Review
Review

Affiliation:
1MRes Artificial Intelligence in Business, University of Hertfordshire de Havilland Campus, AL10 9EU Hatfield, United Kingdom
ORCID: https://orcid.org/0009-0008-6875-7022
Affiliation:
2Department of Pathology, Cholistan University of Veterinary and Animal Sciences, Bahawalpur 63100, Pakistan
ORCID: https://orcid.org/0009-0009-0864-1412
Affiliation:
3Department of Zoology, Cholistan University of Veterinary and Animal Sciences, Bahawalpur 63100, Pakistan
Email: dr.waseemcuvas@gmail.com
ORCID: https://orcid.org/0000-0001-7915-5344
Explor Endocr Metab Dis. 2026;3:101472 DOI: https://doi.org/10.37349/eemd.2026.101472
Received: March 26, 2026 Accepted: April 25, 2026 Published: May 14, 2026
Academic Editor: Gulali Aktas, Abant Izzet Baysal University Hospital, Turkey
The emerging field of immunometabolism has established that the metabolic pathways governing immune cell function—glycolysis, oxidative phosphorylation, fatty acid oxidation, and amino acid metabolism—are fundamental determinants of immune responses in health and disease. This review synthesizes current evidence on how dietary inputs serve as primary environmental modulators of this immunometabolism programming. We detail the mechanisms by which macronutrients (carbohydrates, lipids, proteins), micronutrients, and specific dietary patterns (e.g., Western, Mediterranean, ketogenic) engage key nutrient-sensing pathways (mechanistic target of rapamycin [mTOR], AMP-activated protein kinase [AMPK], hypoxia-inducible factor-1α [HIF-1α], peroxisome proliferator-activated receptor [PPARs]) to rewire immune cell metabolism and influence functional phenotypes. A central role is afforded to the gut microbiota as a critical intermediary, translating diet into immunoregulatory signals, such as short-chain fatty acids. We further explore how obesogenic diets disrupt this network to fuel chronic low-grade inflammation (metaflammation), driving the pathophysiology of common metabolic diseases, including obesity, type 2 diabetes, non-alcoholic fatty liver disease (NAFLD), and atherosclerosis. Finally, we evaluate therapeutic nutritional strategies—from nutraceuticals and probiotics to the promise of precision nutrition—designed to recalibrate immunometabolism. This review underscores that diet is a powerful, modifiable lever of immunity, positioning targeted nutritional intervention as a pivotal strategy for preventing and managing metabolic disease.
The immune system represents a cornerstone of organismal defense and homeostasis, orchestrating complex responses to pathogens, tissue damage, and neoplastic threats. For decades, the study of immunity focused predominantly on signaling pathways, receptor-ligand interactions, and gene expression networks. A paradigm-shifting perspective emerged with the recognition that immune cell function is inextricably linked to its underlying metabolic state.
This intersection gave birth to the field of immunometabolism, which posits that cellular metabolic pathways are not merely passive suppliers of energy but are active, dynamic regulators of immune cell activation, differentiation, and effector function [1, 2].
At its core, immunometabolism examines how immune cells adjust their use of nutrients—including glucose, amino acids, and lipids—to meet the energetic and biosynthetic demands associated with immune activation, proliferation, and effector responses [3, 4].
This metabolic reprogramming is a fundamental hallmark of immune responses. For example, naïve T cells shift from oxidative phosphorylation (OXPHOS) toward glycolysis following antigen stimulation, supporting rapid proliferation and cytokine production [5]. Similarly, pro-inflammatory macrophages increase glycolysis and pentose phosphate pathway activity, whereas anti-inflammatory macrophages rely more heavily on fatty-acid oxidation and mitochondrial respiration [6]. These pathways are controlled by master regulators like the mechanistic target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), and hypoxia-inducible factors (HIFs), which act as sophisticated nutrient and stress sensors [7].
The evolutionary conservation of these links between metabolism and immunity underscores their biological importance. Nutrient-sensing pathways likely evolved to coordinate energy allocation with immune defense, ensuring survival in environments with fluctuating nutrient availability [8, 9]. Dysregulation of these metabolic circuits is now recognized as a contributor to diverse pathologies, including autoimmunity, chronic infection, cancer, and metabolic disease [10, 11].
If metabolism dictates immune function, then the availability of metabolic substrates—dictated by diet—becomes a powerful extrinsic modulator of the immune system. Immune cells are exquisitely sensitive to their nutritional microenvironment, constantly sampling it via the very same nutrient-sensing pathways that govern their intrinsic metabolism [12].
Dietary inputs regulate immune metabolism through several interconnected mechanisms. Macronutrients provide metabolic substrates that influence cellular bioenergetics and signaling pathways, while amino acids and micronutrients act as metabolic regulators and enzymatic cofactors [13]. Nutrient availability can therefore directly alter immune activation thresholds and metabolic programming.
However, dietary regulation of immunometabolism also operates indirectly through the gut microbiota. A major intermediary in this process is the gut microbiota, which converts dietary substrates into immunologically active metabolites. Dietary fiber fermentation generates short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, which influence immune signaling and regulatory T-cell differentiation through receptor-mediated and epigenetic mechanisms [14–16]. In contrast, diets low in fiber and high in saturated fat promote microbial dysbiosis, intestinal barrier dysfunction, and systemic inflammation through increased exposure to microbial products such as lipopolysaccharide (LPS) [17, 18].
The profound impact of diet on immunometabolism casts a harsh light on modern dietary patterns, which are increasingly characterized by excessive caloric density and high consumption of processed foods, refined carbohydrates, and saturated fats [19, 20]. These “Western” or “obesogenic” diets are the primary environmental drivers of the global epidemic of metabolic diseases, including obesity, type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), and cardiovascular disease (CVD) [21–23].
These diet-induced disturbances contribute directly to metabolic inflammation and immune dysregulation. Chronic activation of inflammatory pathways in metabolic tissues such as adipose tissue and liver promotes insulin resistance, β-cell dysfunction, and vascular injury [24–26]. Furthermore, it creates a compromised immune baseline, which may impair host defense while increasing susceptibility to inflammatory and metabolic disease [27–29].
Conversely, protective dietary patterns such as the Mediterranean diet can exert anti-inflammatory and immunometabolism benefits. Diets rich in fiber, unsaturated fatty acids, and polyphenols support beneficial microbial metabolism, reduce inflammatory signaling, and promote metabolic homeostasis [30–32]. Nutritional strategies such as caloric restriction (CR) or intermittent fasting may further enhance metabolic flexibility and immune regulation [33].
While the fields of immunometabolism, nutrition, and metabolic disease are individually vast, this review focuses specifically on the mechanistic pathways through which dietary factors reshape immune metabolism and influence metabolic disease development. Three central themes are emphasized:
Metabolic programming of immune cells.
Dietary and microbiota-derived signals that regulate these pathways.
The role of diet-driven immunometabolism dysregulation in metabolic diseases such as obesity, T2D, NAFLD/non-alcoholic steatohepatitis (NASH), and atherosclerosis.
This review aims to provide a comprehensive synthesis of the current understanding of how diet regulates immune cell metabolism in both health and metabolic disease.
This manuscript is a narrative review that synthesizes the current literature on diet, immune cell metabolism, gut microbiota, and metabolic disease. Rather than conducting a formal systematic review or meta-analysis, we integrate mechanistic studies, preclinical models, observational research, and clinical evidence to provide a conceptual framework for the field.
Evidence discussed in this review includes mechanistic cellular studies, animal models, observational human research, and clinical investigations. These diverse evidence sources are integrated to illustrate how dietary signals influence immune cell metabolism and contribute to metabolic disease development.
A literature search was conducted to identify relevant peer-reviewed articles, reviews, and key seminal papers. Primary databases included PubMed/MEDLINE, Web of Science Core Collection, and Scopus. The search period spanned from 2008 to 2025, ensuring the inclusion of foundational and highly contemporary research within the rapidly evolving fields of immunometabolism and nutritional immunology.
The search strategy combined controlled vocabulary (e.g., MeSH terms) and free-text keywords organized into three conceptual domains relevant to the scope of this review.
Domain 1 (Immunometabolism): “immunometabolism,” “immune cell metabolism,” “metabolic reprogramming,” “glycolysis,” “oxidative phosphorylation,” “fatty acid oxidation,” “macrophage polarization,” “T cell metabolism.”
Domain 2 (Nutrition/Diet): “diet,” “nutrition,” “macronutrient,” “micronutrient,” “Mediterranean diet,” “Western diet,” “ketogenic diet,” “fasting,” “probiotics,” “prebiotics,” “polyphenols.”
Domain 3 (Disease and physiology): “metaflammation,” “obesity,” “type 2 diabetes,” “NAFLD/NASH,” “atherosclerosis,” “insulin resistance,” “adipose tissue inflammation,” “gut microbiota.”
Boolean operators (AND, OR) were used to combine these domains to identify studies linking diet, immune metabolism, and metabolic disease (e.g., (Domain 1) AND (Domain 2)). The reference lists of identified key review articles and seminal papers were manually scanned (backward snowballing) to ensure inclusion of foundational and highly relevant studies not captured by the initial database search.
Identified records were screened for relevance in a two-stage process based on title/abstract and subsequent full-text review.
Studies were included when they met the following criteria:
Publication in English in a peer-reviewed journal.
Primary research (in vitro, in vivo animal models, human clinical/observational studies) or high-quality review articles/meta-analyses.
Direct investigation of the relationship between a dietary component or dietary pattern and immune cell metabolic pathways, or studies examining the role of immunometabolism dysregulation in metabolic disease pathogenesis.
Studies were excluded if they focused exclusively on:
Undernutrition or protein-energy malnutrition without a metabolic disease link.
Pharmacological interventions unrelated to nutritional or metabolic mechanisms.
Infectious disease immunology without a clear metabolic component.
Data from included studies were extracted and organized thematically according to the pre-defined structure of this review (e.g., foundational metabolic pathways, macronutrient effects, gut microbiota axis, disease-specific mechanisms).
For each thematic area, key findings, mechanistic insights, and experimental models were summarized to construct an integrated conceptual framework linking diet to immune metabolic programming.
Given the narrative and integrative scope of this review, a formal quantitative meta-analysis was not performed. Instead, a qualitative synthesis was employed to identify consensus views, elucidate mechanistic frameworks, highlight contradictory findings, and pinpoint emerging trends across the literature.
Particular emphasis was placed on integrating evidence from molecular studies, preclinical models, and human research to bridge mechanistic insights and clinical relevance in metabolic diseases. Figures were created using PowerPoint and Inkscape, which are widely used for scientific diagram preparation and do not require external image licensing.
As a broad-scope narrative review, this work has inherent limitations. The vast and interdisciplinary nature of the topic precludes an exhaustive citation of all relevant literature.
While the literature search followed structured conceptual domains, the selection of studies inevitably reflects an interpretive synthesis rather than a strictly systematic review methodology.
Furthermore, the synthesis of evidence is interpretive, drawing connections across different experimental systems (cell culture, animal models, human studies) with varying levels of physiological relevance. These limitations are acknowledged, and the review aims to provide a conceptual roadmap and a state-of-the-art synthesis to inform future hypothesis-driven research and systematic reviews on more focused questions within this field.
Although many mechanistic insights into immunometabolism originate from cellular and animal studies, translation to human physiology remains challenging because of species differences in immune regulation, diet handling, and gut microbiome structure [34–36].
Immune cells are not passive responders but metabolically dynamic entities whose functional fate—quiescence, activation, proliferation, or execution of effector duties—is intrinsically governed by their metabolic programming. The field of immunometabolism rests on the principle that specific biochemical pathways are recruited and optimized to supply the distinct bioenergetic, biosynthetic, and redox demands of each immunological state [34–37]. This section outlines the major metabolic programs utilized by immune cells and details how their preferential engagement defines the specialized functions of key immune subsets, forming the foundational metabolic lexicon essential for understanding subsequent dietary regulation.
Immune cells dynamically toggle between several core metabolic pathways, with the balance primarily dictated by activation status, environmental cues, and cellular function.
Glycolysis is the cytosolic breakdown of glucose to pyruvate, yielding a rapid but low-efficiency ATP gain. Its critical role in immunity extends far beyond energy production. Rapid glycolytic upregulation is a hallmark of innate immune cell activation (e.g., in macrophages, dendritic cells) and effector T cell responses, a process potentiated by HIFs [38, 39]. This so-called “aerobic glycolysis” or the Warburg effect provides swift ATP and, crucially, generates metabolic intermediates for biosynthetic pathways (pentose phosphate pathway for nucleotides, glycerol for lipids) necessary for cell growth, proliferation, and cytokine production [40]. In T cells, glycolysis is indispensable for robust activation and function; it fuels phosphoinositide 3-kinase (PI3K) signaling to bolster clonal expansion and effector cytokine production [41]. Conversely, its inhibition can lead to functional exhaustion [42, 43]. In innate cells like macrophages and natural killer (NK) cells, glycolytic flux is essential for pro-inflammatory (M1) polarization, antimicrobial activity, and cytotoxic function [44, 45]. However, this pathway is also co-opted in pathological states, contributing to autoimmunity and providing an immunosuppressive milieu in the tumor microenvironment [46–48].
OXPHOS is the high-yield ATP production pathway in mitochondria, coupling the electron transport chain (ETC) with ATP synthase. It is the dominant metabolic mode in quiescent, memory, and anti-inflammatory immune cells, supporting long-term survival and homeostasis. Naïve and memory T cells rely on OXPHOS fueled by fatty acid oxidation (FAO) and pyruvate from glycolysis [49]. Regulatory T cells (Tregs) also depend on robust OXPHOS for their suppressive function and stability. Impairment of mitochondrial OXPHOS, often due to persistent antigen exposure, is a key driver of T cell exhaustion, limiting their proliferative and effector potential [50, 51]. In macrophages, OXPHOS is characteristic of alternatively activated (M2) cells, supporting their tissue-reparative functions [52]. Recent evidence indicates that enhanced mitochondrial OXPHOS in B cells is associated with increased antibody production and disease progression in conditions such as systemic lupus erythematosus [53]. Furthermore, OXPHOS is vital for the function of B cells and plasma cells, supporting antibody production and, when dysregulated, contributing to inflammatory pathologies like lupus and multiple sclerosis [53–55]. Its role is complex in cancer, as it can both support antitumor immune functions and confer therapy resistance [56, 57].
FAO is the mitochondrial process of breaking down fatty acids into acetyl-CoA for entry into the Krebs cycle to fuel OXPHOS. It is a critical metabolic pathway for immune cells requiring sustained energy and resilience. Memory CD8+ T cells and Tregs heavily utilize FAO to maintain their longevity and functional fitness [49]. In macrophages, FAO is a metabolic signature of M2 polarization and is crucial for their anti-inflammatory and tissue-remodeling activities. For instance, peroxisome proliferator-activated receptor gamma (PPARγ)-dependent induction of FAO is central to alternative macrophage activation [58, 59]. Conversely, inhibition of FAO can promote a more pro-inflammatory, antimicrobial macrophage phenotype, as seen in responses to Mycobacterium tuberculosis [60, 61]. In pathological contexts, tumor-associated macrophages (TAMs) and glioblastoma cells can use FAO to support immunosuppression and resistance to therapy [62, 63].
Amino acid metabolism is indispensable for protein synthesis, but specific amino acids also serve as key signaling molecules and fuel for the Krebs cycle (anaplerosis). Glutamine is the most abundant amino acid and a primary anaplerotic substrate. Its metabolism supports nucleotide biosynthesis and redox balance (via glutathione production), making it vital for proliferating effector T cells and activated macrophages [64]. Arginine metabolism creates a critical bifurcation: it can be metabolized by inducible nitric oxide synthase (iNOS) in M1 macrophages to produce nitric oxide (NO, antimicrobial), or by arginase-1 in M2 macrophages and myeloid-derived suppressor cells (MDSCs) to produce ornithine and polyamines, promoting tissue repair and immunosuppression, respectively [65]. Tryptophan catabolism, particularly via the enzyme indoleamine 2,3-dioxygenase (IDO), depletes this essential amino acid and generates immunosuppressive kynurenines, shaping T cell responses and promoting tolerance [66]. The uptake and metabolism of these amino acids are tightly regulated by specific transporters and enzymes, presenting key nodes for immune modulation in cancer and autoimmunity [67, 68].
The differential engagement of these core pathways underlies the functional specialization of immune cell subsets. The key mechanisms underlying these immune cell metabolic states are summarized in Table 1 and supported by prior studies on glycolysis, OXPHOS, FAO, and amino acid metabolism.
Metabolic programming of major immune cell subsets.
| Immune cell and state | Primary metabolic pathway(s) | Key metabolic regulators | Functional rationale |
|---|---|---|---|
| Naïve T Cell | OXPHOS, FAO [49] | AMPK [69] | Long-term survival in circulation; energy efficiency. |
| Activated effector T Cell (Th1, Th17, CD8+) | Aerobic glycolysis, glutaminolysis [41, 70] | mTOR, HIF-1α [71] | Rapid ATP and biomass for clonal expansion and cytokine production. |
| Treg | OXPHOS, FAO [69] | AMPK, PPARγ [72] | Suppressive function; metabolic flexibility for tissue homeostasis. |
| Memory T Cell | OXPHOS, FAO [49] | AMPK, PGC-1α [73] | Long-term persistence and rapid recall upon reactivation. |
| M1 Macrophage | Aerobic glycolysis, PPP [64] | HIF-1α, mTOR [74] | Bactericidal activity; pro-inflammatory cytokine (IL-1β, TNF-α) production. |
| M2 Macrophage | OXPHOS, FAO [75] | AMPK, PPARγ [76] | Tissue repair, wound healing, and anti-inflammatory resolution. |
| Resting DC | OXPHOS, FAO [77] | AMPK | Immune surveillance; maintenance of tissue tolerance. |
| Activated DC | Aerobic glycolysis [77] | HIF-1α, mTOR | Antigen presentation, co-stimulation, and cytokine secretion. |
AMPK: AMP-activated protein kinase; DC: dendritic cell; FAO: fatty acid oxidation; HIF: hypoxia-inducible factor; mTOR: mechanistic target of rapamycin; OXPHOS: oxidative phosphorylation; PGC-1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PPARγ: peroxisome proliferator-activated receptor gamma; PPP: pentose phosphate pathway; Treg: regulatory T cell.
Having established the foundational metabolic programs that govern immune cell function, this section explores how the macronutrient composition of the diet—the supply of carbohydrates, lipids, and proteins—serves as the primary environmental cue for immunometabolic reprogramming. Nutrients are not merely fuel; they function as signaling molecules that engage specific cellular sensors, directly influencing the metabolic pathways described in the previous section and, consequently, skewing immune responses toward pro-inflammatory, anti-inflammatory, or dysregulated states [34, 35]. This nutrient-immune axis operates both directly, by altering the substrate availability and signaling within immune cells, and indirectly, via modulation of systemic hormone levels and the gut microbiota [36, 37]. Understanding these connections elucidates how modern dietary patterns contribute to metabolic inflammation and reveals potential nutritional strategies for immune modulation.
Glucose is the paramount fuel for activated immune cells, and its systemic availability, dictated by dietary carbohydrate intake and quality, is a critical determinant of immune function. Upon activation, T cells, M1 macrophages, and dendritic cells undergo a rapid metabolic switch to aerobic glycolysis, a process dependent on increased glucose uptake through transporters like glucose transporter 1 (GLUT1) [38, 40]. This glycolytic program supports the biosynthetic demands for rapid proliferation and cytokine production. For instance, glycolysis fuels PI3K signaling to bolster T cell immunity, while its inhibition can lead to functional exhaustion [41, 43].
Dietary patterns that chronically elevate blood glucose and insulin, such as high-glycemic diets rich in refined sugars, can pathologically lock immune cells into this pro-inflammatory, glycolytic state. Excessive glucose availability perpetuates the activation of the PI3K/Akt/mTOR signaling axis, a central hub that integrates nutrient signals to promote inflammatory pathways [69, 78]. In macrophages, high glucose environments stabilize HIF-1α, a master transcriptional regulator of glycolysis and pro-inflammatory genes like IL1B [39, 79]. Furthermore, high dietary fructose, a key component of ultra-processed foods, has been shown to metabolically rewire innate immunity by enhancing mTORC1 activity and IL-1β expression in monocytes, reducing their metabolic flexibility and exacerbating inflammatory responses to stimuli like LPS [80]. This state of nutrient excess drives a feed-forward loop of inflammation, contributing to the pathogenesis of metabolic syndrome and associated diseases [81, 82].
Conversely, dietary interventions that reduce carbohydrate availability, such as CR or ketogenic diets, can suppress this pro-inflammatory network. Low energy status activates AMPK and sirtuins (SIRTs, e.g., SIRT1), which inhibit mTOR and deacetylate targets like HIF-1α and nuclear factor κB (NF-κB), thereby promoting oxidative metabolism and dampening inflammatory cytokine production [83]. The resulting ketone bodies, like beta-hydroxybutyrate, can further inhibit the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in phagocytes, illustrating how diet-induced metabolites can directly modulate immune responses [84].
Dietary lipids exert profound and divergent effects on immune cell metabolism and function, largely determined by their saturation state and chain length. The balance between saturated fatty acids (SFAs) and unsaturated fatty acids in the diet is a key regulator of inflammatory tone (Table 2).
Immunometabolism impact of dietary components and patterns.
| Dietary component/Pattern | Key immune/Cellular targets | Immunometabolism effect | Resulting immune phenotype/Outcome |
|---|---|---|---|
| High glycemic carbs/Fructose | mTOR, HIF-1α, NLRP3 [80] | ↑ Glycolysis,↑ Oxidative Stress | Pro-inflammatory M1/Th1/Th17; β-cell dysfunction [85]. |
| Saturated fats (e.g., palmitate) | TLR4, NLRP3, ceramide synthesis [86, 87] | ↑ Inflammatory signaling,↓ OXPHOS | Pro-inflammatory ATMs, hepatic inflammation, and insulin resistance [88]. |
| Omega-3 PUFAs (EPA/DHA) | GPR120, PPARγ, NLRP3 [89, 90] | ↑ SPMs,↓ NLRP3 activation,↑ FAO | Anti-inflammatory M2/Treg polarization; resolved inflammation [91]. |
| Vitamin D | VDR [92] | ↑ Antimicrobial peptides,↑ Treg differentiation | Enhanced barrier defense, tolerogenic immune environment [93]. |
| Zinc | ZIP/ZnT transporters, NF-κB [94] | Antioxidant, inhibits NLRP3 | Proper lymphocyte development, controlled inflammation [95]. |
| Polyphenols (e.g., resveratrol) | AMPK, SIRT1, Nrf2, NF-κB [96] | ↑ OXPHOS/Mitophagy, ↑ Antioxidant defenses | Reduced oxidative stress, anti-inflammatory, enhanced resilience [97]. |
| Western diet | Gut microbiota, TLR4, mTOR [98] | Dysbiosis,↑ Endotoxemia,↑ Glycolysis | Systemic metaflammation: basis for metabolic diseases [99]. |
| Mediterranean diet | Gut microbiota, AMPK, PPARγ [100] | ↑ SCFA production, favorable n-6:n-3 ratio | Reduced CRP/IL-6, improved cardiometabolic health [101]. |
| Ketogenic diet/Intermittent fasting | AMPK, SIRTs, HDACs (via BHB) [102] | ↑ Ketolysis,↑ Autophagy,↑ OXPHOS | Reduced NLRP3 activity, enhanced memory T cell function. |
AMPK: AMP-activated protein kinase; ATMs: adipose tissue macrophages; CRP: C-reactive protein; DHA: docosahexaenoic acid; EPA: eicosapentaenoic acid; FAO: fatty acid oxidation; GPR: G protein-coupled receptor; HDACs: histone deacetylases; HIF: hypoxia-inducible factor; mTOR: mechanistic target of rapamycin; NF-κB: nuclear factor κB; NLRP3: NOD-like receptor family pyrin domain containing 3; Nrf2: nuclear factor erythroid 2-related factor 2; OXPHOS: oxidative phosphorylation; PPARγ: peroxisome proliferator-activated receptor gamma; PUFAs: polyunsaturated fatty acids; SCFAs: short-chain fatty acids; SIRT: sirtuin; SPMs: specialized pro-resolving mediators; TLR4: toll-like receptor 4; Treg: regulatory T cell; VDR: vitamin D receptor; ZIP/ZnT: zinc importer/zinc transporter proteins.
Beyond macronutrients, essential micronutrients and dietary bioactive compounds serve as critical cofactors and signaling molecules that fine-tune immunometabolism, ensuring optimal immune responses while preventing excessive inflammation [103].
Vitamins play important roles in immune regulation. Vitamin D, through its active form 1,25-dihydroxyvitamin D3, binds the vitamin D receptor (VDR) expressed in many immune cells [92, 104]. Vitamin D signaling enhances antimicrobial responses by inducing cathelicidin (LL-37) in macrophages while also suppressing excessive adaptive immune activation [105, 106]. It inhibits dendritic cell maturation, reduces Th1/Th17 differentiation, and promotes Treg development [93, 107]. Vitamin A (retinoic acid) supports mucosal immunity and lymphocyte trafficking to intestinal tissues, while vitamins C and E act as antioxidants that protect immune cells from reactive oxygen species generated during inflammatory responses. B-complex vitamins (B6, B9/folate, and B12) function as coenzymes in one-carbon metabolism, supporting nucleotide synthesis and epigenetic regulation during immune cell proliferation.
Minerals such as zinc, iron, and selenium are integral to immune protein structure and function. Zinc functions as an intracellular signaling molecule in lymphocytes and acts as a cofactor for numerous enzymes involved in immune responses [94]. Zinc deficiency impairs lymphocyte development, Th1 responses, and macrophage phagocytosis, increasing susceptibility to infection [95, 108]. Cellular zinc homeostasis is tightly regulated by zinc importers (ZIPs) and zinc transporters (ZnTs) and contributes to inflammatory control, including inhibition of NLRP3 inflammasome activation [109, 110]. Iron supports immune cell proliferation and mitochondrial metabolism but is also sequestered during infection as part of nutritional immunity [111]. Selenium contributes to antioxidant defense through selenoproteins such as glutathione peroxidases, protecting immune cells from oxidative stress and supporting T cell function.
Polyphenols, a diverse class of plant-derived bioactive compounds, exert immunomodulatory effects through antioxidant activity and modulation of cellular signaling pathways [96]. Many polyphenols suppress pro-inflammatory transcription factors such as NF-κB while activating cytoprotective pathways, including nuclear factor erythroid 2-related factor 2 (Nrf2). Resveratrol activates SIRT1 and AMPK, promoting mitochondrial metabolism and suppressing mTOR-associated inflammatory signaling [97, 112]. Curcumin has been reported to modulate inflammatory signaling, including NF-κB-related pathways, and to affect immune-cell phenotype in a context-dependent manner [113]. Flavonoids such as quercetin and epigallocatechin gallate influence immune signaling and improve gut barrier function and microbiota composition, thereby indirectly modulating systemic immunity [114, 115].
The synergistic and antagonistic interactions between individual nutrients are integrated at the level of whole dietary patterns, which exert sustained effects on systemic immunometabolism.
The Western diet, characterized by high intake of saturated fats, refined sugars, processed meats, and low fiber, is a primary driver of metaflammation [98, 116]. This pattern promotes pro-inflammatory immune activation through excess metabolic substrates and gut microbiota disruption, contributing to chronic low-grade inflammation and metabolic disease development [117].
In stark contrast, the Mediterranean diet, rich in fruits, vegetables, whole grains, legumes, nuts, olive oil, and fatty fish, is a paradigm of an anti-inflammatory dietary pattern [100]. Its benefits are attributed to high fiber intake, favorable fatty-acid composition, and abundant polyphenols, which support regulatory immune responses and reduced inflammatory biomarkers [101, 118].
Ketogenic diets and intermittent fasting represent dietary interventions that impose a metabolic shift from glucose toward fatty acid and ketone metabolism. These interventions promote metabolic flexibility and can suppress inflammatory signaling pathways while supporting mitochondrial function and immune regulation [102, 119].
Plant-based diets, when well planned, are rich in fiber, polyphenols, and unsaturated fats while low in saturated fat. These diets promote beneficial gut microbiota activity and the production of immunoregulatory metabolites that support balanced immune metabolism [120].
CR without malnutrition is another dietary intervention associated with improved metabolic and immune regulation [120]. CR enhances insulin sensitivity and mitochondrial efficiency while reducing inflammatory mediators and age-related immune dysfunction [121–123].
The relationship between dietary patterns, microbial metabolites, and immune cell metabolic programming is illustrated in Figure 1.
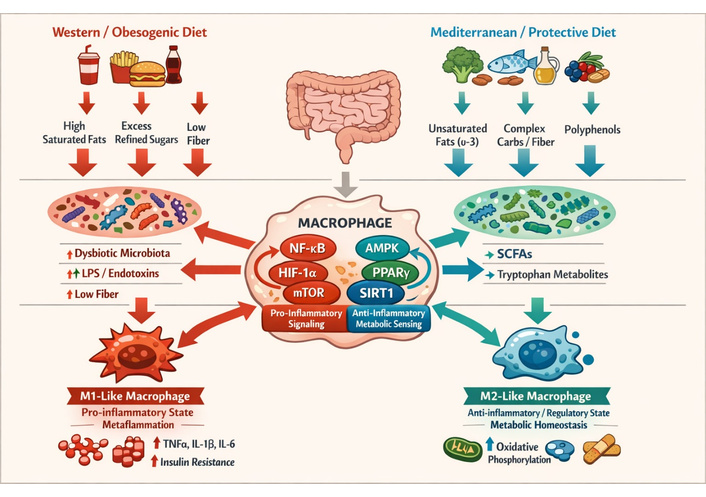
Dietary modulation of immunometabolism and immune-cell functional polarization. AMPK: AMP-activated protein kinase; HIF: hypoxia-inducible factor; LPS: lipopolysaccharide; mTOR: mechanistic target of rapamycin; NF-κB: nuclear factor κB; PPARγ: peroxisome proliferator-activated receptor gamma; SCFAs: short-chain fatty acids.
The gut microbiota, a complex ecosystem of trillions of microorganisms, is a fundamental intermediary that translates dietary patterns into systemic immunometabolic signals. Diet is the primary environmental factor shaping microbiota composition and function, which in turn governs host metabolism and immune homeostasis through the production of microbial metabolites [99].
Long-term dietary patterns imprint distinct microbial signatures. Diets rich in fiber and plant polyphenols, such as the Mediterranean diet, promote the abundance of SCFA-producing bacteria (e.g., Faecalibacterium, Roseburia) and increase microbial diversity [124]. In contrast, the Western diet, high in saturated fats and refined sugars, drives a state of dysbiosis characterized by reduced microbial diversity and expansion of pro-inflammatory microbes [121, 125]. This dysbiotic shift compromises intestinal barrier integrity and facilitates translocation of microbial products such as LPS into circulation, contributing to metabolic endotoxemia and systemic inflammation [126].
The fermentation of dietary fiber by commensal bacteria yields SCFAs, principally acetate, propionate, and butyrate [121]. These metabolites influence immune function by regulating gene expression through histone deacetylase inhibition and activation of G-protein-coupled receptors such as G protein-coupled receptor 43 (GPR43) and GPR109A [62]. Certain vitamins influence the composition of the gut microbiota [103]. SCFAs contribute to immune regulation and intestinal homeostasis while influencing inflammatory responses through microbiota-mediated metabolic pathways [127, 128].
In obesity and metabolic syndrome, dysbiosis is a hallmark feature that contributes to disease pathogenesis [52]. Altered microbial composition is associated with reduced SCFA production, increased gut permeability, and elevated systemic inflammation [99]. This disruption affects the gut-liver axis, promoting hepatic steatosis and inflammatory signaling, and may also influence appetite regulation through the gut-brain axis [129, 130]. Microbiota alterations induced by high-fat diets can further promote inflammatory macrophage accumulation in adipose tissue and impair insulin signaling. Consequently, dietary strategies aimed at restoring microbial balance, including increased fiber intake and microbiota-targeted interventions, may help correct immunometabolism dysfunction [120, 130]. Recent advances in multi-omics approaches have enabled deeper characterization of the microbiome and its association with metabolic and inflammatory diseases [131].
The key immunomodulatory metabolites derived from the gut microbiota, their dietary origins, and their mechanisms of action are summarized in Table 3.
Gut microbiota-derived metabolites: sources and immunometabolism roles.
| Microbial metabolite | Primary dietary precursors | Major receptors/Targets | Immunometabolism effects and relevance |
|---|---|---|---|
| SCAFs (butyrate) | Dietary fiber, resistant starch [132] | GPR109A, HDAC inhibition [122] | Fuels colonocytes; promotes colonic Tregs; enhances gut barrier integrity [122]. |
| SCAFs (propionate, acetate) | Dietary fiber, resistant starch [132] | GPR43/GPR41 [133] | Systemic immune regulation inhibits inflammatory cytokine production [134]. |
| Tryptophan derivatives (indole, IAld) | Dietary tryptophan [121] | AhR [135] | Modulates ILC3, Th17, and Treg balance; maintains mucosal immunity [126]. |
| Secondary bile acids (e.g., LCA, DCA) | Primary bile acids + bacterial enzymes [136] | FXR, TGR5 | Anti-inflammatory in the liver; regulates systemic metabolism and immunity. |
AhR: aryl hydrocarbon receptor; DCA: deoxycholic acid; FXR: farnesoid X receptor; GPR: G protein-coupled receptor; HDAC: histone deacetylase; LCA: lithocholic acid; SCFAs: short-chain fatty acids; TGR: Takeda G protein-coupled receptor 5; Treg: regulatory T cell.
Dietary and microbial signals converge on a set of evolutionarily conserved intracellular signaling hubs within immune cells. These pathways integrate nutrient availability, energy status, and inflammatory cues to direct metabolic reprogramming and functional fate.
Importantly, these nutrient-sensing pathways do not function independently but instead form an interconnected regulatory network controlling immune cell metabolic fate. mTOR promotes anabolic metabolism and glycolytic activation in inflammatory immune cells [137, 138], whereas AMPK functions as an energy sensor that suppresses mTOR signaling and promotes oxidative metabolism and FAO [139, 140]. HIF-1α reinforces glycolytic reprogramming under inflammatory or hypoxic conditions, sustaining pro-inflammatory effector responses [39, 71]. In contrast, PPAR family members, particularly PPARγ, promote lipid metabolism and anti-inflammatory immune phenotypes such as Tregs and M2 macrophages [76, 141, 142]. The balance among these pathways determines whether immune responses shift toward inflammatory or regulatory states [143].
mTOR complex 1 acts as a central metabolic regulator activated by nutrients and growth signals [144]. In immune cells, mTOR signaling promotes glycolytic metabolism and supports differentiation of pro-inflammatory immune subsets such as Th1, Th17, and M1 macrophages. Conversely, reduced mTOR activity, such as during CR, favors oxidative metabolism and regulatory immune phenotypes, including Tregs and M2 macrophages [145].
AMPK functions as a cellular energy sensor activated during nutrient scarcity or energetic stress. It counteracts mTOR signaling and promotes catabolic metabolism, including FAO and mitochondrial biogenesis [146]. AMPK activation also suppresses inflammatory signaling pathways and supports anti-inflammatory macrophage polarization, with potential therapeutic relevance in metabolic diseases such as NASH [147, 148].
HIF-1α is a transcriptional regulator that promotes glycolysis under hypoxic or inflammatory conditions [39, 71]. Stabilization of HIF-1α enhances glucose uptake and glycolytic enzyme expression, thereby promoting pro-inflammatory immune responses in macrophages and effector T cells [71]. Metabolic stress in tissues such as obese adipose tissue can therefore reinforce inflammatory immune activation through sustained HIF-1α signaling [149].
PPAR family members (PPARα, β/δ, and γ) regulate lipid metabolism and inflammatory gene expression. PPARγ promotes fatty-acid metabolism and supports alternative macrophage activation while repressing NF-κB-mediated inflammatory signaling [76]. Disruption of PPAR signaling by unhealthy dietary patterns may therefore contribute to metabolic inflammation [128].
SIRTs are NAD+-dependent deacylases that link cellular metabolic state to transcriptional and epigenetic regulation. SIRT1 activation during energy restriction enhances mitochondrial biogenesis through PGC-1α and suppresses inflammatory pathways, including NF-κB and HIF-1α signaling [144, 145]. These actions connect dietary restriction with improved metabolic and immune regulation [147].
NF-κB is a central transcription factor controlling inflammatory gene expression. It is activated by pattern-recognition receptor signaling and metabolic stress signals such as SFAs [148]. Activation of NF-κB drives production of cytokines, including TNF-α, IL-1β, and IL-6. In contrast, several dietary components—including polyphenols, omega-3 fatty acids, and microbial metabolites—can suppress NF-κB activity and thereby reduce inflammatory signaling [148, 149].
Recent studies (2023–2026) indicate that dietary interventions may modulate immunometabolism through epigenetic mechanisms and microbiota-derived metabolites. Early clinical evidence suggests that targeting these pathways through dietary strategies could improve inflammatory and metabolic outcomes.
Beyond its role in modulating active immune responses, a balanced and sufficient diet provides the essential substrates and signals required to maintain immune homeostasis—a dynamic equilibrium where immune surveillance, tissue repair, and self-tolerance occur without excessive inflammation.
Adequate nutritional intake, including sufficient vitamins, minerals, and omega-3 fatty acids, is essential for maintaining immune cell function and enhancing resistance to infections [127]. Macronutrients supply energy and biosynthetic substrates necessary for lymphocyte proliferation, while vitamins and trace elements such as zinc, selenium, and iron function as enzymatic cofactors that support cellular respiration, antioxidant defense, and immune signaling [128]. Dietary components also influence immune tolerance and tissue repair through interactions with the gut microbiota and metabolic pathways regulating inflammation and resolution [14, 150, 151]. Nutritional balance, therefore, contributes to maintaining immune resilience by supporting metabolic flexibility and preventing chronic inflammatory activation associated with metabolic stress and dietary imbalance.
Collectively, these interactions illustrate how balanced dietary intake sustains immune competence while preserving metabolic and inflammatory homeostasis.
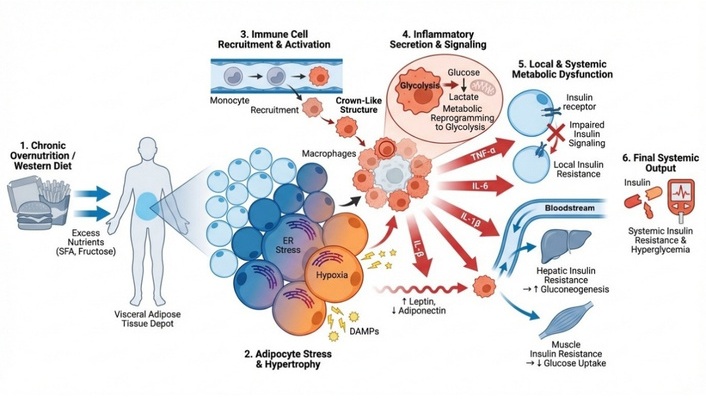
The chronic, low-grade metabolic inflammation (metaflammation) fueled by obesogenic diets is the pathogenic cornerstone of a spectrum of interconnected disorders. Diet-induced immunometabolic reprogramming contributes to the development of major metabolic diseases by altering immune cell activation, inflammatory signaling, and metabolic pathways in key tissues. This cascade of nutrient excess–induced immune activation and metabolic dysfunction leading to systemic insulin resistance is summarized in Figure 2.
Obesity is characterized by chronic low-grade inflammation originating primarily from the expansion and dysfunction of white adipose tissue (WAT). As adipocytes enlarge under nutrient excess, they release inflammatory mediators and stress signals that recruit immune cells to adipose tissue [88, 152]. Circulating monocytes differentiate into pro-inflammatory adipose tissue macrophages (ATMs), which accumulate around damaged adipocytes and secrete cytokines such as TNF-α, IL-6, and IL-1β [153, 154]. This inflammatory signaling disrupts insulin signaling pathways and promotes systemic metabolic inflammation that contributes to insulin resistance and other obesity-associated complications [155, 156].
T2D develops through a combination of insulin resistance and progressive pancreatic β-cell dysfunction, both strongly influenced by immune-mediated inflammation. Macrophage-derived cytokines such as TNF-α and IL-1β impair insulin signaling pathways in metabolic tissues, thereby exacerbating systemic insulin resistance [157, 158]. Persistent inflammatory signaling within pancreatic islets further contributes to β-cell stress, reduced insulin secretion, and eventual β-cell apoptosis [159, 160]. These processes link chronic metabolic inflammation with the progression from insulin resistance to overt diabetes.
The progression from simple steatosis to inflammatory NASH is largely mediated by liver immune cells, particularly Kupffer cells. Dietary lipids and gut-derived endotoxins activate Kupffer cells, which produce inflammatory cytokines and recruit additional monocytes to the liver [161, 162]. These immune responses promote hepatocyte injury and activate hepatic stellate cells, leading to liver inflammation and fibrosis [163, 164]. Persistent immunometabolism dysregulation therefore drives disease progression from benign fat accumulation to more severe hepatic pathology.
Atherosclerosis is a chronic inflammatory disease of the vascular wall characterized by interactions between lipid metabolism and immune activation. Circulating monocytes are recruited to the arterial intima, where they differentiate into macrophages that accumulate modified lipids and form foam cells, a defining feature of atherosclerotic plaques [165]. These foam cells release inflammatory mediators that promote plaque growth and instability [166–168]. Dietary factors that increase circulating lipids can accelerate this process, whereas anti-inflammatory dietary components may help reduce vascular inflammation and improve plaque stability [169, 170].
A comparative overview of the immunometabolism dysregulation in key diet-related diseases is presented in Table 4, which highlights the tissue-specific immune players, dietary drivers, and consequent pathogenic outcomes discussed below.
Immunometabolism dysregulation in key metabolic diseases.
| Disease/Tissue | Key immune players | Driving dietary/Dysbiotic factors | Dysregulated metabolic pathways | Pathogenic outcome |
|---|---|---|---|---|
| Obesity (adipose tissue) | ATMs, CD8+ T cells,↓ Tregs [153] | Excess SFA, fructose [123] | Glycolysis in ATMs; impaired FAO in Tregs | Insulin resistance, chronic systemic inflammation [157]. |
| T2D (pancreatic islets) | Islet-associated macrophages, T cells [159] | Hyperglycemia, AGEs [85] | ↑ Glycolysis & IL-1β production in macrophages | β-cell dysfunction & apoptosis; failure of insulin secretion [160]. |
| NAFLD/NASH (liver) | Kupffer cells, monocyte-derived macrophages [161] | SFA, fructose, gut-derived endotoxins [162] | Glycolytic shift, impaired OXPHOS in KCs [163] | Hepatocyte injury, steatosis, inflammation, fibrosis [164]. |
| Atherosclerosis (vascular wall) | Intimal macrophages (foam cells), T cells [165] | High SFA, cholesterol, trans-fats [171] | Impaired cholesterol efflux, glycolytic shift in foam cells [172] | Plaque formation, progression, and instability [173]. |
AGEs: advanced glycation end products; ATMs: adipose tissue macrophages; FAO: fatty acid oxidation; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; OXPHOS: oxidative phosphorylation; SFA: saturated fatty acid; T2D: type 2 diabetes; Treg: regulatory T cell.
Building upon the understanding of how diet drives immunometabolism dysfunction, nutritional strategies can be used to recalibrate immune metabolism and mitigate metabolic disease.
Targeted supplementation with vitamins, minerals, and bioactive compounds can support immune metabolism and reduce inflammatory signaling when deficiencies are present. For example, vitamin D influences antimicrobial responses and Treg differentiation, while zinc and polyphenols modulate immune signaling pathways linked to inflammation and insulin sensitivity [29, 174].
Dietary patterns such as the Mediterranean diet, ketogenic diets, and intermittent fasting can influence immunometabolism pathways through alterations in nutrient availability and metabolic signaling. These approaches have been associated with improvements in inflammatory markers, metabolic flexibility, and cardiometabolic health outcomes [175, 176].
Probiotics, prebiotics, synbiotics, and postbiotics aim to restore beneficial gut microbial activity and enhance production of immunoregulatory metabolites such as SCFAs, thereby influencing systemic immune and metabolic regulation [177–179]. Nutritional strategies targeting immunometabolism regulation are summarized in Table 5.
Overview of nutritional interventions for immunometabolism recalibration.
| Intervention category | Specific example | Proposed primary mechanism | Targeted conditions/Outcomes |
|---|---|---|---|
| Microbiome-targeted | Probiotics/Synbiotics | Competitive exclusion, barrier enhancement, and direct immune modulation [177] | Improved glycemia, attenuated weight gain, reduced endotoxemia [178, 179]. |
| Dietary pattern | Mediterranean diet | ↑ SCFAs, ↑ ω-3 PUFAs → AMPK/PPARγ activation [100] | Reduced CVD risk, improved glycemic control, and lower inflammation [180]. |
| Nutraceutical | Vitamin D & omega-3 PUFA | VDR signaling; Precursors to SPMs → inflammation resolution [105, 181] | Reduced autoimmune flare risk, improved cardiometabolic markers [182]. |
| Feeding pattern | Time-restricted eating (e.g., 16:8) | ↑ Autophagy, ↑ AMPK, circadian alignment of metabolism [72] | Improved hepatic steatosis, reduced adiposity, and lower inflammation [183]. |
AMPK: AMP-activated protein kinase; CVD: cardiovascular disease; PPARγ: peroxisome proliferator-activated receptor gamma; SCFAs: short-chain fatty acids; SPMs: specialized pro-resolving mediators; PUFA: polyunsaturated fatty acid; VDR: vitamin D receptor.
The effects are often strain-specific, dose-dependent, and subject to high inter-individual variability based on the host’s existing microbiota, underscoring the need for a personalized approach [184].
Precision nutrition seeks to tailor dietary strategies to an individual’s metabolic, genetic, and microbiome profile in order to improve metabolic and immune outcomes [185]. Approaches may incorporate biomarker profiling, microbiome analysis, and individualized dietary planning to optimize metabolic responses and inflammatory regulation [124, 186–192]. Advances in omics technologies, including genomics, metabolomics, and microbiome analysis, have enabled the development of personalized nutrition strategies based on gene–diet interactions and individual metabolic profiles [193].
The intricate relationship between diet, immune metabolism, and metabolic disease presents a promising frontier for therapeutic intervention, yet translating mechanistic insights into universal dietary recommendations remains challenging due to substantial biological complexity.
Individual variability in diet-immune interactions arises from multiple biological factors, including host genetics, metabolic phenotype, gut microbiome composition, sex, and age-related immune remodeling [193–201]. Genetic polymorphisms can influence nutrient sensing and inflammatory signaling pathways, while metabolic phenotypes such as insulin resistance patterns affect dietary responses in clinical interventions [188, 194]. The gut microbiome further acts as a personalized metabolic filter, determining the production of immunomodulatory metabolites that shape systemic immune responses [189, 195]. Sex hormones and aging also influence immune metabolism and inflammatory regulation, contributing to differences in disease susceptibility and therapeutic responses across populations [194, 196–200]. Together, these factors highlight that dietary interventions cannot be universally applied but must account for individual biological variation.
Precision nutrition, therefore, represents an emerging strategy to tailor dietary interventions according to an individual’s genetic, metabolic, microbiome, and immunological characteristics [185]. Integrating multi-omics datasets—including genomics, metabolomics, microbiomics, and immune profiling—with digital health tools and biomarker monitoring may enable personalized dietary strategies capable of improving metabolic and immune outcomes [202–204]. Future research must focus on phenotype-stratified clinical trials, improved biomarkers of immunometabolism health, and microbiome-informed dietary interventions to translate mechanistic discoveries into effective personalized therapies [205–207].
Collectively, the evidence indicates that dietary modulation of immunometabolism offers a plausible strategy for preventing and managing obesity, T2D, NAFLD/NASH, and atherosclerosis. Translating these mechanistic insights into practice will require intervention studies that pair dietary modification with immune and metabolic biomarkers, enabling evidence-based precision nutrition in clinical settings (Table 6).
Key challenges and future directions in precision immunonutrition.
| Source of variability/Challenge | Specific impact on diet-immune response | Future research directions |
|---|---|---|
| Host genetics | Differential nutrient sensing (e.g., VDR, PPAR polymorphisms) [194] | Genotype-stratified dietary intervention trials [186]. |
| Gut microbiome | Personal “metabolic filter” determining SCFA, tryptophan metabolite output [189] | Develop microbiome-based algorithms for dietary response prediction [207]. |
| Sex differences | Hormonal regulation of immune cell metabolism [196, 197] | Mandatory sex-stratified design and analysis in all clinical trials [208]. |
| Aging (inflammaging) | Accumulation of senescent, pro-inflammatory immune cells [198] | Test “senolytic” drugs combined with specific anti-inflammatory nutrients. |
| Measurement and tools | Lack of dynamic, clinically accessible biomarkers of immunometabolism health | Develop wearable sensors for real-time inflammation and metabolite monitoring [191, 204]. |
PPAR: peroxisome proliferator-activated receptor; SCFA: short-chain fatty acid; VDR: vitamin D receptor.
This review has elucidated the profound and mechanistic connections between dietary patterns, immune cell metabolism, and systemic metabolic health. A core principle that emerges is that nutrients are not merely fuel but are potent signaling molecules that instruct immune cell fate, function, and inflammatory potential by engaging conserved nutrient-sensing and metabolic pathways. The chronic overconsumption of refined carbohydrates, saturated fats, and ultra-processed foods—hallmarks of the modern Western diet—pathologically locks innate and adaptive immune cells into pro-inflammatory, glycolytic states. This diet-induced immunometabolism reprogramming, exacerbated by dysbiosis of the gut microbiota, creates a self-perpetuating cycle of metaflammation that is the bedrock of insulin resistance, hepatic steatosis, and vascular dysfunction.
Conversely, evidence robustly supports that protective dietary patterns, such as the Mediterranean diet, and interventions like intermittent fasting, can promote a metabolic switch towards oxidative pathways, bolster regulatory immune populations, and resolve inflammation. The translational potential of this knowledge is significant, paving the way for dietary strategies that extend beyond generic calorie restriction to target specific immunometabolism nodes.
However, the journey from mechanistic insight to effective public health and clinical application is fraught with complexity, primarily due to immense individual variability in genetics, microbiome, metabolic phenotype, sex, and age. These factors dictate personal responses to diet, rendering a universal “one-size-fits-all” prescription inadequate. Therefore, the future of nutritional therapy for metabolic disease lies in precision nutrition—the integration of multi-omics profiling, artificial intelligence, and dynamic monitoring to tailor dietary recommendations to an individual’s unique immunometabolism landscape. By embracing this personalized approach, we can move closer to harnessing diet not just for sustenance but as a precise and powerful tool to reprogram the immune system, restore metabolic homeostasis, and combat the global epidemic of metabolic disease.
AMPK: AMP-activated protein kinase
CR: caloric restriction
FAO: fatty acid oxidation
GPR: G protein-coupled receptor
HIFs: hypoxia-inducible factors
LPS: lipopolysaccharide
mTOR: mechanistic target of rapamycin
NAFLD: non-alcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
OXPHOS: oxidative phosphorylation
PI3K: phosphoinositide 3-kinase
PPARγ: peroxisome proliferator-activated receptor gamma
SCFAs: short-chain fatty acids
SFAs: saturated fatty acids
SIRT: sirtuin
T2D: type 2 diabetes
Treg: regulatory T cell
During the preparation of this work, the authors used Google AI tools for copy-editing purposes to improve readability and correct minor grammar and formatting issues. After utilizing the tool, the authors reviewed and edited the content as necessary and took full responsibility for the final content of the publication.
SK: Methodology, Writing—review & editing. TA: Supervision, Writing—review & editing, Validation. MW: Conceptualization, Writing—original draft, Methodology, Investigation. All authors read and approved the submitted version.
The authors declare that they have no competing interests.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 1599
Download: 53
Times Cited: 0