, activation of microglial mGluR5 has been shown to suppress microglial production of pro-inflammatory mediators in some models, suggesting a complex and context-dependent role [34]. Conversely, microglial mGluR3 is notably important for driving anti-inflammatory and neuroprotective phenotypes, thereby protecting neurons against microglial neurotoxicity [35]. Of note, mGluR4 (group III) has shown selective anti-inflammatory effects in microglia and inhibitory effects on pain behaviors [36]. Evidence for other mGluR subtypes (mGluR2, mGluR7, mGluR8) in microglial pain pathophysiology remains limited or inferential, operating primarily through indirect mechanisms such as reduced glutamate release rather than direct receptor-mediated signaling.
Principal molecular elements in microglial activation during chronic pain are summarized in Table 1 and represented in Figure 1.
Principal molecular elements in microglial activation during chronic pain.
| Target | Upstream activator(s) | Downstream effect(s) | Role in chronic pain | References |
|---|---|---|---|---|
| P2X4 receptor (P2X4R) | ATP released from neurons; transcription factors IRF8, IRF5 | Activation of the p38 MAPK pathway, release of brain-derived neurotrophic factor (BDNF) | Drives neuropathic pain hypersensitivity by downregulating the KCC2 chloride transporter, thereby altering neuronal chloride gradients and excitability | Tsuda et al. [20], 2016Ulmann et al. [23], 2008 |
| P2X7 receptor (P2X7R) | ATP; possibly other DAMPs | Activation of inflammasome (NLRP3), release of IL-1β | Amplifies neuroinflammation and pain persistence | Inoue and Tsuda [8], 2018Xu et al. [29], 2025Grace et al. [30], 2014 |
| Toll-like receptors (TLR2, TLR4) | DAMPs from injured neurons and glia | Activation of pro-inflammatory transcription pathways and cytokine release | Mediation of neuroinflammation and pain hypersensitivity, with a male-specific role for TLR4 in the spinal cord | Kim et al. [21], 2007Liu et al. [22], 2022 |
| Colony-stimulating factor 1 receptor (CSF-1R) | CSF-1 is released from dorsal root ganglion neurons | Microglial proliferation, activation, and gene expression changes | Facilitates microglial activation and chronic pain development | Guan et al. [25], 2016 |
| Interferon regulatory factors (IRF8, IRF5) | Injury signals leading to transcriptional regulation | Upregulate P2X4R and inflammatory mediators including IL-1β, BDNF | Orchestrate the transcriptional program for reactive microglia, essential for pain hypersensitivity | Masuda et al. [27, 28], 2012 and 2014Mapplebeck et al. [80], 2016 |
| NLRP3 inflammasome | Signals integrated from TLRs and P2X7R activation | Maturation and release of interleukin-1β (IL-1β) | Central mediator of local neuroinflammation and chronic pain | Xu et al. [29], 2025 |
| Cathepsin S | P2X7-p38 MAPK signaling | Cleaves neuronal fractalkine, activating the microglial CX3CR1 pathway | Sustains microglial activation and neuroimmune signaling, amplifying nociception | Inoue and Tsuda [8], 2018Lindia et al. [24], 2005 |
| BDNF | Released upon P2X4R activation | Downregulation of the KCC2 chloride transporter in neurons | Causes disinhibition, neuronal hyperexcitability, and pain hypersensitivity | Coull et al. [26], 2005 |
ATP: adenosine 5’-triphosphate; DAMPs: damage-associated molecular patterns; KCC2: potassium-chloride cotransporter 2; MAPKs: mitogen-activated protein kinases.
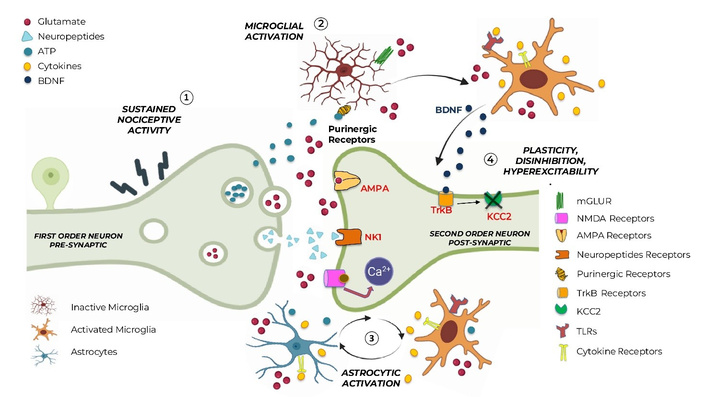
Microglia and astrocytes initiate and maintain the neuroinflammatory cascade. Neuroinflammation involves complex mechanisms and can be triggered by diverse insults, including infections, traumatic injury, neurodegenerative processes, autoimmune reactions, metabolic dysfunction, and persistent nociceptive signaling. (1) Sustained nociceptive input and primary signaling: Persistent peripheral nociceptive activity in first-order neurons leads to repeated action potential firing and enhanced presynaptic release of excitatory neurotransmitters and neuromodulators, including glutamate, ATP, neuropeptides (e.g., CGRP, substance P), and cytokines. These mediators accumulate in the synaptic cleft and surrounding extracellular space, acting not only on postsynaptic neurons but also on resident glial cells, directly and indirectly (glutamate spillover). (2) Microglial activation and neuroimmune amplification: Glutamate triggers ATP release, which activates microglia via purinergic receptors (P2X4/P2X7). Other primary signals activating microglia involve CSF-1 and the p38 MAPK pathway. Activated microglia undergo morphological and transcriptional changes, further amplifying their own activation and propagating inflammation via DAMPs, which initiate the pro-inflammatory cascades by activating TLRs. Activated microglia also release pro-inflammatory cytokines (e.g., IL-1β, TNF-α), as well as BDNF. BDNF acts on neuronal TrkB receptors, contributing to maladaptive synaptic plasticity. (3) Astrocytic activation and dysregulated calcium signaling: Astrocytes contribute to neuroinflammation through bidirectional crosstalk with microglia and are intricately involved in pain processing. They are activated by glutamate spillover, ATP, pro-inflammatory cytokines, and microglia-derived inflammatory mediators. Astrocytic activation persists longer than the microglial one and encompasses intracellular signaling alterations, which appear to be instrumental in the transition from acute to chronic pain states. (4) Postsynaptic neuronal plasticity, disinhibition, and hyperexcitability: At the same time, on second-order dorsal horn neurons, excessive glutamatergic signaling through AMPA activates NMDA receptors, removing the magnesium block, thus leading to increased intracellular Ca2+ and slow depolarization, activating NMDA-mediated synaptic plasticity. Microglia-derived BDNF downregulates KCC2 via TrkB signaling, leading to impaired chloride homeostasis, inverted GABA currents, neuronal disinhibition, and hyperexcitability—key hallmarks of central sensitization and pain chronification. Overall, the figure illustrates how sustained nociceptive signaling initiates a self-reinforcing neuroinflammatory cascade involving microglia and astrocytes, ultimately transforming physiological pain processing into a persistent, centrally sensitized state. ATP: adenosine 5’-triphosphate; BDNF: brain-derived neurotrophic factor; CSF-1: colony-stimulating factor 1; DAMPs: damage-associated molecular patterns; IL-1β: interleukin-1β; KCC2: potassium-chloride cotransporter 2; MAPKs: mitogen-activated protein kinases; mGluR: metabotropic glutamate receptor; NF-κB: nuclear factor kappa B; TLRs: toll-like receptors; TNF-α: tumor necrosis factor-alpha; TrkB: tropomyosin receptor kinase B.
Astrocytes are the most abundant glial cells in the CNS. Distinct from both neurons and immune cells, they play integral roles in multiple functions, including maintenance of the blood-brain barrier, neuroprotection and tissue repair, and modulation of synaptic transmission in a phenotype-dependent manner.
These cells maintain direct neuronal communication through gap junctions [37]. In the context of chronic pain, astrocytes contribute to neuroinflammation through bidirectional crosstalk with microglia and through the release of cytokines and alterations in glutamate transport. Upon PNI, substances like glutamate, ATP, and pro-inflammatory cytokines (including TNF-α, IL-1, and IL-6) are released from afferent neurons or microglia, triggering astrocyte activation. This process involves the gp130-JAK-STAT3 signaling pathway, which plays a role in astrocytic hypertrophy and proliferation, as observed in models of neuropathic pain in rats, suggesting that Janus kinase inhibitors could potentially influence this process and affect central sensitization [38]. When activated, astrocytes produce pro-inflammatory cytokines and chemokines, such as CCL2, which promotes increased sensitivity in second-order neurons [39, 40]. Furthermore, astrocytes can influence synaptic plasticity and neural circuit remodeling through regulation of synaptogenesis and synapse elimination or maintenance [37, 41]. Astrocytic activation typically ensues following microglial cell activation; however, it persists for substantially longer durations, potentially perpetuating chronic pain states. In essence, after an injury stimulates afferent neurons, they release neurotransmitters, cytokines, chemokines, and growth factors that activate microglia, which in turn activate astrocytes. This results in an interactive dialogue between microglia, astrocytes, and neurons, perpetuating neuroinflammation and central sensitization [37]. Recent research has revealed that astrocytic calcium signaling is intricately involved in pain processing, and astrocyte-neuron interactions appear to be instrumental in the transition from acute to chronic pain states. Specifically, astrocytic calcium dynamics regulate synaptic activity and gliotransmitter release, such as glutamate, influencing neuronal excitability and central sensitization [42]. Astrocyte calcium signaling becomes dysregulated following nerve injury or inflammation, contributing to prolonged neuroinflammation and maintenance of chronic pain [43]. This crosstalk between astrocytes and neurons is critical for the rewiring of pain-related circuits underlying chronic pain syndromes.
Furthermore, the formation of glial scars by reactive astrocytes can impede nerve regeneration, potentially exacerbating chronic pain conditions. Liddelow and Barres [44] have delineated the various phenotypes of reactive astrocytes and their diverse functional implications.
Microglia and astrocytes have been the primary focus of glial research in chronic pain, but other glial cell types are gaining recognition for their potential contributions. Oligodendrocytes, responsible for myelination in the CNS, may influence neuropathic pain through alterations in myelin structure and function [45]. Additionally, satellite glial cells (SGCs), unique glial cells that envelop sensory neuron somata in peripheral ganglia, emerge as critical drivers of chronic pain across mechanistically distinct syndromes. Unlike astrocytes that contact multiple neurons, SGCs form exclusive one-to-one perineuronal sheaths with neurons via gap junctions (~20 nm apart), enabling intimate bidirectional neuron–SGC communication. Following nerve injury or inflammation, SGCs undergo stereotyped activation characterized by upregulation of glial fibrillary acidic protein (GFAP), increased connexin-43 (Cx43) gap junctional coupling, downregulation of Kir4.1 potassium channels, enhanced pannexin-1–mediated ATP release, and amplified synthesis of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6). Remarkably, this activation cascade converges across mechanistically disparate pain models—neuropathic pain (nerve injury), post-surgical pain, diabetic neuropathy, and systemic inflammation—suggesting a common peripheral neuroinflammatory sensitization mechanism operating independently of the initiating insult [46]. The convergence of SGC alterations across diverse pain models is particularly significant when considering nociplastic pain—chronic pain without detectable nociceptive activity or somatosensory nerve lesions. Systemic inflammatory triggers such as lipopolysaccharide (LPS) can generate lasting pain hypersensitivity through peripheral sensory ganglia mechanisms without CNS involvement [47]. LPS-induced SGC activation in dorsal root ganglia persists for weeks despite transient LPS exposure, maintained through increased gap junctional coupling and ATP-P2X receptor sensitization. This peripheral neuroinflammatory cascade—involving glial-derived cytokine release and enhanced neuron-SGC calcium wave propagation—mirrors central sensitization mechanisms, suggesting that nocioplastic pain may arise from peripheral SGC-mediated sensitization rather than requiring primary central dysfunction [48]. Thus, systemic inflammation-triggered SGC activation represents a parsimonious explanation for pain hypersensitivity observed without neuropathy or ongoing noxious input, unifying nocioplastic and classical chronic pain mechanisms under a shared peripheral neuroinflammatory framework [46, 48].
While preclinical studies in rodent models have elegantly delineated the molecular mechanisms of glial activation in pain, translation to human chronic pain presents significant challenges. Nonetheless, converging evidence from multiple methodological approaches increasingly supports the relevance of glial mechanisms in human pain conditions.
Positron emission tomography (PET) utilizing radioligands targeting the 18 kDa translocator protein (TSPO) has substantially advanced our capacity to visualize and quantify glial cell activation in vivo in human subjects. TSPO, predominantly expressed on the outer mitochondrial membrane, is dramatically upregulated in activated microglia and reactive astrocytes, making it a sensitive biomarker for neuroinflammation [49]. However, current imaging technology permits the demonstration of supraspinal neuroinflammatory mechanisms in human subjects, whereas the majority of preclinical animal models are limited to investigating neuroinflammatory mechanisms at the spinal level. Loggia et al. [9] first demonstrated elevated TSPO binding in the thalamus and sensorimotor cortex of patients with chronic low back pain compared to healthy controls. The magnitude of the thalamic TSPO signal correlated positively with both pain duration and clinical pain ratings, suggesting that glial activation intensity tracks disease chronicity and severity. Follow-up studies have confirmed these findings and extended them to show bilateral thalamic activation even in unilateral pain, suggesting central sensitization mechanisms [9].
The most comprehensive human glial imaging study to date examined 31 FM patients across three sites using [¹¹C] PBR28 TSPO-PET [15]. Results revealed widespread cortical glial activation affecting multiple brain-related regions, with particularly prominent signals in the dorsolateral prefrontal cortex (dlPFC), which is involved in cognitive-evaluative pain processing; the middle cingulate cortex (MCC), which is critical for pain affect and attention; the parietal cortex, which is associated with sensory-discriminative processing; and the posterior cingulate cortex, part of the default mode network. Critically, TSPO binding in dlPFC correlated with patient-reported fatigue (r = 0.46, p < 0.01), while MCC binding correlated with depression severity (Beck Depression Inventory: r = 0.35, p = 0.05). These associations may provide evidence linking regional glial activation to specific comorbid symptoms, supporting the neuroinflammatory hypothesis of FM-associated cognitive and affective disturbances.
Parkitny et al. [50] reported elevated TSPO binding in the thalamus contralateral to the affected limb in CRPS patients, with signal intensity correlating with mechanical allodynia severity. This somatotopic pattern of glial activation mirrors the lateralized pain distribution and suggests that supraspinal glial responses are organized according to body representation maps.
Finally, neuropathic pain states also display neuroinflammatory patterns in brain imaging studies in rats, although human studies are inconclusive [51].
While TSPO PET has been transformative, important caveats exist. TSPO is expressed by both microglia and astrocytes, precluding cell-type attribution in current studies. Additionally, the rs6971 polymorphism in the TSPO gene affects ligand binding affinity, necessitating genotyping and potentially excluding ~5–10% of individuals (low-affinity binders). Second-generation TSPO ligands and development of cell-type-specific radiotracers (e.g., targeting purinergic receptors for microglia or astrocyte-specific transporters) may overcome these limitations [49].
Beyond brain imaging evidence, multiple studies report elevated CSF levels of neuroimmune mediators in chronic pain. FM patients display a median 2.7-fold increase vs controls of Fractalkine/CX3CL1, 1.7-fold increase of IL-8, and MCP-1/CCL2 elevations [16, 52]. Moreover, elevated levels of IL-1β, IL-6, IL-8, and TNF-α have been shown in patients affected by painful post-herpetic neuralgia and diabetic neuropathy [53]. More recently, specific markers of microglial activation have been explored in the CSF of chronic pain patients. Namely, chitinase-3-like protein 1 (CHI3L1/YKL-40), released by activated microglia and astrocytes, is elevated in CSF of neuropathic pain patients and correlates with pain intensity and neuropathic pain phenotype [54]. Similarly, increased CSF IL‑8 has been described in a majority of patients with postlaminectomy chronic pain, and longitudinal increases in IL‑8 over time have been shown to parallel the clinical evolution of persistent pain, further validating IL‑8 as a dynamic biomarker of central neuroinflammatory activity in humans [18].
Chronic pain rarely exists in isolation. Epidemiological studies consistently demonstrate high rates of co-occurring conditions, including sleep disturbances (50–90% prevalence), fatigue (> 70%), mood disorders (depression: 30–80%; anxiety: 30–60%), cognitive impairment (40–80%), and multisensory hypersensitivity [55, 56]. These comorbidities significantly amplify disability, reduce treatment efficacy, and worsen quality of life, yet their mechanistic basis remains poorly understood.
We propose that neuroinflammation represents a unifying biological mechanism linking pain hypersensitivity with its characteristic comorbidities. This hypothesis is supported by several converging lines of evidence, such as the overlap in brain activation of common areas and the shared inflammatory biomarkers that converge into the paradigm of “sickness behavior”.
Neuroimaging studies reveal that brain regions showing glial activation in chronic pain patients—including the dlPFC, anterior cingulate cortex (ACC), and MCC, insula, and hippocampus—are the same regions implicated in depression, anxiety, cognitive function, and sleep regulation [57]. Human PET studies using TSPO demonstrate that the magnitude of glial activation in these regions correlates not only with pain intensity but also with depression severity (Beck Depression Inventory scores), fatigue levels, and cognitive complaints [15]. This anatomical convergence suggests that a shared neuroinflammatory process affecting these brain networks could simultaneously generate pain, mood disturbance, cognitive dysfunction, and fatigue.
Elevated CSF and serum levels of IL-6, TNF-α, IL-1β, and CRP—commonly observed in chronic pain conditions—are established biomarkers of major depressive disorder and correlate with treatment resistance [58, 59]. Patients with both chronic pain and depression show higher inflammatory marker levels than those with either condition alone, suggesting additive or synergistic neuroinflammatory processes [55].
Fractalkine (CX3CL1), elevated in FM patientsʼ CSF [16], has been implicated in both pain sensitization and depression-like behaviors in preclinical models. CX3CL1–CX3CR1 signaling in the hippocampus contributes to stress-induced depression through microglial activation and synaptic pruning [60]. Similarly, IL-8, elevated in FM CSF, has been linked to cognitive dysfunction in inflammatory conditions [52].
Cytokines influence multiple neurotransmitter systems implicated in both pain and mood regulation. IL-1β and TNF-α activate indoleamine 2,3-dioxygenase (IDO), shunting tryptophan metabolism from serotonin synthesis toward kynurenine pathway metabolites, some of which (quinolinic acid) are NMDA receptor agonists that promote excitotoxicity and have been implicated in depression and pain facilitation [61, 62]. Neuroinflammation also impairs dopaminergic signaling in mesolimbic reward circuits, contributing to anhedonia and motivational deficits characteristic of both chronic pain and depression [63].
The constellation of fatigue, cognitive impairment, sleep disturbance, anhedonia, and hypersensitivity characteristic of chronic pain closely resembles “sickness behavior”—an adaptive response to infection mediated by pro-inflammatory cytokines acting on the brain [64]. In acute infection, peripheral cytokines induce central neuroinflammation, triggering behaviors that promote recovery (rest, social withdrawal, hyperalgesia to prevent further injury). However, in chronic pain, persistent low-grade neuroinflammation may inappropriately activate these same pathways, generating a maladaptive sickness state [65].
In the preclinical experimental setting, when neuropathic pain is induced by sciatic nerve injury, activation of microglia is observed not only at the spinal level but also within the brain. Specifically, Taylor et al. [66] found microglial activation in the thalamus, the ventral tegmental area (VTA), the nucleus accumbens, the prefrontal cortex, and the amygdala, indicating that microgliosis extends beyond the spinal cord to affect various areas involved in cognitive and behavioural aspects of pain processing. For example, microglial activation in the amygdala following partial sciatic nerve ligation underpins anxiety-like behavior in rodent models [67]. Notably, both anxiety-like behavior and mechanical hypersensitivity are reversed by antagonism of the C-C motif chemokine receptor 2 (CCR2), which recognizes the chemokine ligand CCL2 and is essential for microglia activation [68]. Likewise, neuroinflammatory processes following PNI were observed in the ACC, a brain region that plays a key role in the emotional processing of pain. Of note, ACC neuroinflammation was associated with mechanical allodynia [69].
From these observations, it is reasonable to hypothesize that comorbidities that occur with pain hypersensitivity could be the expression of neuroinflammatory phenomena in areas of the pain matrix involved in cognitive and emotional processing of pain. Indeed, pain and depressed mood are often seen as two sides of the same coin and very often co-occur with symptoms such as fatigue, poor refreshment from sleep, problems with concentration and short-term memory, along with hypersensitivity to visual, auditory, and tactile stimuli referred to as multisensory hypersensitivity.
Epidemiological and longitudinal studies indicate a tight and bidirectional relationship between pain, insomnia, and depression: poor sleep predicts subsequent increases in pain intensity while higher pain levels forecast later functional impairment and depressive symptoms [70]. Importantly, sleep deprivation activates microglia and increases pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) in both peripheral blood and the CNS, while also impairing descending pain modulation and increasing experimental pain sensitivity [71, 72]. This creates a vicious cycle: pain → sleep disruption → neuroinflammation → enhanced pain sensitivity and mood disturbance → further sleep disruption.
Fatigue is a highly burdensome comorbidity in chronic pain and is increasingly viewed as a core manifestation of neuroinflammatory processes rather than a purely psychological reaction to persistent nociception [16]. In primary Sjögren’s syndrome, pain, depression, and daytime sleepiness are independently associated with both physical and mental fatigue, even after adjusting for fatigue‑related comorbidities and sedating medications, supporting a shared pathophysiological basis across pain and immune‑mediated conditions [73].
Fibrofog—the cognitive impairment affecting attention, working memory, and executive functions in FM and other chronic pain conditions—correlates with neuroinflammatory markers. dlPFC glial activation, demonstrated by PET imaging in FM [15], affects a region critical for working memory and cognitive control. Pro-inflammatory cytokines impair hippocampal long-term potentiation (LTP), a synaptic mechanism underlying learning and memory [74]. Microglial activation in the hippocampus and prefrontal cortex promotes synaptic pruning and dendritic spine loss, providing a structural basis for cognitive deficits [75].
In surgical cohorts, pre‑existing central sensitization has been associated with a higher incidence and prolonged course of postoperative cognitive dysfunction, in parallel with enhanced pain sensitivity and increased inflammatory markers [76].
Recent work has identified peripheral inflammatory signatures that are preferentially associated with psychological comorbidities rather than with pain intensity per se. In a mixed chronic pain cohort, a panel of pro‑inflammatory and immune‑regulatory proteins—including IL‑7, CD40, CXCL1, CXCL5, and CD244—was significantly associated with anxiety and depressive symptoms measured by standardized scales, whereas none of these markers correlated with self‑reported pain intensity [77]. Moreover, a subset of fifteen inflammatory proteins could distinguish patients with moderate-to-severe psychological burden from those with minimal or no anxiety and depression, supporting the concept that inflammation-linked mechanisms may be particularly relevant to the affective dimension of chronic pain and could guide individualized, mechanism-based treatment strategies [77, 78].
Clinicians often observe secondary signs and symptoms of central sensitization in well-identifiable nociceptive pain of musculoskeletal origin, such as back pain and rheumatic diseases, mechanistically described as secondary nociplastic pain. We assume that peripheral sensitization and increased nociceptive activity are the primary triggers of central sensitization in these cases. Indeed, increased TSPO radioligand binding has been reported in a PET study in patients with chronic low back pain, with elevations in the thalamus, cortical pain-processing regions, spinal cord, and the neuroforamina, supporting the concept that glial activation extends along the neuraxis in chronic pain [9, 17].
However, the mechanistic basis underlying pain syndromes characterized primarily by nociplastic features and widespread pain distribution, such as FM, remains incompletely understood. Albrecht et al. [15] have demonstrated widespread cortical glial activation in FM patients compared to healthy controls, which delineates promising avenues for future investigation.
Taken together, human imaging and CSF studies strongly support the view that glial activation and neuroimmune signaling are not only mechanistic drivers of nociceptive hypersensitivity but also central contributors to the affective and cognitive comorbidities that frequently accompany chronic pain, including depression, fatigue, and sleep disturbances. Consequently, neuroinflammatory pathways emerge as promising therapeutic targets warranting further investigation [79].
While neuropathic pain and chronic pain conditions have a female predominance, experimental models have long focused only on male subjects. Since sex was introduced as a biological variable in basic science, new intriguing insights into pain chronification have emerged.
Notably, sex differences in microglial activation have been observed, potentially explaining disparities in chronic pain prevalence between males and females [80, 81]. Interestingly, in male mice, upregulation of microglial P2X4Rs plays a significant role in generating neuropathic pain through the release of BDNF; in female mice, this microglia-dependent pathway is not utilized. Instead, adaptive immune cells, likely T cells, mediate pain hypersensitivity [82]. TLR4 contributes to pain hypersensitivity in male mice only, while peroxisome proliferator-activated receptors (PPARs) α and γ contribute to pain hypersensitivity in females, with each PPAR having differential expression dependent on testosterone levels [81, 82]. However, both the microglial-dependent and independent pathways converge at the neuronal level.
Evidence further shows that antagonists of glial function reverse allodynia only in males, and that sex-specific expression of transcription factors (e.g., IRF8 and IRF5) underlies divergent regulation of microglial genes [80]. These dimorphic pathways imply that pharmacological strategies targeting microglial activation or specific microglial signaling cascades may show sex‑dependent efficacy, underscoring the need to incorporate sex as a biological variable in the development and testing of glia‑directed analgesics. Clinically, this may help explain the higher burden of chronic pain and associated mood disorders in women. Indeed, women account for the majority of affected individuals across more than 50% of chronic pain disorders [83]. This epidemiological disparity is accompanied by documented sex-based variations in nociceptive processing: women exhibit lower pain thresholds to experimental noxious stimuli [84]. Among those suffering from chronic pain, female patients commonly report heightened symptom intensity, greater functional disruption from pain, and more widespread pain distribution compared to their male counterparts [85]. Sexually dimorphic immune mechanisms suggest that personalized approaches that differentially address glial and adaptive immune components may be required for optimal treatment.
The evolving understanding of glial involvement in chronic pain mechanisms has opened new avenues for therapeutic interventions. Targeting specific signaling pathways involved in glial activation represents a promising approach to modulating pain processing [86]. Moreover, the recognition of sex differences in glial activation underscores the importance of considering sex as a biological variable in pain research and treatment strategies.
Recognizing neuroinflammation as a shared mechanism suggests that treatments targeting neuroimmune processes might simultaneously improve pain, mood, cognition, and sleep. Preliminary evidence supports this: low-dose naltrexone (LDN), which modulates glial activation, has shown benefits for both pain and fatigue in FM [87]. Minocycline, a microglial inhibitor, improves pain, depression-like behavior, and cognitive function in some preclinical models [88]. Anti-cytokine biologics (TNF-α inhibitors) reduce both pain and depression in inflammatory arthritis [89].
In addition to spatially extended glial activation along the neuraxis, temporal dysregulation of neuroimmune processes may further contribute to pain chronification. The co-occurrence of sleep disruption in chronic pain populations reflects dysregulation of circadian physiology, particularly involving altered pineal melatonin production during nocturnal hours and abnormal nocturnal cortisol secretion patterns—both critical for promoting inflammatory resolution during sleep phases [90]. Patients with FM and osteoarthritis demonstrate attenuated nighttime melatonin levels and dysregulated cortisol rhythmicity, indicating that circadian hormone dysregulation constitutes a fundamental pathophysiological feature of chronic pain syndromes [91, 92]. Interestingly, both astrocytes and microglia can synthesize melatonin endogenously; moreover, empirical evidence demonstrates that melatonin facilitates microglial phenotypic transition from a pro-inflammatory (M1) state toward a less activated, anti-inflammatory (M2) phenotype [93]. Since IL-10-dependent NF-κB signaling controls the production of both central and peripheral melatonin, pharmacological or behavioral interventions targeting melatonergic glial pathways and restoring circadian synchronization may offer a promising therapeutic avenue for ameliorating neuroinflammatory cascades and addressing chronic pain alongside its associated comorbid conditions [94].
The neuroinflammatory substrate framework of chronic pain carries important diagnostic and prognostic implications. Patients presenting with chronic pain accompanied by multiple comorbidities—phenotypically characterized as high symptom burden—may represent a distinct neuroinflammatory subtype. This subtype is potentially identifiable through objective biomarkers (CSF cytokines, PET imaging) and may be preferentially responsive to immunomodulatory interventions rather than to conventional analgesics targeting peripheral nociception alone.
Importantly, chronic pain is heterogeneous in its pathophysiological basis. Clinical experience suggests that there are forms of chronic pain (persisting or recurring for more than 12 weeks) provoked by clearly identifiable structural lesions, where pharmacological and/or invasive treatments are effective. In these cases, we assume that the processes of pain hypersensitivity are maintained by nociceptive activity and are confined to the spinal level. Conversely, in presentations where pain is disproportionate to identifiable tissue injury and emotional and cognitive components predominate, pain maintenance shifts from spinal to supraspinal loci, characteristic of primary or secondary nociplastic pain. Conceptualizing nociplastic pain within a neuroinflammatory disease framework fundamentally reshapes the therapeutic paradigm. Both sensory and emotional/cognitive components are reconceptualized as pathological entities with recognizable and measurable biological substrates. Consequently, the primary therapeutic target shifts from tissue or nerve lesion remediation toward direct modulation of neuroinflammatory cascades. This mechanistic approach acknowledges that when nociplastic mechanisms dominate, pain maintenance becomes independent of the original peripheral insult and instead depends upon central neuroinflammatory dysregulation.
ACC: anterior cingulate cortex
ATP: adenosine 5’-triphosphate
BDNF: brain-derived neurotrophic factor
CCL2: C-C motif chemokine ligand 2
CNS: central nervous system
CSF-1: colony-stimulating factor 1
DAMPs: damage-associated molecular patterns
dlPFC: dorsolateral prefrontal cortex
FM: fibromyalgia
IL-1β: interleukin-1β
IRFs: interferon regulatory factors
KCC2: potassium-chloride cotransporter 2
LPS: lipopolysaccharide
MAPKs: mitogen-activated protein kinases
MCC: middle cingulate cortex
mGluRs: metabotropic glutamate receptors
NF-κB: nuclear factor kappa B
P2X4Rs: P2X4 receptors
PET: positron emission tomography
PNI: peripheral nerve injury
PPARs: peroxisome proliferator-activated receptors
SGCs: satellite glial cells
TLRs: toll-like receptors
TNF-α: tumor necrosis factor-alpha
TrkB: tropomyosin receptor kinase B
TSPO: translocator protein
SN: Conceptualization, Project administration, Writing—original draft. DMF: Writing—review & editing. AC: Writing—review & editing. GT: Visualization, Writing—review & editing. LD: Visualization, Writing—review & editing. GLB: Writing—review & editing. MI: Visualization. FF: Visualization. FG: Writing—review & editing. MM: Project administration, Methodology, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1959
Download: 27
Times Cited: 0
Rucha A. Kelkar ... Giustino Varrassi
Marco Cascella ... The TRIAL Group
Mariateresa Giglio ... Filomena Puntillo
Antonella Ciaramella, Giancarlo Carli
Marta Nizzero ... Enrico Polati
Serena Altamura ... Benedetta Cinque
Lucia Daiana Voiculescu ... Sanjay Menghani
