 Review
Review

Affiliation:
1Department of Internal Medicine, Stanford University School of Medicine, Palo Alto, CA 94305, USA
ORCID: https://orcid.org/0009-0000-8816-4605
Affiliation:
2Independent Researcher, Mountain View, CA 94040, USA
Email: moawiahn@hotmail.com
ORCID: https://orcid.org/0000-0003-0451-5901
Explor Immunol. 2026;6:1003255 DOI: https://doi.org/10.37349/ei.2026.1003255
Received: November 30, 2025 Accepted: April 29, 2026 Published: May 29, 2026
Academic Editor: Giuseppe Murdaca, University of Genova, Italy
Autoimmune rheumatic diseases arise when the immune system transitions from a flexible, self-regulating network into a metabolically and epigenetically fixed inflammatory attractor state. This review synthesizes emerging evidence that immune tolerance is governed by a coupled epigenetic-metabolic axis integrating mitochondrial fitness, chromatin accessibility, redox balance, and nutrient flux across lymphoid, myeloid, and stromal compartments. We examine how chronic cytokine signaling, hypoxia, and oxidative stress destabilize regulatory programs, imprint glycolytic effector states, and remodel enhancer landscapes, thereby sustaining autoreactive circuits even after inflammatory pathways are pharmacologically suppressed. Multi-omic and spatial analyses reveal that pathogenic chromatin architectures, persistent mitochondrial dysfunction, and intercellular metabolite exchange cooperate to establish self-sustaining inflammatory ecosystems in rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SSc), and Sjögren’s syndrome. We further highlight therapeutic strategies aimed at tolerance reprogramming, including metabolic correction, chromatin-targeted agents, chimeric antigen receptor regulatory T cells (CAR-Tregs), tolerogenic dendritic cells, and integrative biomarkers that quantify metabolic-epigenetic coherence. By reframing autoimmunity as a disorder of energetic and chromatin desynchronization rather than isolated immune activation, this review outlines a mechanistic path toward durable, drug-free remission through deliberate restoration of the molecular architecture that maintains immune self-recognition.
Autoimmune rheumatic diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and systemic sclerosis (SSc) are chronic, relapsing disorders characterized by a progressive breakdown of immune tolerance. Over the past two decades, major therapeutic advances, including biologics and kinase inhibitors that target inflammatory mediators such as tumor necrosis factor (TNF), interleukin (IL)-6, and the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway, have produced remarkable clinical benefits [1, 2]. Yet these successes have revealed a persistent gap between inflammation control and immune restoration. Most patients relapse once treatment is withdrawn, and even in remission, transcriptional and epigenetic traces of autoreactivity persist within memory T and B cells and within stromal fibroblast compartments that sustain local inflammation [3]. The central challenge for rheumatology is therefore not merely to silence cytokine storms but to re-educate the immune system, reinstating durable self-tolerance at its molecular roots.
Recent insights have reframed tolerance as a dynamic, actively maintained state rather than a passive absence of immune response. In healthy immunity, tolerance is enforced by continuously adaptive networks of regulatory T cells (Tregs), tolerogenic dendritic cells (tolDCs), and B-cell tolerance checkpoints that integrate metabolic, antigenic, and tissue-derived cues. In autoimmune disease, these networks are destabilized by chronic antigenic stimulation, oxidative stress, and inflammatory cytokines, producing transcriptional drift and metabolic exhaustion [4, 5]. Tregs lose their suppressive identity, while Th17 and T-peripheral-helper (Tph) populations expand, and fibroblast-like synoviocytes (FLS) acquire pathogenic phenotypes [6, 7]. Collectively, these shifts transform an adaptable equilibrium into a self-sustaining inflammatory ecosystem—an inflammatory attractor state maintained by interlocking metabolic and epigenetic feedback loops. In this review, we use the term inflammatory attractor state to refer to a self-reinforcing pathological condition in which immune and stromal cells become locked into stable inflammatory metabolic, epigenetic, and transcriptional programs that resist spontaneous return to homeostasis. This transition from dynamic balance to inflammatory attractor state represents the mechanistic essence of tolerance collapse explored in the sections that follow.
A central conceptual advance is the recognition that epigenetic and metabolic processes function as an integrated regulatory system controlling immune cell identity and plasticity [8, 9]. This coupled axis links environmental and intracellular signals to stable transcriptional programs that determine immune tolerance or activation states [10, 11].
This recognition clarifies why current immunotherapies achieve control without cure. Agents that block cytokines or signaling pathways act downstream of the transcriptional and metabolic programs that sustain autoimmunity. They quench inflammation but fail to reset the system’s underlying attractor. Durable, drug-free remission appears to coincide with spontaneous or therapy-induced resetting of these cellular states, manifest as restoration of Treg metabolic competence, reversal of fibroblast activation, and normalization of chromatin accessibility at key immune loci [12, 13]. The future of rheumatology lies in converting such sporadic events into deliberate therapeutic strategies that rebuild regulatory architecture rather than merely dampen effector output.
Parallel advances in immunometabolism and epigenetic regulation now provide new opportunities to therapeutically restore immune tolerance [11, 14]. These approaches highlight the interdependence of cellular metabolic state and transcriptional identity, supporting the concept of a unified epigenetic-metabolic tolerance axis that governs immune stability and plasticity [9, 10].
Viewing rheumatic autoimmunity through this lens carries profound implications. It reframes these disorders as failures of metabolic and epigenetic adaptability rather than purely antigen-driven diseases. It calls for therapies that reprogram rather than suppress by using metabolic and chromatin modulators, either alone or in combination with biologics, to restore homeostatic regulation. It also redefines clinical success beyond inflammatory control, emphasizing measurable biomarkers of tolerance restoration that incorporate mitochondrial function, chromatin accessibility, and the stability of regulatory cells. This systems perspective bridges innate and adaptive immunity, linking macrophage polarization, fibroblast activation, and lymphocyte fate decisions within a single regulatory continuum.
The scope of this review is to synthesize and critically evaluate the expanding evidence connecting epigenetic and metabolic reprogramming to autoimmune pathogenesis and therapy in rheumatology. We analyze how metabolic flux shapes chromatin state, how these mechanisms are disrupted across immune and stromal compartments, and how they can be targeted to rebuild tolerance. Subsequent sections examine the immune-tolerance network, delineate the convergent layers of epigenetic and metabolic control, and highlight emerging therapeutic strategies including small-molecule modulators, cell-based interventions such as chimeric antigen receptor Tregs (CAR-Tregs) and tolDCs, and integrative biomarker frameworks for patient stratification. Together, these analyses outline a path toward programmable immune homeostasis, where remission arises not from suppression but from the deliberate restoration of the epigenetic and metabolic equilibrium that sustains self-recognition.
The maintenance of self-tolerance is the defining property that distinguishes a physiological immune response from pathological autoimmunity. In rheumatic diseases, this balance collapses not through a single defect but through the gradual disintegration of a multicellular, metabolically coordinated tolerance network that normally spans immune and stromal compartments [15]. Classical immunology described tolerance as a binary phenomenon, either maintained or lost. However, contemporary data from single-cell and spatial multi-omics have reframed it as a dynamic systems state governed by continuous feedback among antigen recognition, cytokine signaling, chromatin architecture, and cellular metabolism [16].
Within this framework, lymphoid, myeloid, and stromal populations operate as interdependent modules of immune regulation (Figure 1). As illustrated in Figure 1, immune tolerance is sustained by coordinated interactions among regulatory lymphoid, myeloid, and stromal compartments, rather than by isolated cell-intrinsic pathways. Tregs, B cells, dendritic cells, macrophages, and FLS form a distributed network that integrates environmental signals such as nutrient availability, oxygen tension, redox balance, and microbial metabolites to maintain homeostatic quiescence [17]. When these circuits become metabolically or epigenetically uncoupled, effector and regulatory lineages diverge, stromal cells adopt inflammatory memory, and tolerance collapses into chronic activation. This collapse defines the immunopathology common to RA, SLE, Sjögren’s syndrome, and SSc [13, 18].
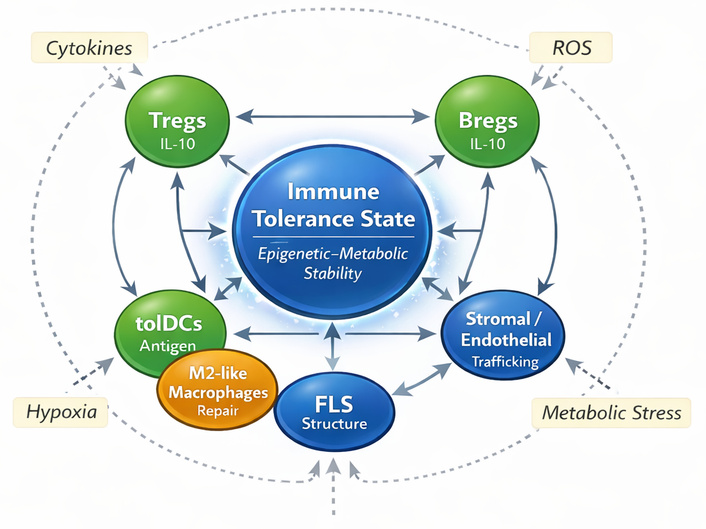
Immune tolerance network architecture. Schematic representation of the multicellular immune tolerance network integrating lymphoid, myeloid, and stromal compartments. Regulatory T cells (Tregs), regulatory B cells (Bregs), and tolerogenic dendritic cells (tolDCs) form core suppressive modules that maintain immune homeostasis through coordinated anti-inflammatory signaling. Anti-inflammatory macrophages support tissue repair and regulate inflammatory tone, while fibroblast-like synoviocytes (FLS) and stromal/endothelial cells provide structural and microenvironmental control of immune-cell trafficking and activation. Bidirectional interactions between these cellular components sustain a dynamic tolerance state. External stressors, including cytokines, hypoxia, reactive oxygen species (ROS), and metabolic stress, can disrupt this network and promote transition toward chronic inflammatory states. IL-10: interleukin-10.
Understanding the architecture of this tolerance network is therefore fundamental to reimagining therapy. It clarifies why conventional cytokine blockade achieves transient remission yet fails to re-establish durable immune homeostasis: The deeper regulatory programs of energy metabolism, chromatin remodeling, and intercellular communication remain misaligned. Mapping how these programs interact provides the mechanistic scaffold for a new generation of interventions aimed at immune re-education rather than immunosuppression.
The following section examines this network in detail, tracing how central and peripheral tolerance mechanisms integrate across T-cell, B-cell, myeloid, and stromal lineages, how metabolic and epigenetic cues sustain or destabilize their interactions, and how their coordinated failure produces the chronic inflammatory states characteristic of autoimmune rheumatology.
Immune tolerance represents the foundational principle of self-non-self-discrimination and the long-term stability of immune homeostasis. It is not a static barrier but a dynamic, multilayered regulatory architecture encompassing central, peripheral, and tissue-level mechanisms that collectively prevent self-reactivity while preserving immune adaptability [19]. The failure of this architecture underlies the pathogenesis of autoimmune rheumatic diseases, transforming transient inflammation into self-sustaining pathology [20, 21]. Understanding tolerance as a distributed and metabolically governed network rather than a series of isolated checkpoints has become a defining shift in modern immunology.
At the central level, tolerance is established during lymphocyte ontogeny in the thymus and bone marrow. In the thymus, developing T cells are exposed to a broad repertoire of self-antigens presented by medullary epithelial cells under the control of the transcription factors autoimmune regulator (AIRE) and FEZ family zinc finger 2 (FEZF2). This process eliminates highly self-reactive clones through negative selection while allowing survival of low-affinity T cells capable of peripheral regulation [22, 23]. Similarly, in the bone marrow, autoreactive B cells undergo clonal deletion or receptor editing through secondary light-chain rearrangements, recalibrating their antigen specificity [24, 25]. However, both systems operate probabilistically, permitting the escape of partially autoreactive clones into the periphery. Thus, central tolerance provides a structural foundation but not a self-sufficient guarantee of immune restraint.
Peripheral tolerance expands this foundation into a context-dependent, adaptive network involving multiple immune and stromal players. Tregs form their keystone, exerting antigen-specific suppression through contact inhibition, IL-10, and TGF-β secretion, and control of dendritic cell maturation. Complementing Tregs are regulatory B cells (Bregs), tolDCs, and anti-inflammatory macrophages that collectively modulate effector activation thresholds [26, 27]. Peripheral tolerance is therefore achieved through active regulatory processes that maintain effector-regulatory equilibrium under fluctuating antigenic conditions.
Recent systems-level studies indicate that tolerance maintenance depends on coordinated regulatory programs across immune cell populations that preserve functional stability and prevent aberrant activation [28–30]. Disruption of these regulatory networks promotes a shift toward persistent inflammatory states, reflecting a breakdown in immune system coordination. The metabolic and epigenetic mechanisms underlying these processes are discussed in detail in Epigenetic-metabolic programming of immune cell fate.
Immune tolerance is also shaped by tissue microenvironments, where stromal and parenchymal cells actively regulate immune behavior. In the synovium, fibroblasts and endothelial cells provide signals that influence immune cell activation and differentiation [31, 32]. Under conditions of chronic stress, these stromal cells can adopt pathogenic phenotypes that promote sustained inflammation and disrupt local immune regulation [33, 34].
Temporal dynamics further define tolerance architecture. Resolution of immune responses requires coordinated resetting of regulatory programs, processes that are often impaired in chronic autoimmune disease [35, 36]. This results in immune cells remaining in partially activated states, a phenomenon referred to as tolerance inertia.
Collectively, these insights position immune tolerance as a self-organizing systems equilibrium regulated by dynamic interactions between immune cells and their environment. Rheumatic autoimmunity arises when this equilibrium loses resilience—when metabolic stress and inflammatory signaling reinforce one another to fix the network in an inflammatory attractor state that resists return to homeostasis. Recognizing tolerance as a dynamic architectural system reframes the therapeutic challenge: rather than suppressing inflammation downstream, interventions must restore the metabolic and epigenetic balance that underpins the architecture of immune self-regulation. This systems-based understanding provides the conceptual groundwork for exploring the cellular and molecular participants of tolerance networks in autoimmune rheumatology (Table 1). Table 1 summarizes these core cellular modules by linking each cell type to its dominant metabolic program, epigenetic features, and functional contribution to immune tolerance.
Core cellular modules of the immune tolerance network.
| Cell type | Key metabolic program | Key epigenetic features | Functional outcome | References |
|---|---|---|---|---|
| Regulatory T cells (Tregs) | Oxidative phosphorylation (OXPHOS); fatty-acid oxidation (FAO); high NAD+; AMPK-SIRT1 signaling | FOXP3 CNS2 demethylation; repressive histone marks at effector loci | Maintenance of immune suppression and tolerance stability | [12, 37] |
| Regulatory B cells (Bregs) | Balanced glycolysis/OXPHOS; low ROS | BLIMP-1-dependent chromatin program | Suppression of humoral autoimmunity and control of germinal-center responses | [38, 39] |
| Tolerogenic dendritic cells (tolDCs) | OXPHOS-dominant; IDO1-kynurenine pathway | Reduced H3K27ac at costimulatory loci; tolerogenic enhancer landscape | Induction of Tregs and peripheral tolerance | [40–42] |
| M2-like macrophages | OXPHOS; low succinate; low HIF-1α | Repressive chromatin at inflammatory gene loci | Tissue repair, anti-inflammatory signaling, and resolution of inflammation | [43–45] |
| Fibroblast-like synoviocytes (FLS) | Increased glycolysis; altered mitochondrial/OXPHOS balance in inflammatory states | Epigenetic activation of inflammatory and tissue-remodeling programs, including EZH2- and non-coding RNA-associated regulation | Stromal activation, inflammatory mediator production, matrix remodeling, and persistence of synovial inflammation | [46–48] |
| Stromal & endothelial cells | Oxidative tissue niche; retinoic acid and kynurenine production | Repressed adhesion molecule loci (ICAM1/VCAM1) | Regulation of immune-cell trafficking and tissue-level equilibrium | [49, 50] |
AMPK: AMP-activated protein kinase; BLIMP-1: B lymphocyte-induced maturation protein 1; CNS2: conserved non-coding sequence 2; FOXP3: forkhead box P3; HIF-1α: hypoxia-inducible factor-1 alpha; ICAM1: intercellular adhesion molecule 1; IDO1: indoleamine 2,3-dioxygenase 1; ROS: reactive oxygen species; SIRT1: sirtuin 1.
Immune tolerance emerges from the coordinated activity of lymphoid, myeloid, and stromal lineages, which together constitute a multicellular regulatory network maintaining self-non-self discrimination and tissue homeostasis. This distributed system integrates antigen sensing, cytokine signaling, and metabolic feedback to preserve immune equilibrium under constant environmental fluctuation [51]. T cells establish regulatory polarity and enforce suppression through forkhead box P3 (FOXP3)-governed transcriptional programs; B cells calibrate humoral memory and self-reactivity thresholds; myeloid antigen-presenting cells (APCs) define the cytokine and costimulatory context that determines whether antigen encounter yields activation or tolerance; and FLS, along with other stromal cells, provide the structural and metabolic scaffolding through which these immune interactions are spatially and energetically integrated [51–53]. The orchestration of these cellular modules depends on synchronized metabolic and epigenetic states, such as oxidative phosphorylation (OXPHOS), redox homeostasis, and chromatin accessibility, that collectively define the tolerogenic milieu. Disruption of any component propagates through the network, dismantling intercellular feedback loops and precipitating the self-reinforcing inflammatory circuits characteristic of autoimmune rheumatic diseases.
T cells constitute the central executors of adaptive immune tolerance, integrating antigenic cues with metabolic and epigenetic programs that determine whether immune responses resolve or perpetuate. Tregs, defined by sustained FOXP3 expression and IL-2 dependence, enforce self-tolerance through multilayered mechanisms including cytokine-mediated suppression (IL-10, TGF-β), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-dependent trans-endocytosis of costimulatory ligands, and adenosinergic control via CD39/CD73 ectoenzymes [53–55].
The epigenetic and metabolic mechanisms that stabilize Treg identity and regulate their functional plasticity are discussed in detail in Epigenetic-metabolic programming of immune cell fate.
Tregs are not a uniform population but comprise functionally and phenotypically distinct subsets whose stability and suppressive capacity are shaped by epigenetic and metabolic programming [56]. Broadly, Tregs can be viewed as including resting or naïve-like Tregs, activated or effector Tregs, and unstable or ex-Treg states that emerge under chronic inflammatory pressure [56].
Resting Tregs are characterized by a relatively quiescent state, sustained FOXP3 expression, and dependence on OXPHOS and fatty-acid oxidation (FAO), metabolic programs that support long-term survival and maintenance of suppressive identity [57]. Epigenetically, these cells maintain stable demethylation at the FOXP3 locus and repressive chromatin architecture at inflammatory genes, preserving lineage fidelity [58].
Activated or effector Tregs arise in inflammatory or tissue-specific environments and display enhanced suppressive function, increased migratory capacity, and greater metabolic flexibility [56]. Compared with resting Tregs, they rely on a more dynamic balance between mitochondrial metabolism and controlled glycolytic engagement to support proliferation, trafficking, and local suppressive activity [59]. These activated Tregs also exhibit adaptive chromatin remodeling at loci involved in tissue homing, cytokine responsiveness, and immune regulation, enabling context-specific function within inflamed tissues such as rheumatoid synovium [59].
In contrast, unstable or ex-Treg states emerge when inflammatory cytokines, hypoxia, oxidative stress, and metabolic disruption overwhelm regulatory programs [60]. These cells show erosion of FOXP3 stability, increased glycolytic bias, mitochondrial dysfunction, and epigenetic reconfiguration toward permissive inflammatory chromatin states, which may allow acquisition of effector-like features including IL-17 or interferon (IFN)-γ production [60]. Such transitions are particularly relevant in autoimmune rheumatic diseases, where persistent inflammatory microenvironments may shift Tregs from stable suppressive states toward dysfunctional or pathogenic phenotypes [61].
Together, these observations indicate that epigenetic and metabolic reprogramming regulate not only global Treg abundance, but also the balance between resting, activated, tissue-adapted, and unstable Treg subsets. A clearer understanding of this subset heterogeneity may help refine therapeutic strategies aimed at selectively stabilizing the most suppressive Treg states in rheumatic disease [58]. In rheumatic diseases such as RA and SLE, this subset-specific instability may help explain why total Treg numbers alone do not reliably reflect regulatory competence or therapeutic responsiveness [61, 62]. Table 2 summarizes how epigenetic and metabolic programming shape distinct Treg states that may differentially contribute to immune regulation or tolerance failure in rheumatic disease.
Distinct Treg subsets and their epigenetic-metabolic features in rheumatic disease.
| Treg subset | Functional state | Dominant metabolic features | Key epigenetic features | Relevance in rheumatic disease |
|---|---|---|---|---|
| Resting/naïve-like Tregs | Baseline suppressive maintenance | OXPHOS, FAO, mitochondrial fitness | Stable FOXP3 demethylation; repressive inflammatory chromatin | Maintenance of systemic tolerance |
| Activated/effector Tregs | Tissue-adapted suppression, proliferation, migration | Mixed oxidative metabolism with controlled glycolytic support | Adaptive chromatin remodeling at trafficking and suppressive loci | Active suppression in inflamed tissues, such as the RA synovium |
| Unstable/ex-Tregs | Loss of suppressive identity; inflammatory conversion | Glycolytic bias, ROS accumulation, mitochondrial dysfunction | FOXP3 instability; permissive inflammatory chromatin | Treg dysfunction and pathogenic plasticity in chronic inflammation |
FAO: fatty-acid oxidation; FOXP3: forkhead box P3; OXPHOS: oxidative phosphorylation; RA: rheumatoid arthritis; ROS: reactive oxygen species; Tregs: regulatory T cells.
The balance between Tregs and effector T-cell subsets, particularly Th17 cells, represents a critical axis governing immune tolerance. Disruption of this balance is a hallmark of autoimmune rheumatic diseases, where pro-inflammatory T-cell responses dominate over regulatory control. In RA, inflammatory cytokine signaling and tissue microenvironmental stress promote sustained effector T-cell activity and limit the stability of regulatory populations, contributing to persistent immune activation. Beyond the Th17/Treg axis, Tph cells, expanded in RA and Sjögren’s syndrome, provide extrafollicular B-cell help through IL-21 and C-X-C motif chemokine ligand 13 (CXCL13) secretion, linking T-cell dysregulation to humoral autoimmunity [63–65]. Restoration of immune tolerance, therefore, requires rebalancing T-cell regulatory and effector functions within the broader immune network.
B cells act as both sensors and executors of immunological tolerance, mediating central deletion, receptor editing, and peripheral anergy to prevent self-reactivity [66]. In the bone marrow, recombination-activating gene (RAG)-dependent receptor editing and clonal deletion purge high-affinity autoreactive clones. In the periphery, anergy is enforced by inhibitory receptors (FcγRIIB, PD-1) and metabolic restraint. Under chronic inflammatory conditions, however, tolerance checkpoints are reprogrammed [67]. Persistent IL-21 and B-cell activating factor (BAFF) signaling from Tph cells induces the transcriptional regulators T-bet and BLIMP-1, in concert with the histone methyltransferase (HMT) enhancer of zeste homolog 2 (EZH2), imprinting a hyper-responsive memory phenotype. Single-cell RNA-seq analyses in RA and SLE reveal expansion of T-bet+ CD11c+ age-associated B cells [68–72].
Within inflamed tissues, ectopic germinal centers emerge, enabling somatic hypermutation and class switching outside secondary lymphoid organs. These structures, sustained by IL-21/CXCL13 axes, produce high-affinity autoantibodies against citrullinated peptides, nucleic acids, and other post-translationally modified self-antigens. Persistent plasma-cell niches, maintained by IL-6 and a proliferation-inducing ligand (APRIL), perpetuate autoreactive memory.
The metabolic and epigenetic mechanisms underlying B-cell activation, differentiation, and persistence are discussed in detail in Epigenetic-metabolic programming of immune cell fate.
Targeted interventions that modulate B-cell regulatory programs, including epigenetic modulators such as EZH2 and bromodomain and extra-terminal (BET) inhibition, have demonstrated the capacity to re-establish B-cell anergy and contract autoantibody pools [5, 73, 74]. These findings highlight the therapeutic potential of restoring humoral tolerance by reprogramming B-cell functional states.
Myeloid APCs determine whether antigen encounter induces immunity or tolerance. Under homeostatic conditions, tolDCs present self-antigens with low costimulation, secrete IL-10 and TGF-β, and induce regulatory T-cell differentiation through mechanisms such as retinoic acid signaling and indoleamine 2,3-dioxygenase 1 (IDO1)-mediated tryptophan metabolism. In autoimmune rheumatic disease, this tolerogenic balance is disrupted, and APCs acquire a pro-inflammatory phenotype characterized by enhanced costimulatory signaling and cytokine production, thereby amplifying effector immune responses [75–77].
Macrophages similarly act as critical regulators of tissue immune tone, with anti-inflammatory and pro-inflammatory states shaping either resolution or persistence of inflammation. In chronic disease settings, this balance shifts toward sustained inflammatory activation, contributing to tissue damage and reinforcement of autoreactive circuits.
These functional transitions in APCs are tightly linked to underlying metabolic and epigenetic reprogramming (see Epigenetic-metabolic programming of immune cell fate), which governs their stability, plasticity, and long-term contribution to immune tolerance or inflammation.
Beyond hematopoietic compartments, stromal cells act as contextual regulators of immune fate. In the synovium, FLS contribute to tissue structure and local immune regulation, but under chronic inflammatory stimulation, they can acquire pathogenic, tissue-invasive phenotypes that sustain synovial inflammation and joint damage [78, 79]. Chronic exposure to TNF, IL-1β, and hypoxia promotes persistent transcriptional and epigenetic remodeling in FLS, leading to stable activation of inflammatory and matrix-remodeling programs. The result is a self-sustaining imprinted phenotype that can continue to secrete cytokines and matrix-degrading enzymes even ex vivo, representing a classic example of stromal inflammatory memory [18, 79].
Single-cell omics have identified specialized FLS subsets: lining FLS enriched in metalloproteinase genes drive pannus invasion, while sublining FLS expressing IFN-responsive chemokines mediate immune-cell recruitment [13, 18]. Crosstalk between these subsets and infiltrating immune cells creates a self-amplifying inflammatory circuit that sustains synovial pathology.
These stromal-state transitions are closely linked to underlying epigenetic-metabolic coupling mechanisms (see Epigenetic-metabolic programming of immune cell fate), which govern their stability and persistence.
Across lymphoid, myeloid, and stromal compartments, a convergent principle emerges: Immune tolerance reflects a coordinated transcriptional equilibrium maintained through stable chromatin regulation and intercellular signaling. Regulatory and tolerogenic populations such as Tregs, tolDCs, and M2-like macrophages maintain repressive chromatin landscapes that enforce quiescence, whereas pathogenic lymphocytes and stromal cells adopt epigenetically permissive configurations that sustain inflammatory gene expression [12, 80]. Autoimmune rheumatic diseases arise when this cross-cellular equilibrium collapses into a fixed inflammatory attractor characterized by persistent epigenetic activation and loss of regulatory control.
The molecular basis of this transition is driven by integrated epigenetic-metabolic programming mechanisms, which are discussed in detail in Epigenetic-metabolic programming of immune cell fate.
Immune cell identity and functional stability are not determined solely by lineage-specific transcription factors but emerge from a tightly integrated epigenetic-metabolic regulatory system that couples cellular energy state to chromatin architecture and gene expression programs [81]. Within this framework, metabolic pathways provide both the energetic substrates and molecular cofactors required for epigenetic enzyme activity, while chromatin configurations reciprocally regulate the expression of metabolic genes, forming a bidirectional control axis that governs immune fate decisions [81, 82].
A central feature of this system is the metabolic bifurcation between regulatory and effector immune states. Regulatory populations, including Tregs, tolDCs, and anti-inflammatory macrophages, preferentially rely on OXPHOS and FAO, metabolic programs that support mitochondrial fitness, redox balance, and sustained energy production [82, 83]. These oxidative states maintain high intracellular NAD+ levels and activate sirtuin-dependent deacetylation pathways, reinforcing repressive chromatin landscapes at inflammatory loci while stabilizing lineage-defining transcriptional programs such as FOXP3 [58, 84]. In Tregs specifically, maintenance of FOXP3 expression depends on DNA demethylation at conserved regulatory elements and controlled histone acetylation, processes that are tightly linked to mitochondrial integrity and oxidative metabolism [58, 84].
In contrast, effector immune cells—including Th1, Th17, cytotoxic T cells, and inflammatory macrophages—adopt glycolytic metabolism characterized by rapid glucose uptake, lactate production, and activation of mechanistic target of rapamycin complex 1 (mTORC1) and hypoxia-inducible factor-1 alpha (HIF-1α) signaling pathways [82, 85]. Glycolysis not only supports biosynthetic demands but also promotes epigenetic activation through increased availability of acetyl-CoA for histone acetylation and through modulation of chromatin accessibility at pro-inflammatory gene loci [82, 86]. This metabolic configuration establishes permissive chromatin states at cytokine and effector genes such as IFN gamma gene (IFNG), IL17A, and TNF, reinforcing inflammatory transcriptional programs and cellular persistence [85].
At the molecular level, key metabolites act as direct regulators of chromatin-modifying enzymes, linking cellular metabolism to epigenetic state. Acetyl-CoA serves as the substrate for histone acetyltransferases (HATs), promoting open chromatin and active transcription, whereas NAD+ regulates sirtuin-mediated deacetylation, contributing to chromatin compaction and transcriptional repression [81, 87]. Similarly, α-ketoglutarate functions as a cofactor for ten-eleven translocation (TET) enzymes and histone demethylases, facilitating DNA and histone demethylation, while succinate and fumarate act as competitive inhibitors of these enzymes, stabilizing methylation marks and reinforcing inflammatory gene expression [81, 87]. S-adenosylmethionine (SAM), generated through one-carbon metabolism, provides methyl groups for DNA and histone methylation, further integrating nutrient availability with chromatin regulation [81, 87].
Mitochondrial function plays a central role in maintaining this epigenetic-metabolic coherence. Intact mitochondrial respiration ensures balanced production of adenosine triphosphate (ATP), NAD+, and metabolic intermediates while limiting reactive oxygen species (ROS) accumulation [88]. In contrast, mitochondrial dysfunction—characterized by impaired electron transport, altered membrane potential, and defective mitophagy—leads to increased ROS production, disruption of NAD+/NADH ratios, and accumulation of metabolic byproducts such as succinate [88, 89]. These changes directly perturb chromatin-modifying enzyme activity, promoting epigenetic drift and destabilization of regulatory gene programs [81, 87]. In T cells, such mitochondrial stress contributes to the loss of FOXP3 stability and the conversion of regulatory cells into effector-like phenotypes, illustrating the metabolic fragility of immune tolerance [84].
Importantly, this epigenetic-metabolic programming operates not only within individual cells but also across multicellular immune networks. Metabolite exchange, redox gradients, and cytokine signaling coordinate metabolic states between lymphoid, myeloid, and stromal compartments, synchronizing chromatin landscapes and functional outputs across tissues [90]. Disruption of this coordination results in loss of system-wide coherence, driving the transition from adaptive immune regulation to fixed inflammatory states.
Collectively, these findings establish epigenetic-metabolic coupling as a core regulatory axis of immune tolerance, in which cellular energy flux, mitochondrial integrity, and chromatin architecture form an integrated system controlling immune identity and plasticity [91]. This framework provides the mechanistic basis for understanding how metabolic perturbations are transcribed into stable inflammatory programs and highlights the potential for therapeutic strategies that restore tolerance by simultaneously targeting metabolic and epigenetic pathways.
Although autoimmune rheumatic diseases share common features of immune tolerance breakdown, the underlying epigenetic-metabolic dysregulation is highly disease-specific, reflecting distinct cellular drivers, tissue environments, and dominant signaling pathways. Understanding these differences is critical for developing targeted and effective therapeutic strategies.
RA is characterized by a synovial-centric pathology in which FLS, together with infiltrating T cells and macrophages, establish a self-sustaining inflammatory microenvironment. A key feature is the metabolic reprogramming of FLS toward glycolysis, accompanied by mitochondrial dysfunction and increased lactate production, which reinforces local inflammation and tissue invasion [6, 92, 93].
Epigenetically, RA FLS exhibit persistent chromatin remodeling, including increased histone acetylation at inflammatory loci and activation of epigenetic regulators such as EZH2, contributing to stable expression of cytokines, chemokines, and matrix-degrading enzymes [6, 94, 95].
At the immune level, a Th17/Treg imbalance is reinforced by metabolic bias toward glycolysis and epigenetic destabilization of FOXP3 expression, promoting chronic inflammation. Given the systemic inflammatory burden of RA and its broader clinical consequences beyond the joint, strategies that reverse these stromal-immune programs may also carry implications for long-term patient outcomes [96].
In contrast to RA, SLE is a systemic autoimmune disease dominated by dysregulation of B cells and plasmacytoid dendritic cells (pDCs), with a central role for type I IFN signaling.
SLE B cells exhibit enhanced metabolic activity, including increased glycolysis and glutaminolysis, supporting autoantibody production and differentiation into plasmablasts [97, 98]. pDCs further amplify disease through sustained IFN production, which reshapes both metabolic and epigenetic landscapes across immune populations.
Epigenetically, SLE is characterized by widespread DNA hypomethylation of IFN-stimulated genes and increased chromatin accessibility at inflammatory loci, creating a transcriptionally permissive state for chronic immune activation [99–101].
This systems-level dysregulation is clinically important because SLE-related immune instability contributes to serious multisystem complications, underscoring the need for earlier and more precise disease control [102].
Sjögren’s syndrome is distinguished by its tissue-specific pathology in exocrine glands, where epithelial cells actively participate in immune dysregulation rather than serving as passive targets.
Salivary gland epithelial cells undergo metabolic reprogramming, including increased glycolysis and oxidative stress, which promotes the release of inflammatory mediators and enhances immune-cell recruitment [103, 104].
Epigenetically, these epithelial cells display activation of IFN-response elements and chromatin remodeling that sustains local cytokine production and antigen presentation capacity [105, 106].
This epithelial activation supports the formation of ectopic germinal centers and persistent B-cell activation within glandular tissue. Thus, Sjögren’s syndrome represents an epithelial-immune interface disease, where targeting epithelial metabolism and epigenetic activation may be necessary to restore tissue-level tolerance.
SSc is defined by progressive fibrosis driven by activated fibroblasts and myofibroblasts, representing a distinct pathogenic trajectory compared to primarily inflammatory diseases such as RA and SLE.
Fibroblasts in SSc exhibit a pronounced shift toward glycolytic metabolism, increased mitochondrial ROS production, and altered lipid metabolism, which collectively support collagen synthesis and extracellular matrix deposition [107–109].
Epigenetically, these cells acquire stable chromatin states characterized by persistent activation of profibrotic genes and repression of regulatory pathways, resulting in a “locked-in” phenotype resistant to reversal [110–112].
This combination of metabolic and epigenetic stabilization underlies the irreversible nature of fibrosis, positioning SSc as a fibrosis-driven epigenetic-metabolic entrenchment disease, where early intervention targeting fibroblast reprogramming is likely critical.
These disease-specific differences highlight that epigenetic-metabolic dysregulation is not uniform across autoimmune rheumatology but instead reflects distinct pathogenic architectures, as summarized comparatively in Table 3.
Disease-specific epigenetic-metabolic dysregulation in autoimmune rheumatology.
| Disease | Key cellular drivers | Dominant metabolic program | Key epigenetic features | Therapeutic implications | References |
|---|---|---|---|---|---|
| RA | FLS, Th17 cells, macrophages | Glycolysis, lactate accumulation | Histone acetylation, EZH2 activation | Target stromal metabolism + epigenetic imprinting | [6, 92–95] |
| SLE | B cells, pDCs | Glycolysis, glutaminolysis | DNA hypomethylation (ISGs), open chromatin | Target interferon axis + B-cell metabolism | [97–101] |
| Sjögren’s | Epithelial cells, B cells | Glycolysis, oxidative stress | IFN-response chromatin activation | Target epithelial activation + local immune crosstalk | [103–106] |
| Systemic sclerosis (SSc) | Fibroblasts | Glycolysis, ROS, lipid dysregulation | Stable profibrotic chromatin states | Target fibroblast reprogramming early | [107–112] |
EZH2: enhancer of zeste homolog 2; FLS: fibroblast-like synoviocytes; IFN: interferon; ISGs: interferon-stimulated genes; pDCs: plasmacytoid dendritic cells; RA: rheumatoid arthritis; ROS: reactive oxygen species; SLE: systemic lupus erythematosus.
RA is dominated by stromal-immune coupling, SLE by IFN-driven systemic activation, Sjögren’s syndrome by epithelial-immune interactions, and SSc by fibroblast-driven fibrosis. Accordingly, effective therapeutic strategies will require disease-specific targeting of dominant cellular compartments and their associated metabolic and epigenetic programs, rather than a one-size-fits-all approach.
The collapse of immune tolerance in autoimmune rheumatic diseases represents a convergent endpoint of molecular, cellular, and metabolic perturbations that erode the stability of immune homeostasis. Rather than a single initiating lesion, these disorders reflect the progressive failure of a multilayered tolerance network in which immune and stromal compartments lose reciprocal regulation. The breakdown process unfolds through intertwined mechanisms: Chronic cytokine exposure and metabolic stress destabilize regulatory lineages; epigenetic drift fixes inflammatory transcriptional states; and tissue stroma acquires autonomous inflammatory memory [12, 80]. These transitions collectively rewire immune ecosystems from dynamically self-regulating networks into an inflammatory attractor state, in which cellular and metabolic activity becomes self-sustaining and resistant to reversal even after inflammatory pathways are pharmacologically suppressed.
In RA, tolerance failure localizes to the synovial microenvironment, where autoreactive T and B cells specific for citrullinated and carbamylated self-antigens accumulate within hypoxic, cytokine-rich niches. Persistent IL-6, TNF, and granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling drives glycolytic reprogramming in T cells, macrophages, and FLS, reinforcing effector differentiation and tissue invasiveness [113, 114]. Metabolomic and single-cell multi-omic profiling reveal increased lactate and succinate levels, mitochondrial depolarization, and histone hyperacetylation at inflammatory loci, indicating that metabolic and chromatin states become coupled to inflammatory persistence [79, 113]. These findings are supported by integrative RA FLS datasets combining chromatin accessibility, histone-state mapping, transcriptomics, and chromosome conformation analyses, which demonstrate that inflammatory activation in fibroblasts is accompanied by stable regulatory rewiring rather than transient cytokine responsiveness [115, 116]. Even under biologic therapy, residual synovial T cells exhibit open chromatin at IFNG and IL17A loci, while FLS maintain hypomethylated, H3K27ac-enriched promoters controlling cytokine and matrix-remodeling genes. These durable transcriptional signatures define a form of tissue-imprinted inflammatory memory, explaining the relapse-prone nature of RA [117].
In SLE, tolerance breakdown is systemic and nucleic acid-driven. Defective clearance of apoptotic debris and aberrant activation of Toll-like receptors 7/9 (TLR7/9) in pDCs sustain type I IFN signaling, leading to continuous activation of autoreactive B and T cells. Epigenetic alterations—including DNA hypomethylation at IFN-stimulated genes and acetylation of histones at IFIT and MX dynamin-like GTPase 1 (MX1) loci—enhance transcriptional responsiveness to IFN signaling. Concurrent mitochondrial dysfunction in T cells and B cells promotes ROS accumulation, loss of NAD+ balance, and activation of cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathways, further amplifying inflammation [118, 119]. These processes yield a self-reinforcing IFN network in which metabolic exhaustion and chromatin permissiveness coalesce into a stable pathogenic state, resistant to standard immunosuppressive therapy.
Sjögren’s syndrome exemplifies the interface between immune tolerance failure and epithelial stress. Salivary gland epithelial cells acquire features of professional APCs under chronic viral mimicry and IFN-driven signaling, expressing major histocompatibility complex (MHC) class II and co-stimulatory molecules. This epithelial transformation is accompanied by metabolic rewiring—enhanced glycolysis and ROS generation—and by epigenetic activation of IFN-response elements. The local microenvironment promotes ectopic germinal center formation, where Tfh/Tph and B cells interact to sustain autoantibody production against Ro/SSA and La/SSB antigens [120, 121]. Similarly, in SSc, endothelial injury and stromal activation initiate fibrosis through TGF-β-driven transcriptional programs. SSc, fibroblasts undergo metabolic reprogramming, including increased glycolytic and glutaminolytic activity, which supports myofibroblast differentiation, collagen production, and progressive extracellular-matrix accumulation [107]. These metabolic changes cooperate with profibrotic signaling pathways to stabilize fibroblast activation and promote persistent tissue remodeling, a clinically important process reflected in SSc progression assessment and trial-design considerations [122]. These disease-specific manifestations illustrate a convergent pathophysiological principle in which tolerance breakdown arises from the disruption of intercellular metabolic-epigenetic coherence, rather than from a single immune-activation event.
Environmental and systemic modifiers further compound this collapse. Hypoxia, oxidative stress, microbiome-derived metabolites, xenobiotic exposure, and nutrient imbalance intersect with genetic susceptibilities in human leukocyte antigen (HLA), PTPN22, DNA methyltransferase 1 (DNMT1), and MECP2 to perturb metabolic cofactors such as acetyl-CoA, α-ketoglutarate, and NAD+—critical regulators of chromatin-modifying enzymes. These perturbations shift global histone acetylation and DNA methylation patterns, reducing the energy and epigenetic flexibility required for immune adaptation [123, 124]. The resulting state is one of immunologic rigidity, in which feedback loops between metabolism, chromatin architecture, and cytokine signaling are locked into chronic activation.
Collectively, the breakdown of tolerance across autoimmune rheumatic diseases reveals a unifying systems pathology in which immune dysfunction emerges when cellular metabolism and epigenetic control lose synchrony across immune and stromal compartments. What begins as a transient inflammatory adaptation gradually becomes fixed as stable transcriptional reprogramming and persistent metabolic change. Reversing this process through metabolic normalization, epigenetic remodeling, and restoration of communication between mitochondria and the nucleus represents the central therapeutic challenge for the next generation of precision immunomodulatory interventions.
This progressive loss of immune tolerance provides the mechanistic context for the epigenetic remodeling discussed in the following section, where transient inflammatory signals become fixed as durable pathogenic transcriptional states.
Epigenetic remodeling represents a central axis through which environmental, metabolic, and inflammatory inputs are transduced into long-lasting changes in gene expression. Within autoimmune rheumatic diseases, these chromatin-level alterations constitute the molecular scaffold by which transient immune activation evolves into persistent pathogenic memory. The epigenome functions simultaneously as a sensor and effector of immune state, converting fluctuations in cytokine exposure, oxidative stress, and metabolite availability into heritable transcriptional programs that stabilize effector differentiation while eroding tolerance [87, 125]. Far from being stochastic or secondary, this remodeling reflects a coordinated recalibration of enhancer landscapes, histone marks, and higher-order chromatin topology, which together reconfigure cellular identity across lymphoid, myeloid, and stromal compartments. Integrative multi-omic studies now position epigenetic drift as a defining hallmark of rheumatic autoimmunity (Figure 2) [13]. As summarized in Figure 2, chronic inflammatory inputs drive layered epigenetic drift across immune and stromal compartments, culminating in transcription-factor entrenchment, pathogenic memory, and stabilization of an inflammatory attractor state. These epigenetic processes are tightly coupled to cellular metabolism (see Epigenetic-metabolic programming of immune cell fate), which provides the necessary substrates and regulatory signals.
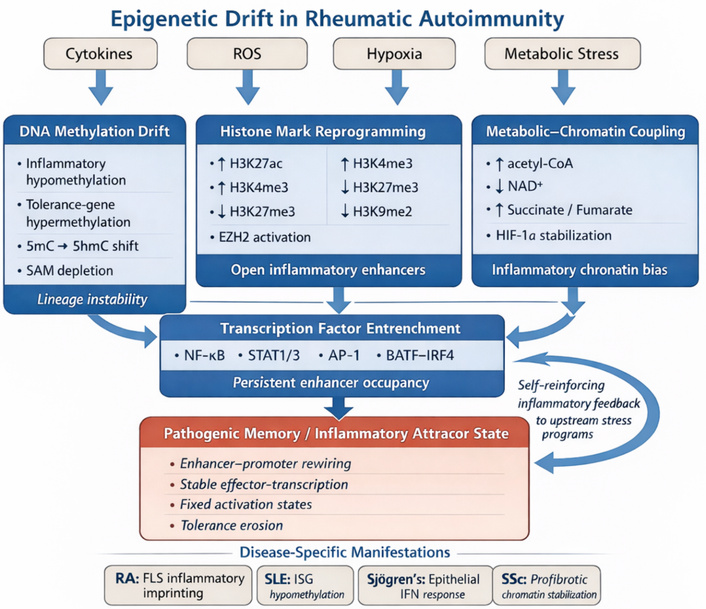
Epigenetic drift in rheumatic autoimmunity. Schematic representation of how chronic inflammatory inputs, including cytokines, ROS, hypoxia, and metabolic stress, drive progressive epigenetic drift across immune and stromal compartments in autoimmune rheumatic disease. DNA methylation abnormalities, histone-mark reprogramming, and metabolite-dependent chromatin regulation converge to destabilize tolerance-associated gene control and promote persistent occupancy of inflammatory transcription factors such as NF-κB, STAT1/3, activator protein-1 (AP-1), and basic leucine zipper ATF-like transcription factor (BATF)-chimeric antigen receptor regulatory T cells (IRF4). These layered changes culminate in pathogenic memory and stabilization of an inflammatory attractor state characterized by persistent effector-gene transcription and resistance to homeostatic resetting. Disease-specific manifestations include inflammatory imprinting in RA FLS, interferon-associated hypomethylation in SLE, epithelial interferon-response activation in Sjögren’s syndrome, and profibrotic chromatin stabilization in SSc. AP-1: activator protein-1; BATF: basic leucine zipper ATF-like transcription factor; EZH2: enhancer of zeste homolog 2; FLS: fibroblast-like synoviocytes; HIF-1α: hypoxia-inducible factor-1 alpha; IRF4: interferon regulatory factor 4; RA: rheumatoid arthritis; ROS: reactive oxygen species; SAM: S-adenosylmethionine; SLE: systemic lupus erythematosus; SSc: systemic sclerosis; STAT1/3: signal transducer and activator of transcription 1/3.
In RA and SLE, these epigenetic shifts manifest differently, with stromal inflammatory imprinting predominating in RA and IFN-associated hypomethylation signatures predominating in SLE.
DNA methylation serves as a long-term stabilizer of transcriptional identity by repressing gene promoters, insulating repetitive elements, and maintaining lineage fidelity. In autoimmune rheumatic diseases, perturbations in methylation patterns have emerged as a unifying mechanism underlying tolerance loss. CD4+ T cells in RA exhibit broad hypomethylation at IFN-responsive and proinflammatory loci, coincident with hypermethylation of genes central to metabolic restraint and immune regulation [e.g., FOXP3, IL-2 receptor alpha (IL2RA)]. This asymmetric remodeling skews transcription toward effector differentiation while impairing regulatory lineage stability [126, 127]. Similarly, B cells in SLE display promoter hypomethylation at CD11a, CD70, and CD40L, conferring autonomous activation potential even in the absence of antigenic stimulation [128].
Mechanistically, inflammatory cytokines such as IL-6, TNF, and type I IFNs converge on DNMT1 and TET dioxygenases, altering their activity through redox-dependent post-translational modifications. Elevated mitochondrial ROS oxidize 5-methylcytosine to 5-hydroxymethylcytosine, disrupting methylation fidelity and propagating transcriptional noise. These oxidative events further disrupt methylation fidelity and reinforce transcriptional instability under chronic inflammatory conditions [129, 130]. The cumulative outcome is a progressive loss of epigenetic precision, whereby gene expression becomes uncoupled from antigenic context and tolerance checkpoints collapse. This methylation drift transforms the adaptive immune system into a metastable state primed for autoreactivity (Figure 2), an effect increasingly recognized as a molecular bridge between chronic inflammation, metabolic stress, and heritable immune mispatterning.
Histone modifications act as rapid yet stable mediators of environmental and metabolic adaptation, determining whether chromatin regions remain transcriptionally accessible or repressed. In rheumatoid arthritis synovial tissue, single-cell transcriptomic and mass-cytometry analyses have identified inflammatory immune and stromal cell states linked to persistent synovial inflammation, while lupus lymphocytes show altered histone-modification patterns, including increased activating marks at immune-response genes [13, 131]. These inflammatory transcriptional and epigenetic patterns are associated with sustained expression of cytokine, chemokine, and tissue-remodeling programs, and may be influenced by metabolite-dependent chromatin-modifying enzymes, including HATs, histone deacetylases (HDACs), and HMTs [132, 133].
In chronically inflamed tissues, sustained inflammatory signaling promotes persistent chromatin accessibility and enhancer activation at pro-inflammatory gene loci, reinforcing stable transcriptional programs [134, 135]. This metabolic encoding of chromatin accessibility integrates mitochondrial dysfunction with nuclear gene regulation, effectively linking bioenergetic imbalance to transcriptional persistence. In parallel, EZH2, the catalytic subunit of the Polycomb repressive complex 2 (PRC2), is aberrantly activated in autoreactive B cells and FLS, resulting in selective silencing of anti-inflammatory and regulatory genes (e.g., SOCS1, CDKN1A) while maintaining proliferation and matrix-degrading activity. The coexistence of localized hyperacetylation and targeted Polycomb repression exemplifies the dual-axis chromatin remodeling that sustains pathogenic transcriptional programs in rheumatic autoimmunity [69, 136]. Collectively, these findings affirm that histone modification patterns in autoimmune rheumatic disease are not passive reflections of inflammation but active, metabolically inscribed determinants of tolerance fate [137].
Beyond linear chromatin modifications, autoimmune inflammation induces profound remodeling of the 3D genome, reconfiguring enhancer-promoter interactions, compartmental organization, and long-range chromosomal topology (Figure 3). This spatial reorganization converts transient transcriptional activation into structural persistence, thereby encoding inflammatory memory within nuclear architecture [138, 139]. High-resolution multi-omic studies now provide more direct support for this concept. In RA FLS, integrated high-throughput chromosome conformation capture (Hi-C), Capture Hi-C, assay for transposase-accessible chromatin with sequencing (ATAC-seq), and RNA-seq analyses have demonstrated TNF-responsive changes in chromatin organization, including altered A/B compartment activity, differential topologically associating domain (TAD) boundary behavior, and enhancer-promoter interactions linked to inflammatory gene regulation [116, 140]. Complementary epigenomic profiling in primary RA FLS, including ATAC-seq, histone-mark mapping, RNA-seq, and whole-genome bisulfite sequencing, has further shown that pathogenic fibroblasts acquire stable enhancer and promoter programs consistent with persistent inflammatory activation [115, 141]. More recent integrated RNA-seq and ATAC-seq studies also support joint-specific chromatin accessibility states in RA FLS, indicating that disease location and inflammatory context shape fibroblast regulatory architecture [6]. These altered contact maps result in the juxtaposition of distal enhancers and promoters previously segregated into repressive compartments, creating aberrant transcriptional hubs that sustain cytokine production independently of external stimuli. Figure 3 illustrates how higher-order chromatin reorganization, including TAD disruption, compartment shifts, enhancer rewiring, and mechanotransduction-linked remodeling, converts transient inflammatory signaling into structurally persistent inflammatory memory.
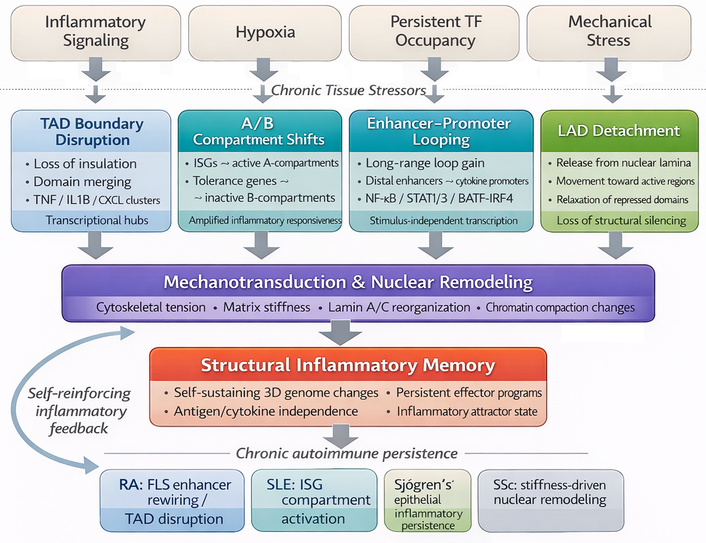
Three-dimensional (3D) genome reorganization and structural inflammatory memory. Schematic representation of how chronic inflammatory and tissue-derived stress reshapes higher-order chromatin architecture in autoimmune rheumatic disease. Inflammatory signaling, hypoxia, persistent transcription-factor occupancy, and mechanical stress promote topologically associating domain (TAD) boundary disruption, A/B compartment shifts, aberrant enhancer-promoter looping, and detachment of lamina-associated domains (LADs), thereby converting transient gene activation into persistent structural remodeling. Mechanotransduction further reinforces these changes through cytoskeletal tension, matrix stiffness, lamin A/C-dependent reorganization, and altered chromatin compaction. Together, these processes generate structural inflammatory memory, in which pathogenic transcriptional programs remain stable even after removal of the initiating stimulus. Disease-specific manifestations include enhancer rewiring in RA FLS, interferon-associated compartment shifts in SLE, epithelial inflammatory persistence in Sjögren’s syndrome, and stiffness-driven nuclear remodeling in SSc [116]. BATF: basic leucine zipper ATF-like transcription factor; CXCL: C-X-C motif chemokine ligand; FLS: fibroblast-like synoviocytes; IRF4: interferon regulatory factor 4; ISGs: interferon-stimulated genes; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; SSc: systemic sclerosis; STAT1/3: signal transducer and activator of transcription 1/3; TNF: tumor necrosis factor.
In SLE, B-cell chromatin exhibits nuclear repositioning of IFN-stimulated genes (ISGs) toward transcriptionally active A-compartments, thereby amplifying responsiveness to type I IFNs and establishing a self-reinforcing IFN signature [142, 143]. Similarly, in synovial and endothelial cells exposed to chronic hypoxia and mechanical stress, lamina-associated domains (LADs) are restructured, weakening their tethering to the nuclear periphery and permitting ectopic enhancer activation. This nuclear reorganization is stabilized by persistent transcription factor occupancy—most notably NF-κB, STAT1/3, BATF-IRF4, and AP-1 complexes—which anchor chromatin loops and maintain open chromatin conformation at inflammatory loci [144, 145]. Concomitantly, actin-lamin A/C coupling transduces mechanical stress to the nucleus, reinforcing pathological chromatin topology through mechanotransductive feedback.
These findings converge on a unifying principle in which inflammatory transcriptional states become structurally encoded once chromatin topology is reprogrammed to favor sustained accessibility of effector loci. The resulting nuclear architecture, marked by altered TAD insulation, loss of compartmental segregation, and pathological enhancer-promoter connectivity, renders effector programs refractory to classical regulatory cues. This form of topological fixation creates a higher-order layer of immune memory that transcends lineage boundaries, embedding the history of inflammatory stress directly into nuclear organization and sustaining autoimmunity even when antigenic stimulation is no longer present.
The recognition that aberrant epigenetic remodeling constitutes a primary driver of autoimmune persistence has reframed therapeutic strategy from transient immunosuppression toward durable immune reprogramming. In rheumatic autoimmunity, pathogenic chromatin landscapes are increasingly viewed as druggable architectures, dynamic and reversible determinants of lineage identity. Broad-spectrum epigenetic modulators, including HDAC inhibitors (vorinostat, givinostat), BET inhibitors (JQ1, PLX51107), and EZH2 inhibitors (tazemetostat), have demonstrated the capacity to compress hyper-accessible chromatin, silence inflammatory super-enhancers, and restore regulatory gene expression in preclinical models of arthritis and lupus [146–148]. These agents suppress cytokine transcription (IL-6, TNF, IFN-γ), attenuate fibroblast invasiveness, and partially reinstate FOXP3-dependent regulatory networks. However, their systemic administration remains constrained by dose-limiting hematopoietic toxicity, interference with antiviral defense, and incomplete cellular specificity, underscoring the need for precision-guided delivery. These approaches primarily target chromatin structure and transcriptional regulation, independent of upstream metabolic modulation.
Contemporary efforts, therefore, focus on cell-type-restricted and combinatorial strategies that exploit the intrinsic coupling between metabolism and chromatin state. Nanoparticle- or liposome-mediated targeting of epigenetic drugs to synovial fibroblasts, macrophages, or autoreactive lymphocytes can achieve locus-specific remodeling while sparing quiescent tissues [149, 150]. Parallel approaches employ ex vivo metabolic conditioning of regulatory T and B cells with cofactors such as NAD+ precursors, α-ketoglutarate, or AMP-activated protein kinase (AMPK) agonists to stabilize FOXP3- and BLIMP-1-dependent enhancer architectures before cellular reinfusion [151–153]. These manipulations enhance mitochondrial oxidative capacity and reinforce repressive chromatin marks, generating epigenetically fortified regulatory populations capable of resisting pro-inflammatory conversion in vivo. In parallel, CRISPR-based epigenome editing platforms using dCas9-fused acetyltransferase or demethylase domains allow locus-selective activation of tolerance genes (e.g., IL10, CTLA4) or silencing of effector loci (IL17A, IFNG), introducing a programmable dimension to immunotherapy [154, 155].
The most transformative advances lie in integrative metabolic-epigenetic interventions, which re-align energetic flux with chromatin control. Activation of AMPK, enhancement of sirtuin-dependent deacetylation, and restoration of NAD+ balance collectively re-establish transcriptional restraint, while inhibition of glycolytic checkpoints [lactate dehydrogenase A (LDHA), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3)] mitigates acetyl-CoA-driven histone hyperacetylation [156, 157]. Combining these metabolic correctives with selective epigenetic modulators yields synergistic tolerance induction, dampening effector transcription while restoring mitochondrial and redox homeostasis. Such frameworks exemplify a paradigm shift from immune inhibition to epigenetic rehabilitation of tolerance, in which therapy seeks not to extinguish immune activity but to recalibrate its regulatory grammar.
Within precision-medicine paradigms, chromatin accessibility, histone-mark dynamics, and circulating metabolomic profiles are emerging as quantifiable biomarkers of therapeutic response, enabling real-time monitoring of tolerance restoration. Collectively, these innovations define an emerging frontier: the epigenetic re-education of the immune system, where targeted manipulation of chromatin-metabolism circuits holds the potential to convert episodic remission into sustained immunologic equilibrium.
Despite promising preclinical efficacy, the translational potential of BET inhibitors remains constrained by safety and tolerability concerns. Early-phase clinical trials in oncology have reported dose-limiting toxicities, including thrombocytopenia and broader hematopoietic suppression, reflecting the global role of BET proteins in transcriptional regulation [158, 159]. In addition, off-target epigenetic effects and limited therapeutic windows raise concerns regarding long-term use in chronic autoimmune diseases such as RA. These limitations highlight the need for improved target selectivity, optimized dosing strategies, and patient stratification to balance efficacy with safety.
Cellular metabolism in autoimmune rheumatic diseases is characterized by coordinated alterations across core biochemical pathways, including glycolysis, OXPHOS, FAO, amino-acid metabolism, and redox regulation. These pathways integrate nutrient availability, mitochondrial function, and environmental stress signals to shape cellular behavior within inflamed tissues. The functional consequences of these metabolic states on immune cell identity and tolerance are described in Epigenetic-metabolic programming of immune cell fate. Here, we focus on the specific metabolic circuits and their contributions to disease pathogenesis and therapeutic targeting (Figure 4).
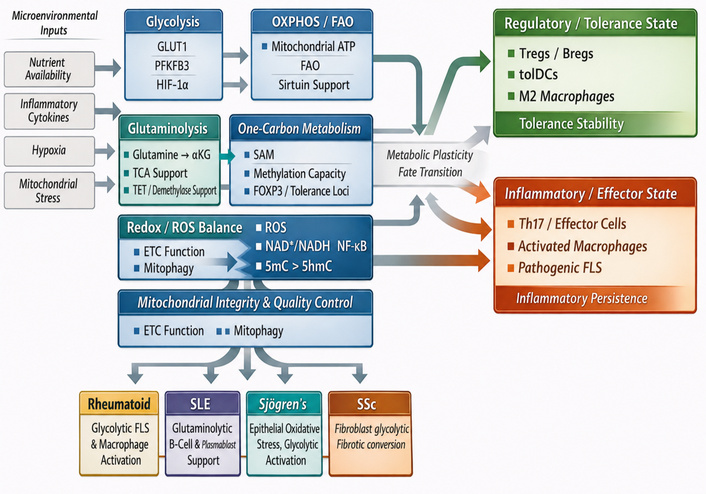
Metabolic circuits governing immune cell fate. Schematic representation of how microenvironmental inputs, including nutrient availability, inflammatory cytokines, hypoxia, and mitochondrial stress, reshape the metabolic circuits that govern immune and stromal cell fate in autoimmune rheumatic disease. The figure highlights major interconnected pathways, including glycolysis, oxidative phosphorylation/fatty-acid oxidation (OXPHOS/FAO), glutaminolysis, one-carbon metabolism, redox/ROS balance, and mitochondrial integrity/quality control. Together, these pathways regulate metabolic plasticity and influence whether cells transition toward a regulatory/tolerance-promoting state or an inflammatory/effector state. Glycolytic and redox-skewed programs favor Th17 and effector-cell differentiation, activated macrophage states, and pathogenic FLS behavior, thereby promoting inflammatory persistence. In contrast, OXPHOS/FAO-dominant programs, supported by mitochondrial fitness and sirtuin-linked regulation, favor Tregs, Bregs, tolDCs, and M2-like macrophages, thereby supporting tolerance stability. Glutaminolysis and one-carbon metabolism are depicted as intermediate metabolic nodes that influence lineage commitment and epigenetic precision through α-ketoglutarate- and SAM-dependent processes. The lower panel highlights disease-specific manifestations of these metabolic states, including glycolytic FLS and macrophage activation in rheumatoid arthritis, glutaminolytic B-cell and plasmablast-supporting programs in SLE, epithelial oxidative stress and glycolytic activation in Sjögren’s syndrome, and fibroblast glycolytic-fibrotic conversion in SSc. Bregs: regulatory B cells; ETC: electron transport chain; FLS: fibroblast-like synoviocytes; FOXP3: forkhead box P3; GLUT1: glucose transporter-1; HIF-1α: hypoxia-inducible factor-1 alpha; PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3; ROS: reactive oxygen species; SAM: S-adenosylmethionine; SLE: systemic lupus erythematosus; SSc: systemic sclerosis; TCA: tricarboxylic acid; TET: ten-eleven translocation; tolDCs: tolerogenic dendritic cells; Tregs: regulatory T cells; 5mC: 5-methylcytosine; 5hmC: 5-hydroxymethylcytosine.
As illustrated in Figure 4, metabolic circuits do not merely supply bioenergetic support, but actively bias immune and stromal cells toward either regulatory/tolerance-promoting states or inflammatory/effector states.
The immune system relies on dynamic metabolic programming to balance energy expenditure with biosynthetic demand.
Immune activation in autoimmune rheumatic disease is associated with increased glycolytic flux, enhanced glucose uptake via glucose transporter-1 (GLUT1), and activation of key metabolic regulators, including mTORC1 and HIF-1α [160]. These changes support biosynthetic demand and inflammatory signaling. Concurrently, suppression of oxidative pathways, including OXPHOS and FAO, is frequently observed, particularly under conditions of hypoxia and chronic cytokine exposure. Disruption of AMPK signaling and mitochondrial regulatory networks further contributes to metabolic imbalance across immune and stromal compartments. These pathway-level alterations establish a bioenergetic environment that favors sustained inflammatory activity (Figure 4) [83, 161, 162]. As illustrated in Figure 4, these metabolic shifts are not uniform across diseases: glycolytic dominance is especially prominent in rheumatoid synovial fibroblasts and inflammatory macrophages, whereas glutaminolytic and mitochondrial rewiring are more prominent in lupus B cells and plasmablasts.
Multi-omic profiling of RA, SLE, and SSc reveals a convergent pattern of mitochondrial dysfunction and glycolytic bias. RA T cells exhibit diminished respiratory-chain capacity and increased mitochondrial ROS, which activate HIF-1α and sustain glycolytic flux despite ATP inefficiency. In RA synovial fibroblasts, TNF-related mitochondrial stress and altered mitophagy have been implicated in synovial inflammation, whereas mitochondrial abnormalities and ROS accumulation have been described in SLE lymphocytes [163, 164]. More broadly, mitochondrial DNA release can activate cGAS-STING-type I IFN pathways in inflammatory contexts, although the specific cell-type and disease-context relationships require careful distinction [165].
In SLE, B cells and plasmablasts rely on enhanced glutaminolysis and tricarboxylic acid (TCA)-cycle anaplerosis, producing excess α-ketoglutarate and fumarate that modulate histone demethylase activity and stabilize antibody-secreting programs. Systemic-sclerosis fibroblasts undergo a glycolytic conversion mediated by pyruvate dehydrogenase kinase 1 (PDK1)-dependent pyruvate shunting and TGF-β-driven repression of mitochondrial genes, culminating in fibrotic matrix overproduction [107, 166]. Collectively, these lesions define metabolic reprogramming as a core convergent mechanism of chronic inflammation, in which impaired mitochondrial quality control translates energetic instability into transcriptional fixation of pathogenic states.
Specific metabolites accumulate within inflamed tissues and directly modulate inflammatory signaling pathways. Lactate, abundant in hypoxic synovium, can signal through GPR81 and influence immune-cell function, including dendritic-cell maturation and inflammatory responses [167]. Succinate stabilizes HIF-1α by inhibiting prolyl hydroxylases, thereby amplifying IL-1β production in inflammatory macrophages [168]. In contrast, itaconate, generated via immune-responsive gene 1 (IRG1), exerts counter-regulatory control by alkylating KEAP1 and activating nuclear factor erythroid 2-related factor 2 (NRF2)-dependent antioxidant transcription, limiting ROS-driven cytokine cascades [169, 170].
Beyond carbohydrates and lipids, amino-acid metabolism forms a third regulatory tier that links nutrient flux to epigenetic fidelity. The methionine-one-carbon cycle generates SAM, the universal methyl donor for DNA and HMTs. Limitation of methionine or serine availability, a common feature of nutrient-restricted or inflamed microenvironments, reduces SAM pools and undermines methyltransferase activity, which promotes transcriptional noise. At the same time, glutamine-derived α-ketoglutarate fuels TET and lysine demethylase (KDM) dioxygenases and supports demethylation and chromatin plasticity [171, 172]. These amino-acid-dependent pathways illustrate the way nutritional context becomes epigenetically encoded within immune and stromal compartments.
Collectively, these metabolite-mediated processes integrate environmental sensing with nuclear regulation, allowing metabolic perturbations to be transcribed into chromatin landscapes. The reciprocal nature of this system explains why metabolic correction—through restoration of NAD+/NADH ratios or mitochondrial redox balance—can re-establish tolerance even without direct genomic intervention (Table 4).
Metabolic pathways → chromatin effects → immune fate outcomes.
| Pathway | Key metabolite | Chromatin effect | Immune fate effect | Disease context | References |
|---|---|---|---|---|---|
| Glycolysis | Lactate, pyruvate | Increased H3K27ac via acetyl-CoA availability; HIF-1α-driven enhancer activation | Promotes Th17 differentiation, effector T-cell expansion, inflammatory macrophages, and invasive FLS | RA synovium shows high glycolytic flux; Th17-FLS inflammatory loops | [173–175] |
| Oxidative phosphorylation (OXPHOS) | NAD+, ATP | Sirtuin-dependent histone deacetylation; repression of effector loci; maintenance of FOXP3 enhancer integrity | Supports Tregs, Bregs, tolerogenic DCs, and M2 macrophages | NAD+ depletion in RA/SLE reduces Treg stability and mitochondrial fitness | [176–178] |
| Fatty-acid oxidation (FAO) | Acetyl-CoA, NADH | Promotes SIRT1/3 activity; enhances repressive chromatin landscapes | Stabilizes Treg phenotype; supports long-lived regulatory programs | FAO impairment contributes to Treg instability in autoimmunity | [56, 178, 179] |
| Glutaminolysis | α-Ketoglutarate (αKG) | αKG supports TET-mediated DNA/histone demethylation; maintains open chromatin at regulatory genes | Enables Treg and Breg epigenetic stability; excessive glutaminolysis drives effector expansion | High glutamine flux in RA FLS; αKG dysregulation affects Treg tolerance | [60, 180, 181] |
| One-carbon metabolism | SAM, SAH | SAM availability regulates DNA/histone methylation; SAM depletion causes global hypomethylation | Controls FOXP3 methylation status; impacts lineage fidelity | SAM: SAH imbalance seen in RA and SLE; influences T-cell differentiation | [182, 183] |
| Redox/ROS regulation | ROS, NAD+/NADH | ROS oxidizes 5mC → 5hmC; alters methylation fidelity; NAD+ levels dictate sirtuin activity | High ROS favors inflammatory programs; balanced redox supports regulatory phenotypes | Excess ROS in RA/SLE fuels inflammatory memory in T cells and macrophages | [184–186] |
| TCA cycle dysfunction | Succinate, fumarate | Succinate/fumarate inhibit αKG-dependent demethylases → hyperacetylated, pro-inflammatory chromatin | Enhances IL-1β, TNF expression, and effector persistence | Elevated succinate in RA macrophages drives pathologic cytokine output | [43, 187] |
| Mitochondrial integrity & mitophagy | NAD+ | Healthy mitochondria support epigenetic precision; dysfunctional mitochondria increase ROS & chromatin noise | Supports Treg stability and prevents exhaustion; dysfunction drives inflammatory cell fate | Mitochondrial fragmentation in FLS and T cells reinforces chronic inflammation | [188–190] |
ATP: adenosine triphosphate; Breg: regulatory B cell; FLS: fibroblast-like synoviocytes; FOXP3: forkhead box P3; HIF-1α: hypoxia-inducible factor-1 alpha; IL-1β: interleukin-1 beta; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1 alpha; RA: rheumatoid arthritis; ROS: reactive oxygen species; SAH: S-adenosylhomocysteine; SAM: S-adenosylmethionine; SIRT1/3: sirtuin 1/3; SLE: systemic lupus erythematosus; TCA: tricarboxylic acid; TET: ten-eleven translocation; TNF: tumor necrosis factor; Treg: regulatory T cell; 5mC: 5-methylcytosine; 5hmC: 5-hydroxymethylcytosine.
Therapeutic redirection of cellular metabolism is emerging as a promising strategy for restoring immune homeostasis. Metformin, through AMPK activation, re-establishes mitochondrial integrity, limits ROS, and promotes FAO-driven Treg persistence [191]. Peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists (pioglitazone, rosiglitazone) enhance oxidative metabolism and reduce fibroblast glycolysis [192], while NAD+ precursors (nicotinamide riboside, nicotinamide mononucleotide) reactivate sirtuin-dependent deacetylation and transcriptional restraint. mTOR inhibitors (rapamycin, everolimus) suppress anabolic signaling and have demonstrated clinical benefit in refractory lupus and vasculitis [193, 194].
Combination frameworks are under investigation that merge metabolic correction with epigenetic modulation. For example, SIRT1 activation paired with BET or HDAC inhibition can work synergistically to recalibrate immune networks [178]. Parallel translational approaches include bioenergetic conditioning of adoptive cell therapies in which ex vivo programming of CAR-Tregs or tolDCs under oxidative culture conditions increases mitochondrial mass and improves in vivo stability [195, 196]. Together, these interventions signal a shift from symptomatic immune suppression to metabolic restoration of tolerance architecture and align molecular therapy with systems-level homeostasis.
However, the clinical translation of NAD+-targeting strategies has yielded mixed results. While preclinical models suggest restoration of immune regulation, early clinical studies in RA have reported limited or no significant therapeutic benefit, with inconsistent effects on Treg function in human cohorts [177, 197]. These discrepancies likely reflect differences in disease stage, metabolic heterogeneity, and insufficient target engagement in vivo, underscoring the need for biomarker-guided patient selection and improved pharmacodynamic monitoring.
Across immune and stromal lineages, tolerance stability arises from the synchronization of energetic and redox states across tissues. Immune, endothelial, and mesenchymal cells engage in continuous exchange of metabolites, oxygen, and signaling intermediates that collectively maintain systemic homeostasis. In health, balanced oxidative metabolism preserves chromatin restraint and transcriptional adaptability. In disease, compartmentalized hypoxia, lactate buildup, and mitochondrial fragmentation disrupt this coherence, producing spatially heterogeneous yet functionally unified inflammation [198, 199].
Emerging spatial metabolomics and single-cell fluxomics have revealed that rheumatic tissues function as metabolic ecosystems, where dysregulation in one cellular subset propagates through shared NAD+, succinate, or ROS pools [46, 200]. Restoring these intercellular networks requires integrative strategies that align metabolic, epigenetic, and biomechanical correction. From a systems perspective, autoimmune rheumatic tissues function as interconnected metabolic environments in which alterations in one cellular compartment propagate through shared metabolite pools, including lactate, succinate, and ROS. Restoring metabolic balance across these networks represents a key requirement for durable disease control. These findings further support the concept that metabolic reprogramming is not cell-intrinsic alone but operates at the tissue and system levels.
Immune tolerance is sustained by the continuous alignment of metabolic and epigenetic programs. Metabolism provides the energetic and redox infrastructure for transcriptional regulation, while chromatin organization preserves and transmits the cell’s metabolic experience as transcriptional memory [201]. In autoimmune rheumatic diseases, disruption of this reciprocal coupling manifests as a bidirectional feed-forward loop in which inflammatory metabolism alters chromatin accessibility, and remodeled chromatin reinforces a metabolic configuration that perpetuates inflammation [46, 202]. Conversely, loss of this coordinated regulation drives the system toward an inflammatory attractor state, where pathogenic metabolic and chromatin programs become mutually reinforcing. Tolerance therefore emerges as a system-level property arising from dynamic coupling between metabolic flux and chromatin architecture across interacting immune and stromal compartments, rather than from any single regulatory pathway (Figure 5). Figure 5 integrates these relationships into a unified systems model in which metabolism, epigenetic enzyme activity, chromatin-state remodeling, transcriptional circuitry, and nuclear-mitochondrial signaling jointly determine whether tolerance is maintained or chronic inflammatory fixation emerges.
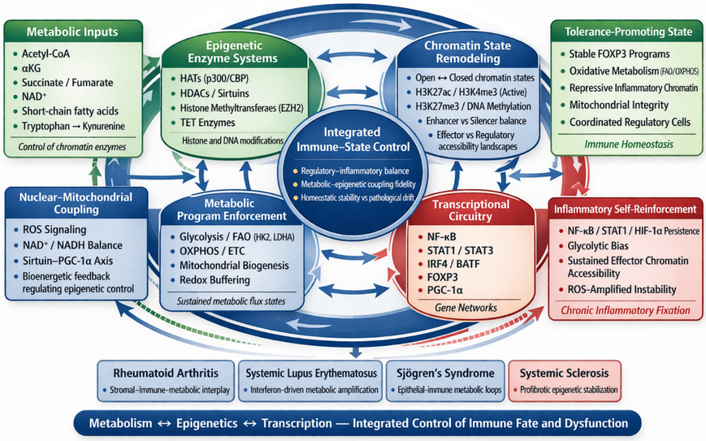
Unified epigenetic-metabolic tolerance axis in autoimmune rheumatology. Integrated systems-level schematic of the epigenetic-metabolic tolerance axis governing immune-cell stability and dysfunction in autoimmune rheumatology. Metabolic inputs, including acetyl-CoA, α-ketoglutarate, succinate/fumarate, NAD+, microbiome-derived metabolites, and redox cues, regulate epigenetic enzyme systems that shape chromatin accessibility and transcriptional circuitry. In turn, these programs reinforce metabolic pathway selection, mitochondrial integrity, and nuclear-mitochondrial coupling, creating a bidirectional network that stabilizes either regulatory immune homeostasis or chronic inflammatory self-reinforcement. The figure highlights how coordinated metabolism-epigenetics-transcription coupling maintains tolerance-promoting states, whereas breakdown of this coupling drives inflammatory fixation across diseases such as rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, and systemic sclerosis. BATF: basic leucine zipper ATF-like transcription factor; ETC: electron transport chain; EZH2: enhancer of zeste homolog 2; FAO: fatty-acid oxidation; FOXP3: forkhead box P3; HATs: histone acetyltransferases; HDACs: histone deacetylases; HIF-1α: hypoxia-inducible factor-1 alpha; IRF4: interferon regulatory factor 4; OXPHOS: oxidative phosphorylation; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1 alpha; ROS: reactive oxygen species; STAT: signal transducer and activator of transcription; TET: ten-eleven translocation.
Metabolism and chromatin form an interdependent regulatory circuit in which shared intermediates act as biochemical currencies that couple cellular energetics to gene regulation. At this interface, metabolic intermediates function not as isolated biochemical inputs but as integrative signals that couple cellular energy state to gene-regulatory architecture. Their significance lies in coordinating transcriptional stability across cells rather than in their individual enzymatic roles. Acetyl-CoA fuels HATs, SAM provides methyl donors for DNA and HMTs, α-ketoglutarate supports TET and Jumonji-domain demethylases, and NAD+ activates sirtuin deacetylases, collectively coupling metabolic flux to the chromatin landscape. In parallel, the promoters and enhancers of metabolic genes are themselves regulated by DNA methylation and histone marks, generating bidirectional feedback loops that integrate energy metabolism with transcriptional control [132, 203, 204].
Several metabolic enzymes are chromatin-associated. ATP-citrate lyase (ACLY) generates local acetyl-CoA at sites of active transcription; SIRT6 and poly(ADP-ribose) polymerase 1 (PARP1) function as chromatin-bound redox sensors; and EZH2, the catalytic core of PRC2, integrates SAM abundance to modulate histone methylation. Perturbations in nutrient availability, oxidative stress, or mitochondrial signaling destabilize this biochemical synchrony, producing transcriptional noise and lineage infidelity [205–207].
Emerging evidence further highlights nuclear-mitochondrial coordination as a stabilizing layer of this interface. Retrograde communication through NAD+/sirtuin-peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) pathways and mitochondrial transcription factor A (TFAM) links nuclear chromatin acetylation to mitochondrial gene expression, ensuring bidirectional alignment between nuclear and organellar epigenomes (Figure 5). When this coordination fails—as observed in T cells and FLS from RA and lupus—metabolic stress responses decouple from chromatin regulation, initiating persistent inflammatory transcription [208–210].
Fluctuations in cellular energy flux reshape chromatin accessibility in real time. Elevated acetyl-CoA promotes histone hyperacetylation at cytokine and chemokine loci, whereas NAD+ depletion limits sirtuin-mediated deacetylation, collectively sustaining open, transcriptionally active chromatin [211, 212]. Restoration of OXPHOS increases NAD+ availability, re-engaging sirtuin activity and favoring chromatin compaction. AMPK and mTOR function as metabolic rheostats for this process [213, 214]. AMPK activation promotes mitochondrial biogenesis and histone deacetylation, while mTORC1 activity maintains anabolic flux and supports chromatin relaxation [213, 215]. Importantly, these relationships operate as dynamic control systems, where fluctuations in energy flux continuously recalibrate chromatin accessibility rather than statically determining cell identity.
Redox signaling and oxygen tension exert additional control. ROS modify DNA and histone residues, perturb nucleosome positioning, and disrupt LADs. Chronic hypoxia promotes the migration of inflammatory gene clusters into transcriptionally active A-compartments [216, 217]. Transcription factors such as HIF-1α, PGC-1α, FOXP3, and c-Myc integrate metabolic sensing with chromatin remodeling, linking energy utilization to lineage-specific transcription [12, 173]. Through these coupled circuits, energy flux does not merely dictate cell fate but continuously reshapes the probability landscape of immune-state transitions.
Within inflamed rheumatic tissues, immune, stromal, and endothelial cells form interconnected metabolic-epigenetic ecosystems. Metabolites such as lactate, succinate, and NAD+ diffuse across the microenvironment and act as paracrine cues that synchronize chromatin states among neighboring cells [218]. Lactate derived from glycolytic fibroblasts suppresses dendritic-cell maturation yet stabilizes Th17 cells, while macrophage-derived succinate amplifies HIF-1α activity and IL-1β production in adjacent fibroblasts [168, 219]. Spatial metabolomics and single-cell ATAC-seq studies demonstrate coordinated enhancer activation across cellular compartments, revealing cross-cell synchronization of metabolic and chromatin landscapes [185, 220]. This intercellular exchange creates a distributed regulatory system in which no single cell type controls inflammation; instead, collective metabolic-epigenetic synchronization determines tissue-level behavior.
Tolerance failure arises when this synchronization collapses, fragmenting the system into locally stable but globally pathological states. Once immune and stromal cells become locked into mutually reinforcing glycolytic and inflammatory programs, the tissue transitions into a pathological attractor state that is self-sustaining, metabolically polarized, and epigenetically stabilized, persisting independently of antigenic stimulus.
Integration of ATAC-seq, chromatin immunoprecipitation sequencing (ChIP-seq), metabolomics, and transcriptomics now enables quantitative modeling of metabolic and epigenetic coupling. Dynamic Bayesian inference and network-entropy analyses identify energy-regulatory attractors that correspond to tolerant or inflammatory states. Machine-learning models applied to single-cell multi-omic datasets can predict fate transitions based on metabolic and epigenetic signatures. Machine-learning models applied to integrated transcriptomic, epigenomic, and metabolomic datasets may help identify immune-metabolic patterns, disease-associated cellular programs, and treatment-responsive states. Although broader multi-omic studies illustrate the value of computational integration across metabolism, immune features, and gene-regulatory datasets [221, 222], direct validation of specific metabolite ratios, chromatin marks, and effector-cell persistence in autoimmune rheumatic diseases remains an important area for future investigation.
These computational frameworks conceptualize tolerance as a low-entropy basin within immune-state space, characterized by energy efficiency and chromatin order, while inflammation corresponds to a high-entropy regime of metabolic inefficiency and transcriptional chaos. Such modeling provides a quantitative basis for forecasting how targeted interventions might shift cellular ensembles back toward the low-entropy, tolerant state. These models collectively support a view of immune regulation as a dynamic systems process governed by state transitions rather than fixed cellular identities.
At the systems level, therapeutic intervention can be reframed as the deliberate re-synchronization of coupled metabolic-epigenetic networks rather than the suppression of isolated inflammatory pathways. Agents that concomitantly modulate both metabolic and chromatin axes such as AMPK activators in combination with BET or HDAC inhibitors, NAD+ augmentation coupled with EZH2 modulation, or PPAR-γ agonists integrated with chromatin re-educators, embody rational designs aimed at restoring durable immune equilibrium [223, 224]. Rather than transiently silencing cytokine effector cascades, these interventions seek to reconstruct the energetic and epigenomic architectures that physiologically uphold tolerance, thereby enabling stable remission through the re-establishment of regulatory setpoints.
At the diagnostic frontier, the convergence of epigenomic and metabolomic profiling is giving rise to a new generation of integrative biomarkers capable of capturing the multidimensional state of immune regulation. Quantitative indices such as the acetyl-CoA/NAD+ ratio, SAM/S-adenosylhomocysteine (SAH) flux, mitochondrial redox potential, chromatin accessibility signatures, and single-cell transcriptional states offer mechanistic readouts of metabolic-epigenetic coherence [59, 225]. However, most of these candidate biomarkers remain research tools rather than clinically validated assays, and none are currently standardized for routine clinical decision-making in autoimmune rheumatology [225, 226]. Their current limitations include inter-platform variability, lack of assay harmonization, uncertain threshold definitions, and insufficient prospective validation across disease stages and treatment settings [59, 226, 227]. These constraints underscore the need for multicenter validation studies and clinically deployable biomarker panels before precision tolerance-reprogramming strategies can be broadly implemented.
At the conceptual level, this framework reframes autoimmunity as a disorder of epigenetic-metabolic desynchronization, wherein the loss of temporal and functional coherence between cellular energy homeostasis and gene-regulatory architecture entrenches the system within a chronic, self-sustaining inflammatory state. Reinstating tolerance thus necessitates the coordinated realignment of oxidative metabolism, redox equilibrium, and chromatin restraint across immune and stromal compartments. Within this systems-theoretic framework, immune homeostasis emerges not as a static equilibrium but as an actively regulated, programmable attractor state, a coupled configuration of metabolic efficiency and epigenetic stability that can, in principle, be rationally recalibrated through precision interventions informed by the logic of metabolic-epigenetic integration.
From this perspective, durable remission represents not the elimination of inflammation, but the restoration of system-wide coherence across metabolic and epigenetic dimensions.
Contemporary immunotherapy is shifting from nonspecific immunosuppression toward immune re-education, a strategy that seeks to retrain rather than silence pathogenic circuits. Conventional agents that block cytokine signaling or lymphocyte proliferation attenuate inflammation transiently but rarely restore the regulatory architecture required for durable tolerance. In contrast, tolerance reprogramming refers to the deliberate recalibration of immune homeostasis through coordinated modulation of metabolic, epigenetic, and transcriptional checkpoints that determine cellular identity and plasticity.
At the mechanistic level, tolerance reprogramming realigns the molecular interfaces linking energy metabolism with chromatin state. The AMPK-mTOR-sirtuin axis serves as a central rheostat integrating nutrient and redox cues to dictate whether immune cells adopt glycolytic effector profiles or oxidative, quiescent phenotypes [228, 229]. Parallel remodeling of histone acetylation, DNA methylation, and enhancer accessibility consolidates these metabolic states into heritable transcriptional programs [230, 231]. Restoring alignment across these layers re-establishes oxidative metabolism, NAD+ sufficiency, and repressive chromatin landscapes that stabilize FOXP3+ Tregs, IL-10+ Bregs, and reparative stromal subsets.
From a systems perspective, tolerance reprogramming constitutes an integrative therapeutic philosophy that unifies pharmacologic, biologic, and cellular interventions under a shared mechanistic framework (Figure 6). As summarized in Figure 6, translational implementation of immune tolerance reprogramming requires coordinated integration of therapeutic strategies, tissue-level target engagement, multi-omic biomarker frameworks, and computational modeling to guide adaptive treatment optimization. Rather than targeting single cytokines or receptors, emerging strategies act on the network topology of the immune system, coordinating chromatin accessibility, metabolic flux, and signaling feedback loops to restore systemic equilibrium. This conceptual transition—from suppression to re-education—defines the translational frontier of modern rheumatology and immunotherapy.
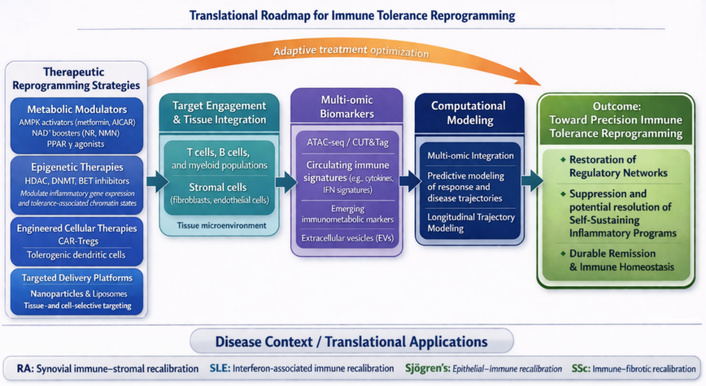
Translational roadmap for immune tolerance reprogramming. Schematic overview of the translational pathway from mechanistic insight to precision immune tolerance reprogramming in autoimmune rheumatology. Therapeutic reprogramming strategies, including metabolic modulators, epigenetic therapies, engineered cellular therapies, and targeted delivery platforms, must be integrated with tissue-specific target engagement, multi-omic biomarker frameworks, and computational modeling of disease trajectories and treatment response. Adaptive treatment optimization links these components to the ultimate goal of restoring regulatory networks, suppressing self-sustaining inflammatory programs, and achieving durable immune homeostasis. The lower panel highlights potential disease-specific applications across RA, SLE, Sjögren’s syndrome, and SSc. AMPK: AMP-activated protein kinase; ATAC-seq: assay for transposase-accessible chromatin with sequencing; BET: bromodomain and extra-terminal; CAR-Tregs: chimeric antigen receptor regulatory T cells; CUT&Tag: Cleavage Under Targets and Tagmentation; DNMT: DNA methyltransferase; HDAC: histone deacetylase; IFN: interferon; NMN: nicotinamide mononucleotide; NR: nicotinamide riboside; PPAR-γ: peroxisome proliferator-activated receptor gamma; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; SSc: systemic sclerosis.
The successful translation of these strategies will depend not only on mechanistic efficacy but also on validated biomarkers, rational patient stratification, long-term safety assessment, and clinically feasible delivery systems.
A new generation of therapeutics exploits epigenetic and metabolic coupling to re-establish tolerance (Figure 6). Epigenetic modulators such as HDAC inhibitors (e.g., givinostat, vorinostat) suppress pro-inflammatory gene networks and favor expansion of Tregs. BET inhibitors (e.g., PLX51107) selectively dismantle super-enhancers controlling TNF, IL6, and CXCL loci, thereby dampening sustained transcriptional activation. BET inhibitors have been investigated for suppressing inflammatory transcriptional programs, whereas EZH2 inhibitors and low-dose DNMT-targeting strategies are being explored as approaches to modulate pathogenic chromatin states and regulatory gene programs in selected experimental contexts [232–235]. Collectively, these agents exemplify how selective chromatin remodeling can re-educate immune networks toward regulatory stability.
In parallel, metabolic immunomodulators are being repositioned as tools to tune energy flux and redox homeostasis. Metformin activates AMPK and enhances OXPHOS, indirectly augmenting sirtuin-mediated histone deacetylation. PPAR-γ agonists recalibrate lipid metabolism and repress NF-κB-driven transcription, while NAD+ precursors such as nicotinamide riboside and nicotinamide mononucleotide restore mitochondrial function and sustain regulatory-cell persistence [213, 236, 237]. Agents targeting the mTOR-AMPK-SIRT network thus provide metabolic leverage points through which cellular bioenergetics can be re-aligned with transcriptional tolerance programs.
Increasingly, combinatorial regimens are being designed to exploit this reciprocity. Metabolic pre-conditioning through AMPK activation or NAD+ supplementation may augment the durability of chromatin-directed therapies by stabilizing repressive histone marks and limiting effector relapse [11, 35]. Nevertheless, rational combination therapy design remains a major translational challenge. Key unresolved issues include optimal sequencing of metabolic and epigenetic agents, avoidance of synergistic toxicities, and alignment of specific combinations with disease-specific pathogenic mechanisms [238, 239]. For example, combinations that may be suitable in stromal-dominant RA may not translate directly to IFN-driven SLE or fibrosis-dominant SSc [240, 241]. These considerations highlight the need for biomarker-guided trial design and adaptive dosing strategies rather than empiric combination approaches (Table 5). Figure 6 should be read in conjunction with the therapeutic limitations summarized in Table 5, as translational feasibility depends not only on mechanistic rationale but also on safety, scalability, and disease-specific applicability. Table 5 summarizes the major therapeutic classes currently under investigation, together with their mechanistic rationale, development stage, and key translational constraints relevant to clinical implementation.
Therapeutic classes, development stage, and translational limitations in tolerance reprogramming.
| Modality | Target pathway (metabolic/epigenetic/cellular) | Mechanism of tolerance reprogramming | Disease evidence | Stage (preclinical/phase I/phase II/clinical use) | Limitations/challenges | References |
|---|---|---|---|---|---|---|
| AMPK activators (e.g., metformin, AICAR) | Metabolic | Restore OXPHOS, reduce glycolysis, lower ROS; support Treg survival and mitochondrial fitness | RA, SLE, vasculitis (immunometabolic and clinical data) | Clinical use (repurposed); formal tolerance-focused trials mostly phase II /exploratory | Indirect effects; variable efficacy across patients; limited specificity for immune subsets | [242, 243] |
| NAD+ boosters (NR, NMN, nicotinamide) | Metabolic | Increase NAD+ → enhance sirtuin activity; promote histone deacetylation; improve mitochondrial quality and regulatory-cell stability | Preclinical models of arthritis, lupus, and inflammatory aging | Preclinical; early human studies in metabolic/aging contexts | Limited clinical efficacy in RA; inconsistent effects on Treg function; unclear target engagement | [244–246] |
| PPAR-γ agonists (e.g., pioglitazone) | Metabolic | Reprogram lipid metabolism; suppress NF-κB; favor oxidative and anti-inflammatory phenotypes in immune and stromal cells | Experimental arthritis and fibrosis models; limited rheumatic clinical data | Clinical use in diabetes; preclinical/early translational in rheumatology | Systemic metabolic effects, weight gain, and limited immune-specific targeting | [247] |
| HDAC inhibitors | Epigenetic | Increase histone acetylation at regulatory loci; enhance Treg function; compress inflammatory chromatin accessibility | Arthritis and lupus models; small early-phase human data | Preclinical and phase I/II | Off-target epigenetic effects; toxicity with chronic use; limited selectivity | [248, 249] |
| BET inhibitors | Epigenetic | Disrupt BRD4/super-enhancer complexes at TNF, IL6, CXCL loci; reduce sustained inflammatory transcription | Preclinical RA and tissue-inflammation models | Preclinical; some early oncology trials, limited immune-tolerance trials | Hematopoietic toxicity (e.g., thrombocytopenia); off-target transcriptional effects; narrow therapeutic window | [146, 158, 250] |
| EZH2 inhibitors | Epigenetic | Reduce H3K27me3 at silenced regulatory genes (e.g., SOCS3, CDKN1A); relieve repression of tolerogenic programs | B cells and FLS in RA and SLE; strong preclinical rationale | Preclinical and early clinical (mainly oncology); rheumatic use exploratory | Risk of global chromatin dysregulation; unclear long-term safety in non-malignant disease | [94, 147] |
| Low-dose DNMT inhibitors | Epigenetic | Partially reverse pathological DNA hypermethylation at tolerance genes; re-open FOXP3 and IL10 loci | Preclinical models of autoimmunity; conceptual alignment with epigenetic drift data | Preclinical; early clinical experience in oncology | Risk of global hypomethylation; potential oncogenic effects; dose optimization challenges | [251, 252] |
| CAR-Tregs | Cellular | Antigen-specific suppression at inflamed sites; stable FOXP3 expression and local IL-10/TGF-β delivery | Strong preclinical efficacy in arthritis, colitis, and transplantation models | Preclinical; early phase I trials in other immune contexts | Limited in vivo persistence; phenotypic instability; high cost and manufacturing complexity | [253, 254] |
| Tolerogenic dendritic cells (tolDCs) | Cellular | Present self-antigen with low costimulation; induce and expand Tregs; dampen effector priming | Phase I/II trials in RA and other autoimmune diseases are showing safety and biological activity | Phase I/phase II | Variable durability; scalability challenges; patient-to-patient variability | [255, 256] |
| MSC-based and EV-based therapies | Cellular/paracrine | Deliver tolerogenic cytokines, metabolites, and miRNAs; remodel immune and stromal metabolic-epigenetic states | Early trials in RA, SLE, and systemic sclerosis (SSc); preclinical evidence of regulatory reprogramming | Phase I/phase II; some compassionate/controlled clinical use | Heterogeneity of preparations; unclear mechanism consistency; regulatory complexity | [257, 258] |
| Nanoparticle-targeted metabolic/epigenetic agents | Delivery/metabolic/epigenetic | Cell- or tissue-specific delivery of metabolic/epigenetic drugs to synovium or lymphoid organs; limit systemic toxicity | Robust preclinical data in arthritis and systemic inflammation | Preclinical | Limited tissue penetration; delivery efficiency challenges; manufacturing scalability | [259, 260] |
| Integrated multi-omic/digital-twin–guided regimens | Systems/computational | Use epigenomic, transcriptomic, and metabolomic signatures to tailor and adapt tolerance-reprogramming therapies over time | Emerging computational and pilot translational studies | Conceptual and early translational; not yet in routine clinical practice | Lack of validated clinical biomarkers; high cost; limited clinical integration | [261–263] |
AICAR: 5-aminoimidazole-4-carboxamide ribonucleotide; AMPK: AMP-activated protein kinase; BET: bromodomain and extra-terminal; BRD4: bromodomain-containing protein 4; CAR-Tregs: chimeric antigen receptor regulatory T cells; CDKN1A: cyclin dependent kinase inhibitor 1A; CXCL: C-X-C motif chemokine ligand; DNMT: DNA methyltransferase; EV: extracellular vesicle; FLS: fibroblast-like synoviocytes; FOXP3: forkhead box P3; HDAC: histone deacetylase; IL: interleukin; MSC: mesenchymal stem cell; NMN: nicotinamide mononucleotide; NR: nicotinamide riboside; OXPHOS: oxidative phosphorylation; PPAR-γ: peroxisome proliferator-activated receptor gamma; RA: rheumatoid arthritis; ROS: reactive oxygen species; SOCS3: suppressor of cytokine signaling 3; SLE: systemic lupus erythematosus; TGF-β: transforming growth factor beta; TNF: tumor necrosis factor.
Despite this promise, targeted delivery platforms face important clinical limitations. These include variable synovial penetration, low bioavailability in inflamed tissues, batch-to-batch manufacturing complexity, and high production costs [264, 265]. In addition, delivery efficiency may differ substantially across disease sites and tissue compartments, limiting the consistency of therapeutic exposure [239, 264, 266]. Next-generation biomaterial and nanoparticle systems may improve localization and reduce systemic toxicity, but their clinical scalability and regulatory feasibility remain incompletely established.
Importantly, therapeutic strategies must be aligned with disease-specific epigenetic-metabolic architectures (Disease-specific epigenetic-metabolic dysregulation in autoimmune rheumatology), as targeting stromal metabolism in RA, IFN signaling in SLE, epithelial activation in Sjögren’s syndrome, or fibroblast reprogramming in SSc may require distinct intervention approaches.
Table 6 summarizes how epigenetic and metabolic therapeutic strategies may be differentially prioritized across RA, SLE, Sjögren’s syndrome, and SSc according to dominant disease mechanisms.
Disease-specific roles of epigenetic and metabolic therapeutics in autoimmune rheumatology.
| Disease | Dominant pathogenic axis | Most relevant therapeutic approaches | Rationale for therapeutic relevance | Key translational considerations |
|---|---|---|---|---|
| RA | Stroma-immune coupling; Th17/Treg imbalance; FLS inflammatory memory | AMPK activators, BET inhibitors, EZH2 inhibitors, nanoparticle-targeted stromal delivery, CAR-Tregs/tolDCs | Target synovial glycolysis, stromal epigenetic imprinting, and effector-regulatory imbalance | Synovial penetration, chronic-use safety, biomarker-guided patient selection |
| SLE | Interferon-driven immune activation; B-cell and pDC dysfunction | mTOR inhibitors, NAD+-targeting strategies, HDAC inhibitors, EZH2 inhibitors, B-cell-directed metabolic modulation | Target interferon-associated chromatin activation, B-cell metabolic rewiring, and systemic immune instability | Heterogeneous disease activity, systemic toxicity risk, stratification by interferon/metabolic signatures |
| Sjögren’s syndrome | Epithelial-immune crosstalk; glandular inflammatory activation | Metabolic modulators targeting epithelial stress, epigenetic modulators, B-cell/tolDC-based strategies | Target epithelial glycolytic activation, interferon-responsive chromatin states, and local ectopic immune activation | Limited tissue-specific delivery options, fewer disease-specific trials, need for gland-focused biomarkers |
| SSc | Fibroblast-driven fibrosis; profibrotic chromatin lock-in | PPAR-γ agonists, AMPK activators, antifibrotic metabolic modulators, EZH2/epigenetic targeting, targeted delivery platforms | Target fibroblast glycolytic-fibrotic reprogramming and stable profibrotic epigenetic states | Need for early intervention, fibrosis reversibility limits, and long-term safety in chronic disease |
AMPK: AMP-activated protein kinase; BET: bromodomain and extra-terminal; CAR-Tregs: chimeric antigen receptor regulatory T cells; EZH2: enhancer of zeste homolog 2; FLS: fibroblast-like synoviocytes; HDAC: histone deacetylase; mTOR: mechanistic target of rapamycin; pDC: plasmacytoid dendritic cell; PPAR-γ: peroxisome proliferator-activated receptor gamma; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; SSc: systemic sclerosis; tolDCs: tolerogenic dendritic cells.
Cellular immunotherapies embody tolerance reprogramming in a living form. Treg therapies, particularly CAR-Tregs, are engineered to recognize disease-specific antigens and deliver localized immune control through contact-dependent suppression and IL-10 or TGF-β secretion [267]. Preclinical models of RA and SLE show that CAR-Tregs maintain FOXP3 expression and resist pro-inflammatory conversion when metabolically preconditioned to favor OXPHOS and adequate NAD+ reserves [268, 269]. Engineered tolDCs, modified to express IL-10 and IDO1, promote peripheral tolerance by presenting self-antigens under metabolically quiescent conditions that favor Treg induction [270, 271].
Beyond cellular re-engineering, mesenchymal stem cell (MSC)-derived exosomes and extracellular vesicles function as nanoscale mediators of metabolic and epigenetic recalibration. These vesicles deliver microRNAs, metabolites, and chromatin-modifying enzymes that remodel the metabolic landscape of recipient immune cells, fostering regulatory phenotypes within inflamed tissues. Metabolic pre-conditioning of MSCs through AMPK activation or NAD+ enrichment enhances the tolerogenic composition of their secreted vesicles by reinforcing FOXP3 and BLIMP-1 chromatin programs [272, 273]. Early-phase clinical studies in RA, SLE, and type 1 diabetes have confirmed safety and feasibility, signaling a shift toward living biologics capable of implementing immune reprogramming in situ [274].
Despite encouraging preclinical results, significant challenges remain for CAR-Treg therapies. Recent studies indicate that CAR-Tregs may exhibit limited in vivo persistence and phenotypic instability within inflammatory microenvironments, with potential conversion toward effector-like states under sustained cytokine exposure [84, 275]. These risks raise concerns regarding durability, safety, and functional fidelity, particularly in chronically inflamed tissues such as rheumatoid synovium. Addressing these barriers will require improved CAR design, stabilization of FOXP3 expression, and microenvironment-aware engineering strategies.
Rapid advances in multi-omic profiling are redefining how immune tolerance can be quantified, monitored, and predicted. The next generation of biomarkers moves beyond cytokine titers or autoantibody levels toward integrated indicators of the epigenetic-metabolic architecture that defines immune equilibrium.
Chromatin accessibility profiling by ATAC-seq and Cleavage Under Targets and Tagmentation (CUT&Tag) enables the derivation of tolerance signatures that reflect the re-closure of inflammatory enhancers and the stabilization of FOXP3- and BLIMP-1-associated regulatory elements. Integration of these data with histone-mark landscapes (H3K27ac/H3K27me3 balance) and DNA-methylation clocks yields dynamic indices of epigenomic restraint and functional maturity of regulatory networks [276, 277].
Metabolomic and redox biomarkers complement these readouts by linking bioenergetic efficiency to chromatin control. Ratios such as acetyl-CoA: NAD+, SAM: SAH, and mitochondrial ROS flux provide quantitative proxies for histone-acetyltransferase and demethylase activity. Circulating metabolites that reflect NAD+ salvage, β-oxidation, and tricarboxylic-acid-cycle activity correlate with T-regulatory-cell frequency and remission probability in early interventional trials, underscoring their translational potential [278, 279].
Minimally invasive liquid-biopsy approaches extend these tools to clinical practice. Exosomes and extracellular vesicles isolated from plasma or synovial fluid contain microRNAs, histone fragments, and metabolic cofactors that mirror the transcriptional and energetic state of tissue-resident immune and stromal cells. Longitudinal profiling of vesicle cargo enables dynamic assessment of tolerance restoration without invasive sampling [280].
Emergent computational biomarkers employ machine-learning models trained on integrated epigenomic, transcriptomic, and metabolomic datasets to infer chromatin openness, metabolic flux, and treatment responsiveness. These predictive systems support individualized modeling of tolerance trajectories. Their adoption, however, requires rigorous regulatory standardization, encompassing sample harmonization, normalization pipelines, and cross-platform reproducibility. As these digital readouts mature, they are poised to become integral to adaptive clinical-trial design and real-time immunotherapy monitoring.
Tolerance reprogramming converges with the principles of systems medicine, which interprets immune function as an emergent property of dynamically coupled molecular and cellular networks. Within this framework, AI-driven patient stratification and multi-omic immune atlases allow mechanistic subtyping of autoimmune disorders that were previously defined solely by clinical phenotype, supporting biomarker-guided therapeutic decision-making and precision immunomodulation [281].
By jointly mapping metabolic state and chromatin configuration, clinicians can distinguish patient subsets dominated by glycolytic inflammation from those exhibiting mitochondrial insufficiency or epigenetic rigidity. This precision enables rational matching of metabolic modulators or chromatin-targeted agents to underlying molecular pathophysiology. The resulting concept of immune-network re-entrainment envisions therapy as restoration of synchronized metabolic and transcriptional oscillations across immune and stromal compartments, rather than unidirectional cytokine blockade.
This systems perspective also aligns tolerance reprogramming with the biology of regeneration and aging. Restoration of redox equilibrium, mitochondrial quality control, and chromatin fidelity parallels the rejuvenation of immune plasticity observed in caloric-restriction and sirtuin-activation paradigms. Integrating metabolic and senolytic interventions with tolerance reprogramming could therefore unify the therapeutic treatment of chronic inflammation, autoimmunity, and age-associated immune decline within a single conceptual scaffold of immunologic rejuvenation.
Translation of such complexity demands robust ethical and regulatory governance. Off-target chromatin effects, potential epigenetic inheritance of therapeutic modifications, and the persistence of engineered or metabolically conditioned cells necessitate proactive oversight [282]. In parallel, digital biomarkers introduce additional challenges concerning algorithmic transparency, data privacy, and interpretability [283]. Establishing validated standards and oversight mechanisms will be essential to ensure that precision immunology evolves responsibly and maintains public trust.
Emerging technologies now make it feasible to design immune tolerance as a programmable property of living systems. Synthetic-biology platforms are constructing gene-circuit modules capable of sensing inflammatory metabolites and autonomously activating regulatory transcriptional programs. These self-regulating constructs enable cells to maintain homeostasis within defined biochemical thresholds [284], offering an unprecedented degree of precision in controlling immune dynamics.
Simultaneously, longitudinal digital-twin frameworks are being developed to simulate immune reprogramming in silico. By integrating continuous omic, metabolic, and clinical data streams, these models can predict therapeutic efficacy, optimize dosing, and anticipate relapse before clinical manifestation. The resulting feedback systems transform immunotherapy into an adaptive, data-driven process of continual recalibration.
Future progress will depend on cross-disciplinary integration spanning metabolism, neuroscience, synthetic biology, and regenerative medicine. Such collaboration will clarify how systemic energy balance, neuronal signaling, and tissue repair interface with immune homeostasis. The synthesis of these domains will define the foundations of tolerance engineering. This includes the deliberate design of immunologic stability through coordinated manipulation of metabolic, epigenetic, and signaling networks.
Ultimately, immune tolerance should be viewed not as a passive outcome but as an actively maintainable physiological state that can be induced, stabilized, and monitored through rationally designed interventions. Achieving this vision would signal a paradigm shift in rheumatology and immunotherapy, transforming disease management from symptomatic control to the proactive cultivation of immune resilience.
While emerging epigenetic, metabolic, and cellular therapies offer promising avenues for restoring immune tolerance, several translational challenges remain. Across therapeutic classes, discrepancies between preclinical efficacy and clinical outcomes highlight the complexity of human autoimmune disease.
Epigenetic modulators, including BET and HDAC inhibitors, demonstrate potent anti-inflammatory effects in preclinical models; however, their broad regulatory roles introduce risks of off-target effects, hematologic toxicity, and limited therapeutic windows [158, 285]. Achieving selective modulation of pathogenic programs without disrupting essential cellular functions remains a central challenge.
Metabolic interventions, such as NAD+ augmentation or AMPK activation, have shown the capacity to rebalance immune cell function in experimental systems. However, clinical responses have been variable, likely reflecting inter-patient metabolic heterogeneity, disease chronicity, and incomplete target engagement [177]. These findings emphasize the need for biomarker-driven stratification and real-time metabolic monitoring.
Cellular therapies, including CAR-Treg approaches, provide high specificity but face challenges related to stability, persistence, and scalability. Inflammatory microenvironments may compromise regulatory phenotypes, while manufacturing complexity and cost remain significant barriers to widespread implementation [286–288].
Importantly, these emerging strategies must be evaluated in the context of existing standard-of-care therapies, including biologics and JAK inhibitors, which offer well-established efficacy and safety profiles. While next-generation approaches may provide deeper or more durable immune reprogramming, their comparative effectiveness, long-term safety, and cost-efficiency remain to be fully established.
Together, these considerations highlight that successful translation will require not only mechanistic innovation but also careful integration of safety, patient selection, and therapeutic positioning within current clinical frameworks.
The translation of epigenetic-metabolic tolerance reprogramming into clinical practice faces several unresolved barriers. First, biomarker validation remains limited. Although candidate measures such as chromatin accessibility profiles, metabolic ratios, and multi-omic signatures offer mechanistic resolution, most remain research-grade tools without standardized clinical thresholds, assay harmonization, or prospective validation [289, 290].
Second, patient stratification remains underdeveloped. It is still unclear how best to identify patients most likely to benefit from metabolic correction, epigenetic modulation, or cellular therapies. Distinguishing glycolysis-dominant, mitochondrial-dysfunction-dominant, IFN-driven, or fibrosis-dominant disease states will likely be essential for successful therapeutic selection [289, 291].
Third, rational combination therapy design remains a major challenge. While combined metabolic and epigenetic interventions are mechanistically attractive, their implementation requires careful optimization of dose, sequence, timing, and toxicity management. Matching specific combinations to disease-specific pathogenic architectures will be critical to avoid empiric and potentially harmful regimens [238, 239, 290].
Fourth, long-term safety requires far greater attention, particularly for epigenetic modulators. Chronic manipulation of chromatin regulators raises concerns regarding off-target transcriptional remodeling, malignant transformation, impaired host defense, and potentially durable epigenetic effects in non-target tissues [238, 239, 292]. These concerns are especially relevant in non-malignant rheumatic diseases, where therapies may be used for prolonged periods.
Finally, targeted delivery remains a major practical obstacle. Nanoparticle and ligand-directed platforms may reduce systemic toxicity, but limitations in tissue penetration, bioavailability, manufacturing complexity, and cost continue to constrain clinical translation [265, 293]. Together, these barriers indicate that successful translation will require not only mechanistic innovation but also clinically scalable biomarker platforms, disease-informed patient stratification, safety monitoring, and practical delivery solutions.
Beyond mechanistic and trial-design challenges, real-world implementation of tolerance-reprogramming strategies will depend on practical clinical considerations. Compared with established standard-of-care therapies such as biologics and JAK inhibitors, many emerging epigenetic, metabolic, and cellular interventions currently carry greater uncertainty regarding cost-effectiveness, scalability, and health-system integration [294, 295].
Cellular therapies such as CAR-Tregs and tolDCs may offer highly specific and durable immunoregulation, but their manufacturing requirements, individualized production workflows, and reimbursement challenges may limit broad accessibility [288, 295, 296]. Similarly, widespread use of multi-omic profiling for patient stratification remains constrained by cost, turnaround time, technical complexity, and uneven access across care settings [289, 290].
These realities suggest that the future adoption of tolerance-reprogramming therapies will depend not only on efficacy and safety, but also on whether they can be deployed in a cost-conscious, scalable, and equitable manner within routine rheumatology practice.
Autoimmune rheumatic diseases can be understood as disorders of disrupted epigenetic-metabolic coordination, in which immune and stromal compartments become locked into self-sustaining inflammatory states. Across the evidence reviewed here, a common principle emerges: durable restoration of tolerance is unlikely to be achieved through suppression of isolated inflammatory mediators alone, but instead will require re-establishing coordinated control over metabolism, chromatin architecture, and intercellular regulatory networks.
This framework has important translational implications. It supports the development of therapeutic strategies that combine metabolic correction, epigenetic modulation, and, where appropriate, cell-based approaches such as CAR-Tregs or tolDCs. At the same time, the field must move beyond conceptual promise toward practical implementation. Key priorities include validating disease-specific biomarkers of epigenetic-metabolic coherence, improving patient stratification, optimizing rational combination therapies, strengthening delivery platforms, and addressing long-term safety for chronic use in non-malignant disease [297, 298].
Future progress will depend on integrating mechanistic precision with clinical feasibility. This includes aligning therapeutic design with disease-specific pathogenic architecture, incorporating multi-omic and systems-level monitoring into trial design, and developing scalable, cost-conscious strategies that can ultimately be implemented in routine rheumatology care [289, 299].
In this view, the central goal of next-generation immunotherapy is not simply to suppress inflammation, but to restore immune resilience through deliberate reprogramming of the molecular and cellular systems that sustain self-tolerance. If successfully translated, this approach may redefine remission in autoimmune rheumatology as the active maintenance of programmable immune homeostasis rather than temporary control of inflammatory activity.
3D: three-dimensional
AMPK: AMP-activated protein kinase
AP-1: activator protein-1
APCs: antigen-presenting cells
ATAC-seq: assay for transposase-accessible chromatin with sequencing
ATP: adenosine triphosphate
BATF: basic leucine zipper ATF-like transcription factor
BET: bromodomain and extra-terminal
Bregs: regulatory B cells
CAR-Tregs: chimeric antigen receptor regulatory T cells
cGAS-STING: cyclic GMP-AMP synthase-stimulator of interferon genes
CXCL13: C-X-C motif chemokine ligand 13
DNMT1: DNA methyltransferase 1
EZH2: enhancer of zeste homolog 2
FAO: fatty-acid oxidation
FLS: fibroblast-like synoviocytes
FOXP3: forkhead box P3
HATs: histone acetyltransferases
HDACs: histone deacetylases
Hi-C: high-throughput chromosome conformation capture
HIF-1α: hypoxia-inducible factor-1 alpha
HMT: histone methyltransferase
IDO1: indoleamine 2,3-dioxygenase 1
IFN: interferon
IFNG: interferon gamma gene
IL: interleukin
IRF4: interferon regulatory factor 4
JAK: Janus kinase
LADs: lamina-associated domains
MSC: mesenchymal stem cell
mTOR: mechanistic target of rapamycin
mTORC1: mechanistic target of rapamycin complex 1
OXPHOS: oxidative phosphorylation
pDCs: plasmacytoid dendritic cells
PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1 alpha
PPAR-γ: peroxisome proliferator-activated receptor gamma
PRC2: Polycomb repressive complex 2
RA: rheumatoid arthritis
ROS: reactive oxygen species
SAH: S-adenosylhomocysteine
SAM: S-adenosylmethionine
SLE: systemic lupus erythematosus
SSc: systemic sclerosis
STAT: signal transducer and activator of transcription
TAD: topologically associating domain
TET: ten-eleven translocation
TNF: tumor necrosis factor
tolDCs: tolerogenic dendritic cells
Tph: T-peripheral-helper
Tregs: regulatory T cells
OAA: Conceptualization, Investigation, Visualization, Writing—original draft. MMN: Conceptualization, Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
The authors declare no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 663
Download: 25
Times Cited: 0
