Review
Review

Affiliation:
Department of Organic Chemistry, Faculty of Sciences, University of Malaga, 29071 Malaga, Spain
Email: mgcastro@uma.es
ORCID: https://orcid.org/0000-0001-6141-2648
Affiliation:
Department of Organic Chemistry, Faculty of Sciences, University of Malaga, 29071 Malaga, Spain
Affiliation:
Department of Organic Chemistry, Faculty of Sciences, University of Malaga, 29071 Malaga, Spain
Affiliation:
Department of Organic Chemistry, Faculty of Sciences, University of Malaga, 29071 Malaga, Spain
Explor Drug Sci. 2023;1:180–209 DOI: https://doi.org/10.37349/eds.2023.00013
Received: February 13, 2023 Accepted: April 23, 2023 Published: June 30, 2023
Academic Editor: Jean-Marc Sabatier, Aix-Marseille University, France
The article belongs to the special issue Exploring Potential Drugs from Natural Products
Bacterial infections constitute one of the major cases of primary medical incidences worldwide. Historically, the fight against bacterial infections in humans has been an ongoing battle, due to the ability of bacteria to adapt and to survive. Indeed, bacteria have developed various mechanisms of resistance against several therapeutic agents. Consequently, the scientific community is always interested in search of new therapeutic agents, which are able to efficiently kill resistant-bacterial strains. This article covers the most recent antibacterial molecules approved by the Food and Drugs Administration (FDA) and European Medicines Agency (EMA) from 2012 to 2022 and intends to focus on synthetic derivatives to give a pedagogical view, with the goal of highlighting the importance of organic synthesis to obtain greater efficacy. A focus will be made on studies describing the structure and activity of the organic molecules and their interactions with their respective biological targets.
According to the professional version of the Merck Sharp & Dohme (MSD) Manual (also known as Merck Manual), “Antibacterial drugs are derived from bacteria or molds or are synthesized de novo. Technically, ‘antibiotic’ refers only to antimicrobials derived from bacteria or molds, but is often used synonymously with the term ‘antibacterial drug’.” [1]. The urgent need to search for alternative sources of antimicrobial substances also includes planting secondary metabolites, named phytochemicals [2, 3]. It is well known that there are different classifications for antibacterial drugs based on various criteria, a key one being their mechanism of action. According to this criterion, antibiotics are capable of inhibiting cell wall synthesis, increasing cell membrane permeability, or interfering with protein synthesis, nucleic acid metabolism, or other metabolic processes. Another relevant classification corresponds to their molecular structure. Accordingly, we can find aminoglycosides, cephalosporines, penicillins, macrolides, tetracyclines, lipoglycopeptides, sulfonamides, and others. For a deeper discussion and understanding of these types of classification, we refer the reader to excellent books written on this topic [4–6].
After a century of engagement in the fight against bacterial infections, the scientific community continues to intensively search for new therapeutic agents, especially those active against resistant bacteria. According to a World Health Organization (WHO) report approximately 700,000 people die from drug-resistant infections globally each year [7]. The use and abuse of antibiotics for decades have strengthened the rapid adaptation of pathogens to develop resistance to these weapons. Furthermore, an important issue to take into consideration is the degradation of antibiotics to prevent the formation of multi-drug resistant bacteria in the environment. Pharmaceutical industry discharges have been identified as important supporters to antibiotic-related aquatic contamination, having damaging consequences on biota at various trophic levels as well as human health [8]. As an important challenge, antibacterial agents themselves, even those novel bactericidal nanomaterials, can induce mutation of bacteria into resistant bacteria [9]. Consequently, the need to identify new antibiotics with novel modes of action capable of circumventing the current mechanisms of resistance and novel antimicrobial nanomaterials which show antimicrobial resistance independence represents a major priority in our society [9].
Although there are reviews in the literature covering classical and recent advances in this matter [9, 10], in the present review, we wish to deal with the field of approved antibacterial drugs during the last ten years, highlighting the importance of synthetic designs towards the final molecules. Thus, we will focus on the synthetic aspects directed to the preparation of the targeted antibiotics, discussing the relevant organic reactions and synthetic methodologies involved in their syntheses. Obviously, from a synthetic standpoint, the molecular structures of the drugs will occupy a central position in this review, focusing on the critical functional groups and pharmacophores and discussion of their novelty. Conversely, this review will not focus on taking a deep discussion on the biological mechanisms of action of these approved drugs. For that purpose, we direct the reader to important primary literature.
Firstly, we will list all the newly approved antibacterial drugs from 2012 to August 2022 by the Food and Drugs Administration (FDA) and European Medicines Agency (EMA). Secondly, we will analyze each drug according to its chemical structures, classifying them according to their key structural features. Finally, we will describe the most important and representative aspects of their syntheses.
According to our investigation by searching on Scifinder (PubMed, Scopus, Web of Science, ScienceDirect...) [11], from 2012 to August 2022, there have been 22 newly approved antimicrobial drugs by the main worldwide medicament agencies, the FDA and EMA. They have been classified according to their main structural features and into the three different categories proposed by WHO: “critically important” (“CI”), “highly important” (“HI”), and “important” (“I”) antimicrobials, following the criteria reported in a 2018 report [7]. We summarize these approved antimicrobial drugs in Table 1, including their names, major classifications, and main medical uses.
Names, major classifications, and main uses of the 22 approved antimicrobials (2012–2022)
| Entry | Antibacterial drug | Approval year | Structural classification (categorization)a | Main use |
|---|---|---|---|---|
| 1 | Bedaquiline | 2012 | Diarylquinoline (“CI”) | Combination therapy with pulmonary multi-drug resistant tuberculosis (TB) |
| 2 | Dalbavancin | 2014 | Lipoglycopeptide (“CI”) | Acute bacterial skin and skin structure infections (ABSSSI) caused by designated susceptible strains of Gram-positive microorganisms |
| 3 | Tedizolid | 2014 | Oxazolidinone (“CI”) | ABSSSI caused by designated susceptible bacteria |
| 4 | Ceftolozane-tazobactamb | 2014 | Cephalosporine and β-lactamase inhibitor (BLI; “CI”)b | Combination for: complicated intra-abdominal infections (cIAIs); complicated urinary tract infections (cUTIs); hospital-acquired bacterial pneumonia (HABP) |
| 5 | Oritavancin | 2015 | Lipoglycopeptide (“CI”) | ABSSSI by susceptible isolates of designated Gram-positive microorganisms |
| 6 | Ceftazidime-avicactamc | 2015 | Cephalosporine and BLI (“CI”)c | cIAI; cUTI; HABP |
| 7 | Obiltoxaximab | 2016 | Monoclonal antibody | Directed against the protective antigen of Bacillus anthracis |
| 8 | Bezlotoxumab | 2016 | Human monoclonal antibody | Reducing recurrence of Clostridium difficile infection (CDI) |
| 9 | Secnidazole | 2017 | Nitroimidazole (“I”) | Bacterial vaginosis in female patients; treatment of trichomoniasis |
| 10 | Delafloxacin | 2017 | Fluoroquinolone (“CI”) | ABSSSI, community-acquired bacterial pneumonia (CABP) |
| 11 | Meropenem-vaborbactamd | 2017 | Penem antibacterial and BLI (“CI”)d | cUTI |
| 12 | Ozenoxacin | 2017 | Quinolone (“CI”) | Topical treatment of impetigo due to Staphylococcus aureus or Streptococcus pyogenes |
| 13 | Plazomicin | 2018 | Aminoglycoside (“CI”) | cUTI |
| 14 | Eravacycline | 2018 | Tetracycline (“HI”) | cIAI |
| 15 | Sarecycline | 2018 | Tetracycline | Inflammatory lesions of non-nodular moderate to severe acne vulgaris |
| 16 | Omadacycline | 2018 | Tetracycline (“HI”) | CABP, ABSSSI |
| 17 | Rifamycin | 2018 | Rifamycin (“CI”) | Travelers’ diarrhea caused by noninvasive strains of Escherichia coli |
| 18 | Imipenem-cilastatin-relebactame | 2019 | Penem antibacterial, renal dehydropeptidase inhibitor, and BLI (“CI”)e | cUTI; cIAI; HABP/ventilator-associated bacterial pneumonia (VABP) |
| 19 | Pretomanid | 2019 | First-in-class oxazine | Pulmonary extensively drug-resistant TB (XDR-TB) |
| 20 | Lefamulin | 2019 | First-in-class pleuromutilin antibacterial | CABP |
| 21 | Cefiderocol | 2019 | Cephalosporin | cUTI, including pyelonephritis and HABP/VABP |
| 22 | Vonoprazan | 2022 | A potassium-competitive acid blocker (PCAB) | Treatment of Helicobacter pylori infection |
a Categorization according to 2018 WHO’s inform; b both drugs are suministred in combination under the name Zerbaxa® and both are “CI”; c both drugs must be together administered under the name Avycaz® and both are “CI”; d both drugs are administered in combination under the name Vabomere® and both are “CI”; e sold under the brand name Recarbrio® is a fixed-dose combination medication
First-in-class drugs are defined as drugs that modulate an as-yet unprecedented drug target or biological pathway [12]. Based on this definition, only two new antimicrobial molecules corresponding to this category have been approved during the last ten years, pretomanid and lefamulin, which were both approved in 2019.
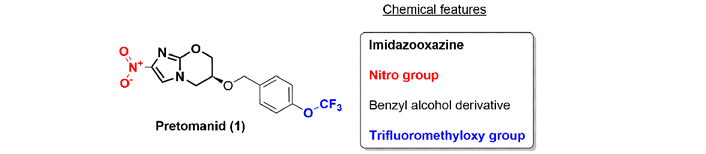
Pretomanid (1), also known as PA-824, was first identified in 1995 [13]. A key structural feature of the compound is the presence of a nitroimidazooxazine system (Figure 1). Pretomanid is an orally administrated drug that inhibits mycolic acid biosynthesis and kills actively replicating Mycobacterium tuberculosis by blocking cell wall production. It also acts as a respiratory poison and inhibits the protein synthesis of the bacteria. Pretomanid is also metabolized to form highly reactive nitrogen intermediates, including nitric oxide [14].
Pretomanid is indicated for pulmonary XDR-TB and must be used only in combination with bedaquiline (116, see “Quinoline: bedaquiline”) and linezolid [15].
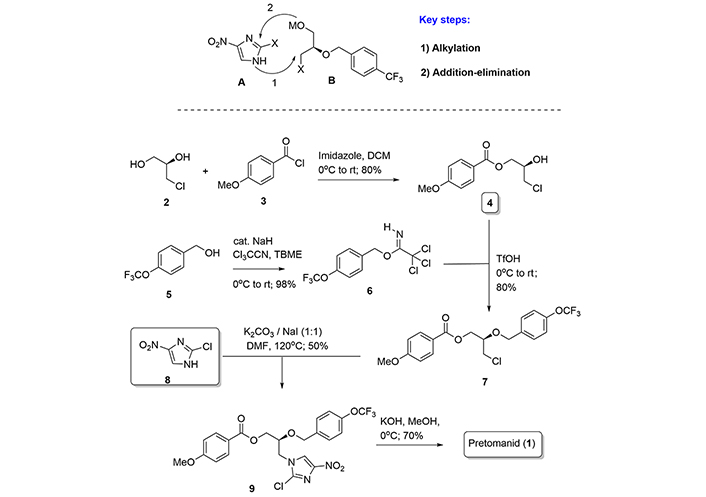
Among the multitude of patents and academic articles regarding the synthesis of pretomanid [16–18], we have selected two interesting examples where we describe the divergent synthetic plans to produce the final molecule. Initial improvements for large-scale production of this compound have been proposed in the concise and convergent synthesis of PA-824 by Marsini et al. [19] in 2010.
The hallmark of their synthesis lies with the convergent synthetic design from fragments A and B, the employment of a safe imidazole derivative (8), and the use of the key chiral building block (7) derived from (R)-α-glycerol chlorohydrin (2). Briefly, the selection of a p-methoxybenzoyl group, introduced by reaction of (2) with acid chloride (3) in the first step, was key in the synthesis because of the stability and resistance to migration of this protecting group during the synthetic route. The group is removed in the final step from (9), allowing the oxazine ring construction to present in the final product by a basic treatment (Figure 2). Thus, according to synthetic Figure 2, the coupling of fragments A and B, in the form of products (7) and (8), afford the key precursor (9), which was efficiently converted into pretomanid (1) by basic treatment.
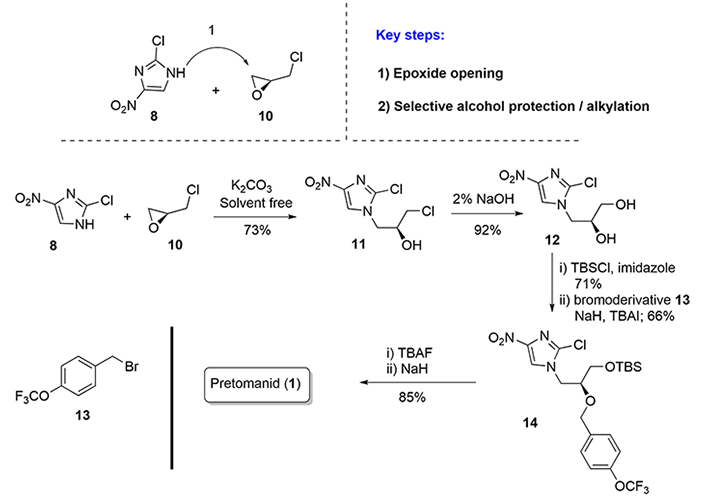
In 2020, an efficient and practical protocol for the production of pretomanid (1) was published by Chen et al. [20], featuring the employment of cheap and readily available raw materials, mild experimental conditions, and a one-pot procedure. Key steps in this route are the selective oxirane-ring opening of (S)-epichlorohydrin (10) by the imidazole derivative (8), and the selective protection of the primary alcohol of product (12) as a silyl ether versus the contiguous secondary alcohol. The potential of the route for scale-up in an industrial capacity via this synthetic route was demonstrated by the preparation of (1) in 50 g batches on a laboratory scale (Figure 3).
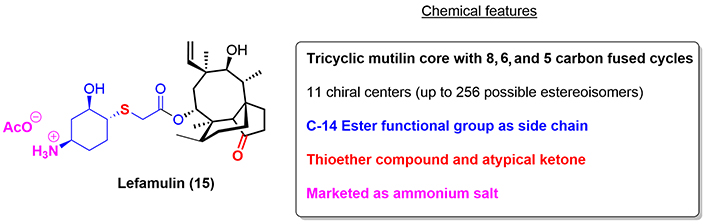
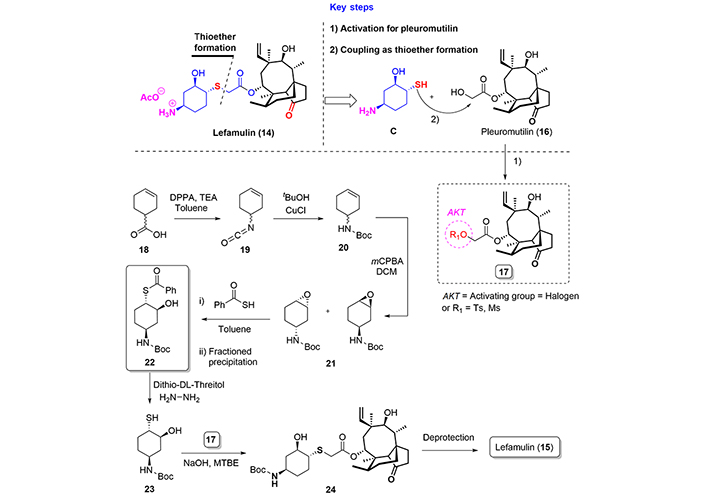
Lefamulin (15) is a novel semisynthetic pleuromutilin antibacterial agent and is the first antibiotic with a novel mechanism of action to obtain FDA approval for the treatment of pneumonia in nearly two decades [21]. The pleuromutilin class of antimicrobials was first discovered in the 1950s, but the first pleuromutilin was not brought to market until 1979 (tiamulin) for exclusive veterinary use. In 2006, lefamulin was synthesized and began undergoing testing for systemic human use (Figure 4) [22].
The mechanism of action of lefamulin is related to its interaction with the central part of domain V at the 23S ribosomal RNA (rRNA) through hydrophobic interactions, Van der Waal forces, and hydrogen bonds, with the side chain being the main driver for its biological activity, leading to inhibition of the biosynthesis of key bacterial proteins [23, 24].
In the literature, only seven patents have been published regarding the preparation of lefamulin [25, 26]. For this review, we have decided to include here one significative route developed by Riedl and co-workers [27].
The full process is based on a semisynthetic route where the side chain is attached to pleuromutilin (16), which is a naturally ocurring antibiotic produced by basidiomycetes Pleorotus mutilus and Pleorotus passeckerianus. After the preparation of pleuromutilin (16) for the introduction of the side chain, via activation of the hydroxyl group of the 2-hydroxy acetyl unit as tosylate, mesylate group, or even as a chloride, the coupling with the enantiomerically pure thiol (23) under basic conditions in methyl-tert-butyl ether, affords the protected lefamulin (24), which is finally transformed into lefamulin in a final deprotection step.
For the synthesis of stereoisomerically pure (23), Riedl’s group [27] starts from a racemic mixture of 3-hexenoic acid (18), which is transformed into isocyanate (19) by the employment of diphenylphosphorazide. Isocyanate (19) is then treated with Cooper(I) chloride in the presence of tert-butanol to obtain the protected amine (20). Epoxidation of compound (20), using m-chloroperbenzoic acid, furnished the epoxide (21) as a mixture of enantiomers, which, after a regioselective oxirane-ring opening reaction with thiobenzoic acid, followed by a selective precipation process, provided enantiomerically pure (22), which was finally transformed into the thiol (23) in good overall yield (Figure 5).
Cephalosporins, also known as carbapenems, are very well-known bactericidal β-lactam antibiotics [28]. They inhibit key bacterial enzymes (penicillin-binding proteins), including the transpeptidase enzyme, which is involved in the construction of the bacterial cell wall, and crucial for the structural strength and shape of the bacteria. Currently, there are five generations of cephalosporins.
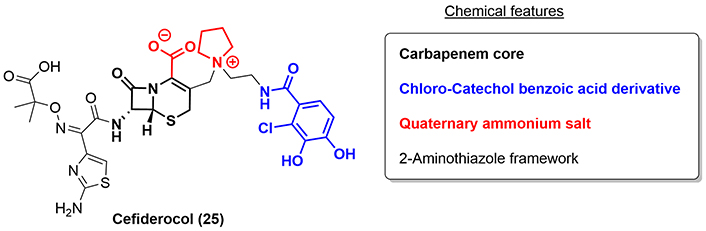
Cefiderocol (25) is an injectable siderophore cephalosporin discovered and developed by Shionogi & Co., Ltd., Japan. As with other β-lactam antibiotics, the main antibacterial-bactericidal activity of cefiderocol is due to its inhibition of the cell wall synthesis of Gram-negative bacteria by binding to penicillin-binding proteins [29]. In 2019, cefiderocol was approved by the FDA for the treatment of cUTI, including pyelonephritis, for HABP, and for VABP. Chemically, it is related to ceftazidime (41; see “Ceftazidime”) and cefepime. The main difference structurally for cefiderocol compared to ceftazidime and cefepime is that in the 3-position of the carbapenem core, a catechol framework is present in the side chain (2-chloro-3,4-dihydroxybenzoic acid moiety) and a covalently bound pyrrolidine ring, which forms a quaternary ammonium salt (Figure 6). According to recent investigations, this side chain confers increased periplasmic concentrations of the drug due to a chelation effect between the catechol moiety with ferric iron present in the outer regions of Gram-negative bacilli, commonly known as the siderophore effect [30].
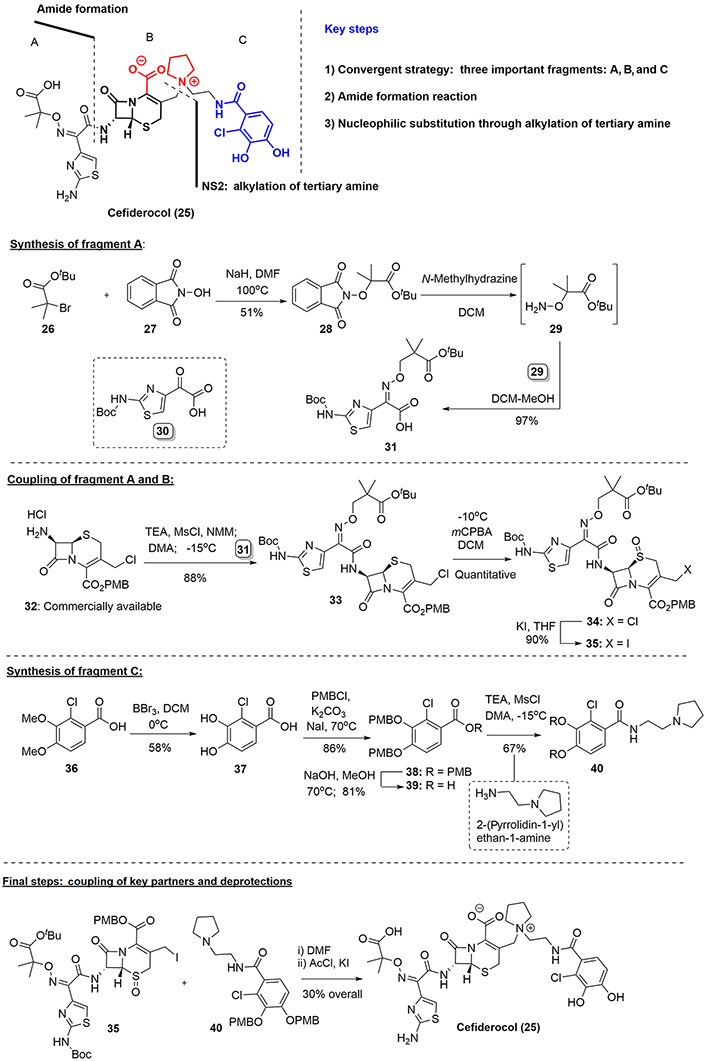
Among the seven patents found for the preparation of cefiderocol [31, 32], we include herein the specific synthetic route that includes the catechol framework in the cephalosporin antibacterials, registered by Shionogi & Co., Ltd. and led by Nishitani and co-workers [33]. The successful total synthesis of cefiderocol was based on the syntheses of the key moieties, fragments A, B, and C, followed by subsequent couplings to join them. As depicted in Figure 7, several steps were needed to build each scaffold independently. Thus, the acid (31) was obtained in a 49% overall yield after treatment of bromoester (26) with hydroxyphtalimide (27), which provided the nitrogen atom necessary for the subsequent imine formation. Here we point out the importance to obtain the imine compound (31) through coupling between compound (29) and ketoacid (30), which contains a free acid. On the other hand, the protected carbapenem (32), which can be readily purchased from commerical vendors was coupled with acid (31), via activation of the acid group with Mesyl (Ms) chloride in the presence of triethylamine, to furnish the amide (33) in excellent yield. Oxidation of the sulfur atom to the sulfoxide was then required to protect the sulfide group during the final coupling of compounds (35) and (40), as described later [34].
In parallel, the preparation of catechol compound (40) required a conventional strategy of protection-deprotection reactions starting from o-chloro-acid compound (36). Having accomplished the synthesis of both coupling partners, the final key step was the alkylation of tertiary amine (40) through a bimolecular substitution reaction of iodo-compound (35). The mixture of both partners in dimethylformamide and subsequent treatment of the resulting ammonium salt with acetyl chloride and potassium iodide afforded cefideracol (25) as a white powder in a 30% yield, over two final steps (Figure 7).
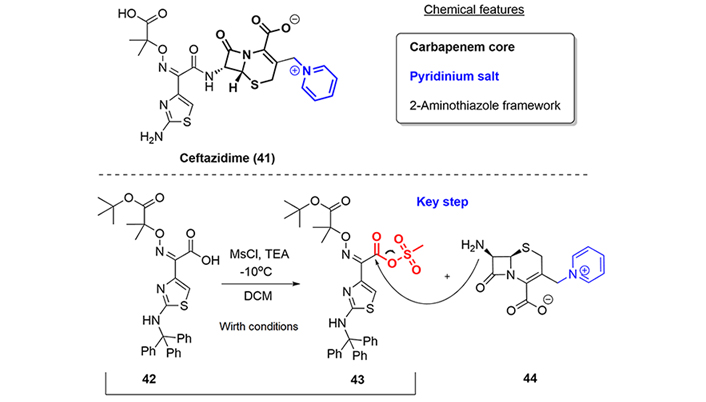
Ceftazidime (41) is a third-generation injectable cephalosporin antibiotic related to the previously described cefideracol (25). Since the only difference between the compounds is the pendant side chain at the 3-carbon, we will only highlight the clinical uses and the early solution provided by Wirth [35], working at Eli Lilly and Co. to prepare the amide function using carboxylic sulfonic mixed anhydrides, a reaction that, as we have already mentioned, was later employed in the syntheses of 4th- and 5th-generation cephalosporins.
Ceftazidime was approved by the FDA in 2015 for the treatment of cUTI, for HABP, and for cIAI, where it must be used in combination with avibactam (53, see “BLI class: tazobactam, avibactam, vaborbactam, and relebactam” section), which is a BLI (Figure 8).
As we have discussed, the key reaction in the ceftazidime synthesis was the improvement introduced by Wirth [35] in the amide formation, consisting of the activation of the acid group as a mixed anhydride. Thus, compound (43) was easily prepared by treatment of acid (42) with methanesulfonyl chloride in the presence of triethylamine. Compound (43) showed no degradation over the course of several h at 0℃ and could be used directly as a solution, without the removal of by-products, which was important from the scale-up point of view. Hence, Wirth’s conditions and later modifications have been applied to the syntheses of subsequent generations of cephalosporins (Figure 8) [35].
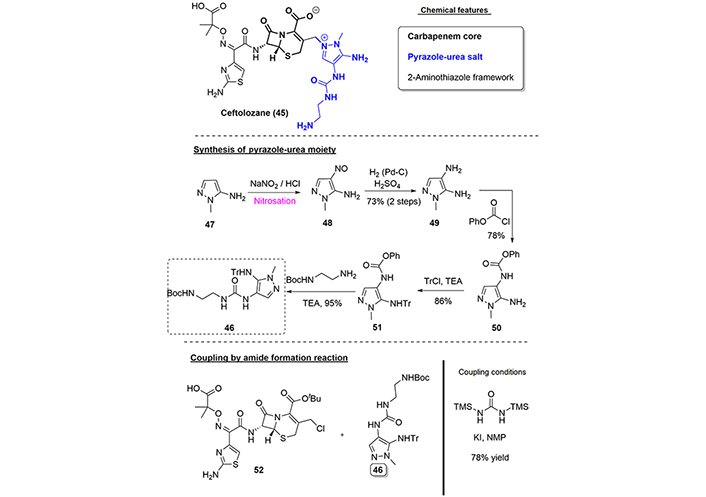
Ceftolozane (45) is a fifth-generation cephalosporin discovered by Astellas Pharma and Wakunaga Pharmaceuticals, which has improved efficacy against resistant microorganisms [36]. Under the commercial name Zerbaxa®, we find a combination of ceftolozane (45) and Tazobactam that is indicated to treat cIAI, cUTI, and HABP, which was approved in 2014 and 2015 by the FDA and EMA, respectively.
Since the key structural difference between ceftolozane (45) and the previously studied cephalosporins is the identity of the side chain in the 3-position of carbapenem ring, we will focus on the challenge to synthesize the pyrazole moiety.
For a full description of all published routes to obtain ceftolozane (45), we recommend the review written by Hughes [37] in 2017. We describe here the synthetic route toward the pyrazole scaffold (46). So, starting from 5-amino-1-methylpyrazole (47), the introduction of the amine group in the 4-position was achieved via a classical nitrosation reaction, followed by reduction of the nitrosocompound (48) to the bis amino compound (49). Subsequently, selective protection of the 4-amino versus the 5-amino group was accomplished by reaction with phenylchloroformate. Next protection of the 5-amino group as the trityl derivative (51) and followed by urea formation using tert-butyloxycarbonyl (Boc)-protected ethylenediamine furnished the desired pyrazole-urea (46) in a 47% overall yield (over 5 steps).
Interestingly, the final coupling of the fully functionalized carbapenem (52) and the pyrazole-urea (46) was achieved using 1,3-bis(trimethylsilyl)urea (BSU) and potassium iodide, providing the protected ceftalozane in 78% yield, in contrast to the previously discussed alternative used by Nishitani et al. [33] in the total synthesis of cefiderocol [through compounds (35) and (40), see Figure 7]; (Figure 9).
The overexpression of β-lactamase enzymes by resistant bacterial strains is one of the main mechanisms of resistance against the family of β-lactam antibiotics, resulting in the degradation of the β-lactam ring of the antibiotic [38]. To surmount this mechanism of resistance, clavulanic acid was developed and combined with the antibiotic as the first BLI in the late 1970s, followed by the launches of tazobactam as a BLI, among others, as described below [39].
In this section, we wish to describe and discuss the new BLIs introduced to the market, as represented by avibactam, vaborbactam, and relebactam.
Avibactam (53) is a BLI, which in combination with ceftazidime (41, Zavicefta®) has been approved by the EMA in 2015 for the treatment of cIAI, cUTI, and HABP [40].
From the structural point of view, avibactam (53) is comprised of a bicyclic urea core of 5- and 6-membered fused rings that contain an amide functional group and a sodium sulfate as pendants groups at the C-2 and N-6 positions, respectively (Figure 10).
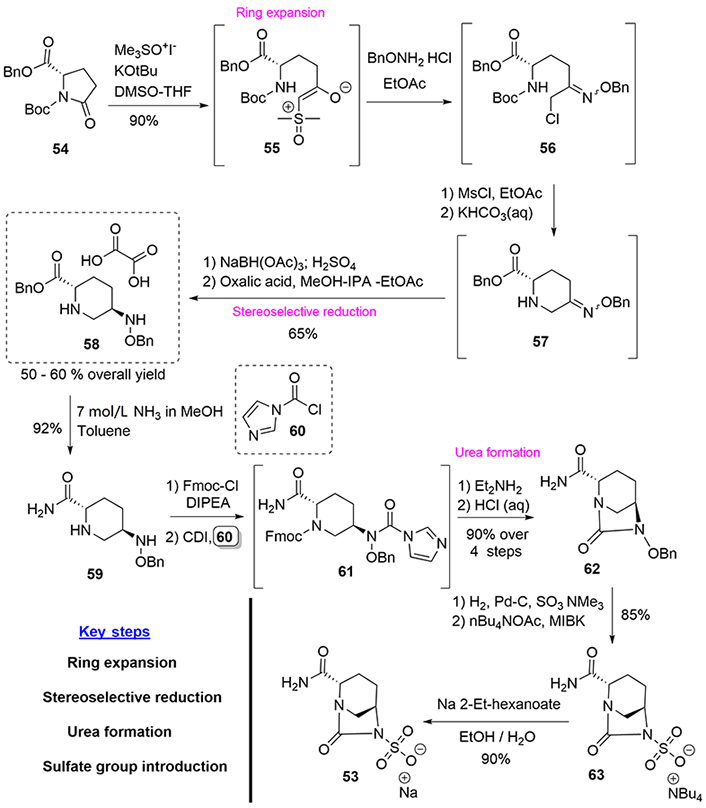
Syntheses of avibactam (53) have remained unchanged throughout its development history. We will discuss here the optimized route currently in practice (Figure 11) [40].
Starting from the glutamic acid derivative (54), the key intermediate (58) was obtained in good overall yield according to the synthetic sequence in which the resulting products (55–57) were not isolated, which is important from the industrial point of view, as it avoids the isolation of reactive intermediates. Opening of the pyrrolidine ring using trimethylsulfoxonium iodide in basic media was followed by oxime formation and displacement of the chlorine atom to yield intermediate (57), which was stereoselectively reduced by treatment with sodium triacetoxyborohydride. The product was then cleanly crystallized as an oxalate salt by using a ternary solvent mixture consisting of methanol isopropyl alcohol (IPA)-ethyl acetate to obtain the salt (58) [41].
Then, after a series of selective protection-deprotection reactions of (58) and the coupling of the resulting product (59) with the imidazole acid chloride (60), the introduction of the sulfate group in compound (62) was successfully achieved by the employment of a sulfur trioxide-trimethylamine complex. Finally, the cation exchange of the tetrabutylammonium counterion with sodium was achieved, employing sodium 2-ethylhexanoate (Figure 11).
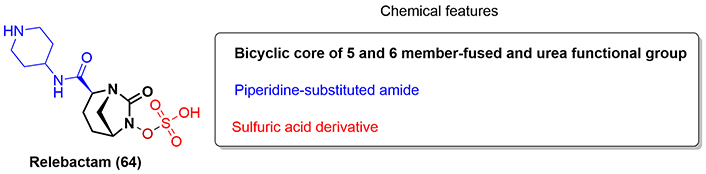
Relebactam (64) is a structural analogue of avibactam (53) approved in 2019 and used in combination with imipenem and cilastatin for the treatment of cUTI, cIAI, and HABP/VABP. The only difference between the antibiotics is that relebactam contains a 4-amino piperidine in the pendant amide group (Figure 12) [41].
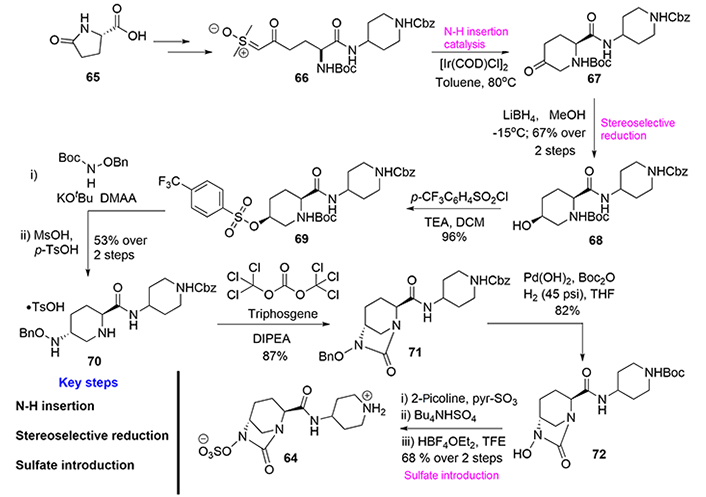
Despite the structural core of relebactam being identical to avibactam (53), we have selected here an early alternative route to obtain relebactam (64) that highlights the versatility of organic synthesis to achieve quite similar objectives (Figure 13) [42].
Thus, Mangion’s group [42] published in 2011 a concise and multi-kilogram scalable synthesis of relebactam to supply clinical trials. The synthesis utilizes the inexpensive and readily available starting material, L-pyroglutamic acid (65). The first steps are quite similar to those used in the avibactam synthesis, most notably the ring expansion via sulfoxonium ylide formation [43]. We highlight here, a novel iridium-catalyzed N-H insertion reaction to afford the chiral core 3-piperidinone moiety (67). Then, a diastereoselective ketone reduction followed by alcohol activation and displacement with a hydroxyl amine derivative furnished the compound (70) in good overall yield. Triphosgene-mediated urea formation provided the bicyclo[3.2.1]urea (71), which through functional group interconversion, yielded relebactam (64, Figure 13).
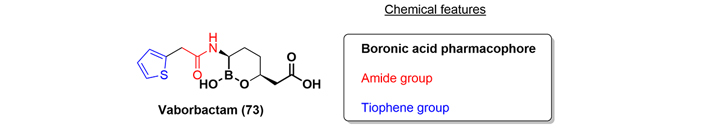
Vaborbactam (73) is a new BLI featuring acyclic boronic acid pharmacophore approved in 2017 to be used in combination with meropenem (a penicillin compound derivative), indicated for the treatment of cUTI and pyelonephritis [44].
Chemically, vaborbactam (73) is unique because it is the only antibiotic approved thus far in the last decade that contains a boronic acid as a pharmacophore. Furthermore, it contains an amide and a thiophene group (Figure 14).
The initial reports of aryl boronic acids as BLIs were in 1978 by Kiener and Waley [45]. Since then, only a few examples in the literature have been reported, with vaborbactam being of the biggest significance [46, 47].
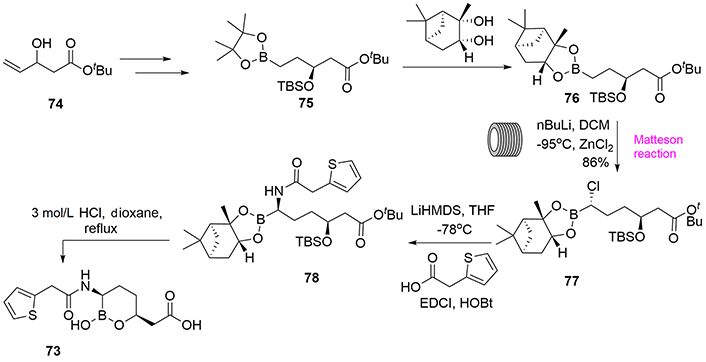
Among the syntheses reported of vaborbactam (73) [48, 49], we shall discuss here a recent development that highlights the importance of continuous flow chemistry for the production of pharmaceutical compounds (Figure 15) [50]. Specifically, the development of the Matteson reaction for full-scale production of the important intermediate (77) reported by Stueckler et al. [51] and Hughes [52] represents a key breakthrough in the field of flow chemistry. Therefore, starting from the racemic pentanoate (74), and after classical multi-step processes, boronic compound (75) was obtained as an enantiomerically pure compound. Transesterification using (+)-pinanediol afforded the precursor for the Matteson reaction [53], compound (76). The flow chemistry design facilitated the homologation reaction to provide the chloro compound (77), which can participate in a bimolecular substitution reaction with the corresponding amide in basic media to form the compound (78), which is finally converted to the final product (73) by acidic media and heating.
The tetracycline class of antibiotics was discovered almost a half-century ago and has been widely used for the treatment of bacterial infections. Structurally, they are characterized by a core of four fused six-membered rings, with one of them completely aromatic (D-ring). Biologically, the tetracyclines inhibit the elongation phase of protein synthesis by binding to the 30S ribosomal subunit of bacteria and blocking the attachment of the aminoacyl transfer RNA (tRNA) to the acceptor site in the messenger RNA (mRNA)-ribosome complex [54].
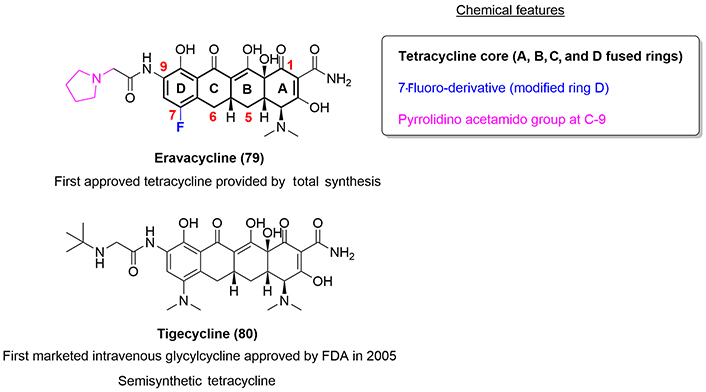
Eravacycline (79) is the first fully synthetic fluorocycline that has been approved by the FDA in 2018 for the treatment of cIAIs. Chemically, it is similar to the newly developed semisynthetic tetracycline, tigecycline (80) [55].
Structure-activity relationship (SAR) studies of the fluorocyclines have shown that more polar or basic substituents attached to the C-9 position, such as the pyrrolidine analogue in the case of eravacycline (79), result in improved antibacterial activity. Specifically, in case of eravacycline, which possesses a pyrrolidine moiety, exhibited 4–64-fold more potency versus other derivatives with substituents such as azetidine or piperidine (Figure 16) [55].
While almost all these complex antibiotics are prepared by semisynthesis, Charest et al. [56] published in 2005 a general, enantioselective, and convergent route to access a group of tetracyclines with the D-ring as a site of structural diversity. Taking advantage of the platform designed by Ronn and co-workers [55], working at Tetraphase Pharmaceuticals, achieved in 2013 the synthesis of a series of 7-fluoro-9-substituted tetracycline analogues, including the total synthesis of eravacycline (79).
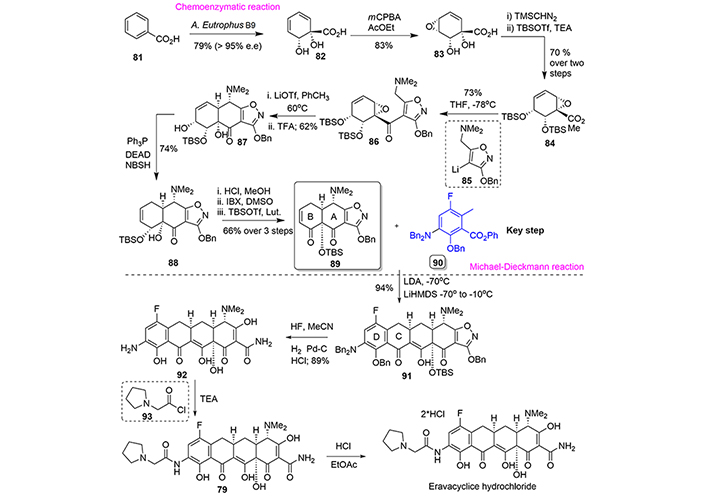
Taking advantage of the oxidative enzymes in the organism Alcaligenes eutrophus B9, the benzoic acid (81) was converted to the enantioenriched dihydroxy acid (82), which was regioselectively epoxidized using meta-chloroperbenzoic acid to provide compound (83). After esterification of (82), the resulting ester was treated with TBSOTf resulting in the isomerization of the vinylogous epoxy alcohol and silyl protection of the resulting diol to afford compound (84). In order to construct the A-ring, compound (84) was treated with the lithium derivative (85), and the resulting ketone (86) subjected to an intramolecular oxirane-ring opening by treatment with lithium triflate at 60℃ to obtain (87). The last steps were addressed to stabilize the α,β-unsaturated ketone in the B-ring and entailed isomerization of a double bond and further oxidation of corresponding alcohol to obtain the precursor of the Michael-Dieckmann reaction, compound (89).
Thus, a Michael-Dieckmann reaction [57] between the tricyclic compound (89) and fluoroderivative (90) was employed to form the C-ring of the tetracycline core. A key feature of this chemical transformation is the high degree of stereoselectivity exhibited, yielding the scaffold (91) in 94% of yield. Two additional steps of protecting group removal provided the fully deprotected compound (92), which was reacted with acid chloride (93) to form the amide and form eravacyclice (79). Finally, the free base was converted to its hydrochloride salt (Figure 17).
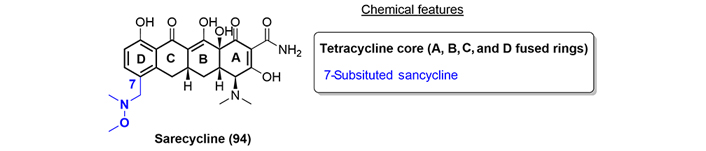
Sarecycline (94) is a new oral tetracycline-class antibiotic developed by Paratek and Allergan [58]. Structurally, sarecycline is a 7-substituted [[methoxy(methyl)amino]-methyl] tetracycline (7-substituted sancycline, Figure 18). It was approved by the FDA in 2018 for inflammatory lesions of non-nodular moderate to severe acne vulgaris. This improved activity may be due to higher lipophilicity at physiologically relevant pH, allowing for better penetration of the compound into the lipid-rich sebaceous follicular tissue [59].
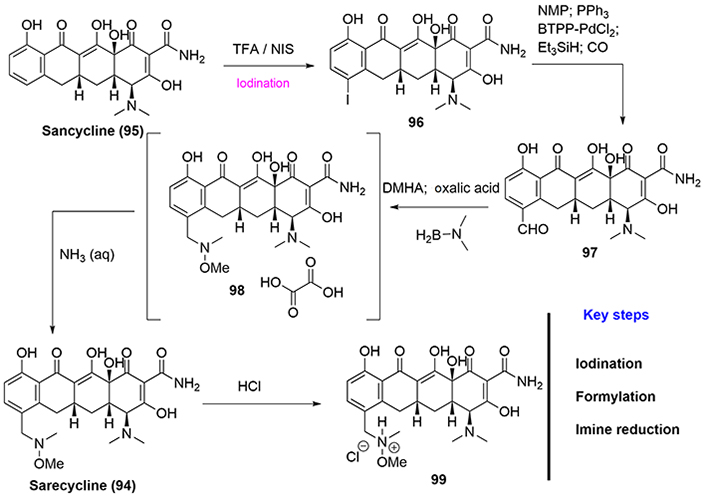
All reported methods to synthesize sarecycline (94) are intellectually protected by patents [60]. Among them, the most interesting method corresponds to the preparation of sarecycline via semisynthesis starting from sancycline (95) [59], which can be prepared by various routes, such as those utilizing 6-demethyltetracyclines by esterification with formic acid, followed by hydrogenolysis [60]. Thus, iodination of sancycline introduces an iodine atom in the C-7 position of the D-ring, which is accomplished by reaction with N-iodosuccinimide (NIS) in the presence of trifluoroacetic acid to obtain (96). Then, a palladium catalyzed formylation introduces the formyl group (compound 97) using bis-triphenylphosphinepalladium dichloride as the catalyst. Compound (97) is then subjected to a reductive amination employing dimethylhydroxylamine (DMHA) and dimethylaminoborane in the presence of oxalic acid to form the oxalate salt (98). The final steps are neutralization and salt formation using hydrochloride acid (Figure 19).
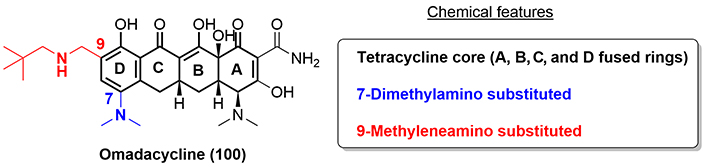
Omadacycline (100) was discovered in 2015 in a full study of the SARs of aminomethylcycline (AMC) by scientists working at Paratek Pharmaceuticals (Boston, USA) [61]. The compound was approved by the FDA in 2018 to treat CABP and ABSSSI (Figure 20).
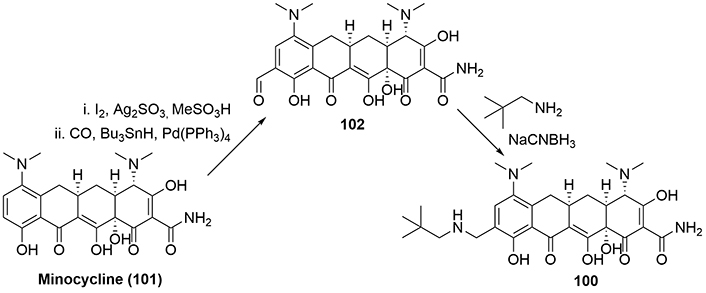
For the synthesis of omadacycline (100), the route for its preparation uses a semisynthesis, starting from minocycline (101), as it possesses the key structural framework required for activity. So, from (101), iodination in the of silver sulfate in acidic media [62] introduces an iodine atom in the D-ring, which is then converted to a formyl group utilizing carbon monoxide and palladium(0) catalysis [63] to afford (102), which participates in a conventional reductive amination reaction with isobutylamine to provide omadacycline (100, Figure 21).
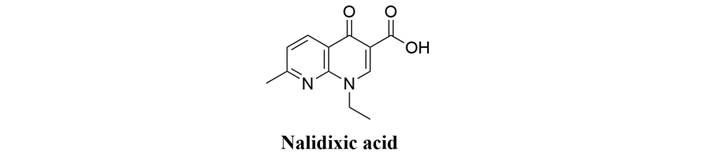
The quinolone class of compounds are antimicrobial molecules discovered in the 1960s, with the initial compound being nalidixic acid. Later introduction of the fluoroquinolones in the 1970s added to the armamentarium of clinically useful antimicrobials [64]. From a structural point of view, quinolones are molecules that contain an aromatic ring fused with a pyridinone ring (Figure 22).
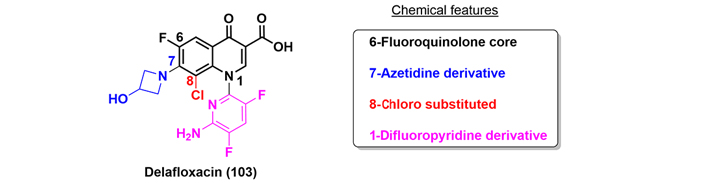
Delafloxacin (103) is a fluoroquinolone approved by the FDA in 2017 to treat ABSSSI and CABP. Delafloxacin has shown greater antibacterial efficicacy than other fluoroquinolones, but maintains the same inhibitory activity against bacterial DNA topoisomerase. The improved biological action is due to the appended groups introduced at the N-1, C-7, and C-8 positions. The azetidine substitution at C-7 results in a weak acid, resulting in an increase in its activity in an acidic medium. The chlorine in C-8 is an electron-withdrawing group, reducing the reactivity of the heterocycle and stabilizing the molecule, and finally, the difluoropyridine substitution at N-1 increases the molecular surface versus other quinolones (Figure 23) [65].
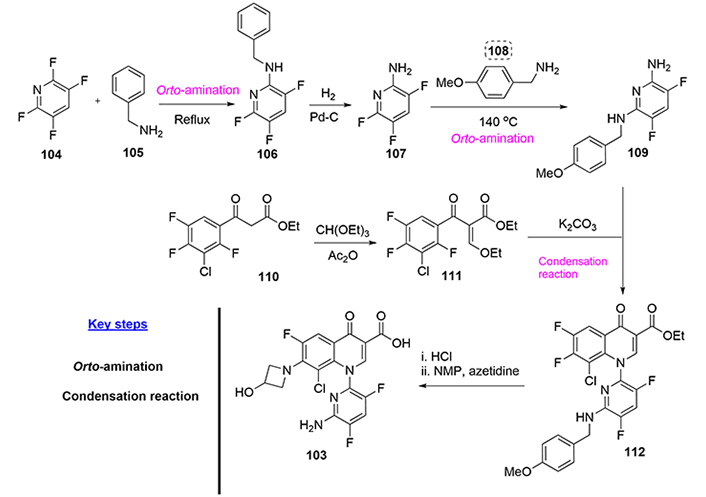
Delafloxacin, also known as ABT-492, was licensed by Abbott Labs. Scientists at Abbott have described two related ways to synthesize delafloxacin [66]. Herein we report the most interesting pathway, which starts with a reaction between tetrafluoropyridine (104) with benzylamine (105) at reflux (ortho-amination reaction is a key step). Then, compound (106) is debenzylated with hydrogen over Pd/C in methanol to yield the amine (107). Reaction of amine (107) with 4-methoxybenzylamine (108) in N-methylpyrrolidone at 140°C affords compound (109, another ortho-amination reaction is key for the introduction of nitrogen atoms in suitable positions), which is then coupled with 2-(3-chloro-2,4,5-trifluorobenzoyl)-3-ethoxyacrylic acid ethyl ester (111) to yield (112). Ester (111) is obtained by condensation of 2-(3-chloro-2,4,5-trifluorobenzoyl)acetic acid ethyl ester (110) with triethyl orthoformate and acetic anhydride. Final deprotection and attachment of the hydroxy azetidine sidechain provide delafloxacin (103, Figure 24).
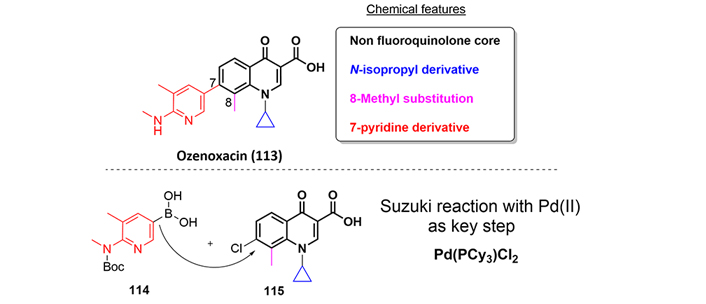
Ozenoxacin (113) is a quinolone approved by the FDA in 2017 for topical treatment of impetigo due to Staphylococcus aureus or Streptococcus pyogenes. Impetigo is one of the most common bacterial skin infections in children, particularly between the ages of 2–5 and can be classified as an ABSSSI primarily affecting the uppermost layers of the epidermis [67].
To date, there are not publications from academic labs regarding the synthesis of ozenoxacin, however, several patents covering its preparation are published [68]. One of them employs the Suzuki reaction [69] as the key step transformation in the cross-coupling of two fragments as depicted in Figure 25, the boronic acid (114) and advanced intermediate (115) [70].
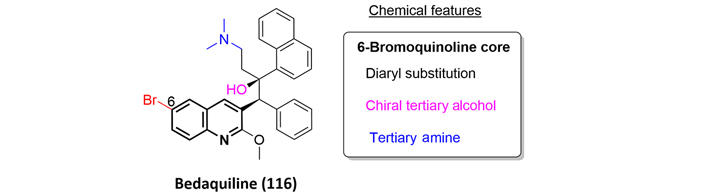
Bedaquiline (116) is a diarylquinoline that was approved in 2012 by the FDA as part of combination therapy for the treatment of pulmonary multi-drug resistant TB. It is important to note that bedaquiline is the first new drug identified to combat TB in forty years. From the structural point of view, bedaquiline possesses two contiguous chiral centers, one of them a quaternary center and containing a tertiary alcohol and tertiary amine (Figure 26). From the biological point of view, this drug exhibits a novel mechanism of action, specifically the inhibition of mycobacterial ATP synthase [71].
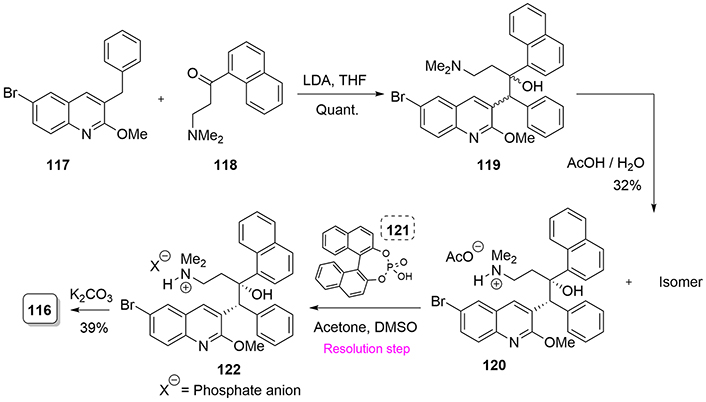
For a comprehensive review covering all reported syntheses of bedaquiline, we strongly recommend the excellent article written by Calvert and co-workers [71] in 2020, where bedaquiline and several derivatives are discussed. Nevertheless, we highlight the industrial synthesis of bedaquiline because of its low cost of preparation, and this is despite the fact that the route affords large amounts of undesired bedaquiline isomers. The synthesis begins with bromoquinoline (117), which is treated with lithium diisopropylamide to generate the corresponding carbanion, which then attacks the electrophilic ketone (118), providing quantitatively compound (119), but without any stereoselectivity. Further resolution of ammonium salt (122), obtained after treatment of (120) with phosphoric acid (121), allows the isolation of the isomer with the correct stereochemistry. Finally, basification employing potassium carbonate affords bedaquiline (116) a modest 39% yield (Figure 27).
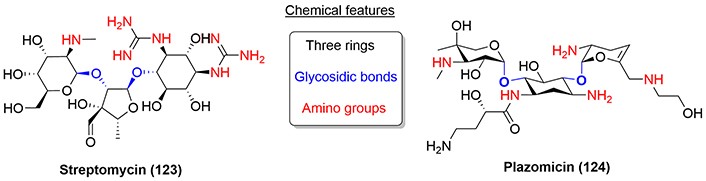
Aminoglycoside antibiotics were discovered in the 1940s and are still one the most commonly used antibiotics worldwide due to the combination of their high efficacy and low cost. Originally, they were a long-sought remedy for TB and other serious bacterial infections, for example, streptomycin (123), a drug that still nowadays is part of the regimen against multi-drug-resistant TB bacteria. From a chemical point of view, aminoglycosides usually contain three rings that are cyclitols, a saturated six-carbon ring, and five- or six-membered sugars that are linked via glycosidic bonds. A key feature of aminoglycosides is the presence of amino groups attached to the various rings of the structure (Figure 28).
From a biological point of view, aminoglycosides are highly efficacious against Gram-negative bacteria because of their ability to kill bacteria and not only prevent their growth [72].
Plazomicin is a semisynthetic aminoglycoside approved by the FDA in 2018 to treat cUTI. Chemically, it is derived from sisomicin (125), the dehydro analog of gentamicin C1a, which can be prepared by fermentation of marine Streptomyces sp. [73]. The primary intracellular site of action of the aminoglycosides is the 30S ribosomal subunit where they disrupt ribosomal protein synthesis [74].
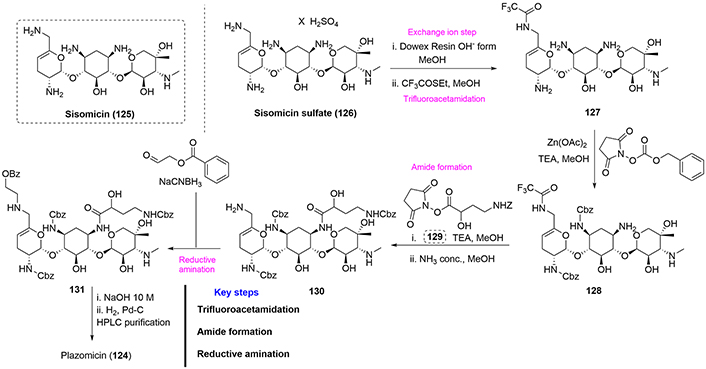
Plazomicin (124) has been prepared by semisynthesis. Sisomicin sulfate (126) was rendered basic by treatment with an ion exchange resin, followed by a reaction with ethyl trifluorothioacetate to selectively form the trifluoroacetamide (127). Treatment of compound (127) with Zn(II) acetate and benzyloxycarbonyl (Z) succinimide results in selective protection of both the 2’ and 3’ positions to form compound (128). N-1 acylation was achieved by reaction with the active ester (129) to yield the corresponding amide, which was deprotected using concentrated ammonia to afford compound (130). Reductive amination of (130), affords compound (131), and final protecting group removals provide plazomicin (124) in poor yield, taking into consideration that several steps required high performance liquid chromatography (HPLC) purification of intermediates (Figure 29) [75].
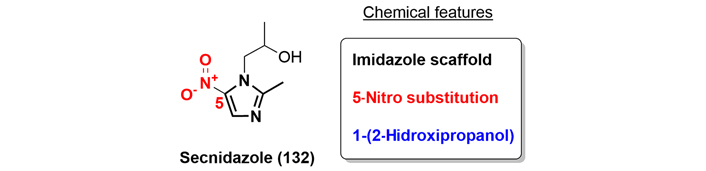
Secnidazole ([1-(2-hydroxypropyl)-2-methyl-5-nitroimidazole]), is an orally available antimicrobial drug, belonging to a group of next-generation agents of the 5-nitroimidazole class. Although secnidazole (132) has been known for over three decades, the FDA only recently approved it in 2017 to treat bacterial vaginosis in female patients and for the treatment of trichomoniasis (Figure 30) [76].
Antimicrobial and antiprotozoal activity of the 5-nitroimidazoles is primarily a function of the nitro group in the imidazole ring which is shared by all the agents. The mechanism of action of secnidazole is similar to other 5-nitroimidazoles, such as metronidazole, where the parent nitro group is reduced to cytotoxic metabolites, which leads to DNA helix damage, disruption of bacterial protein synthesis and replication, and ultimately, cell death [77].
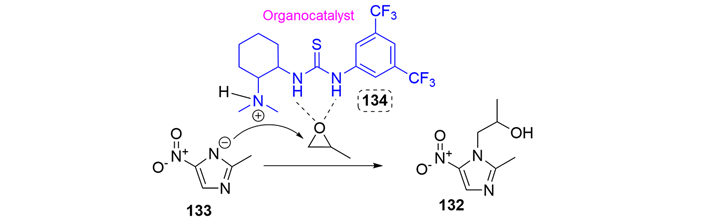
Secnidazole is usually prepared as described in the patent literature starting from 2-methyl-5-nitroimidazole, and an interesting organocatalytic reaction using the thiourea (134) to introduce the alcohol moiety in N-1 was patented by Hu and co-workers in 2020 [78]. The thiourea organocatalyst activates the non-substituted position in the epoxide to be attacked by the nucleophilic nitrogen (Figure 31).
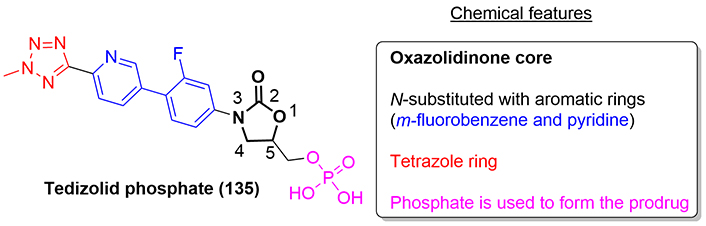
Tedizolid phosphate is commercialized under the name Sivextro® and was approved by the FDA in 2014 for the treatment of ABSSSI caused by susceptible Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA). This compound (135) is a prodrug, being rapidly converted to the active form tedizolid (136) in the presence of endogenous phosphatases. It inhibits bacterial protein synthesis by binding to the 23S rRNA of the 50S subunit of the ribosome, preventing the formation of the 70S ribosomal initiation complex (Figure 32) [79].
Chemically, tedizolid (136) corresponds to the second-generation of oxazolidinone antibiotics, which contains a chain of consecutive aromatic rings (m-fluorobenzene, pyridine, and methyltetrazole) attached to the nitrogen atom of the oxazolidinone core and a methyl dihydrogenphosphate group attached to carbon 5.
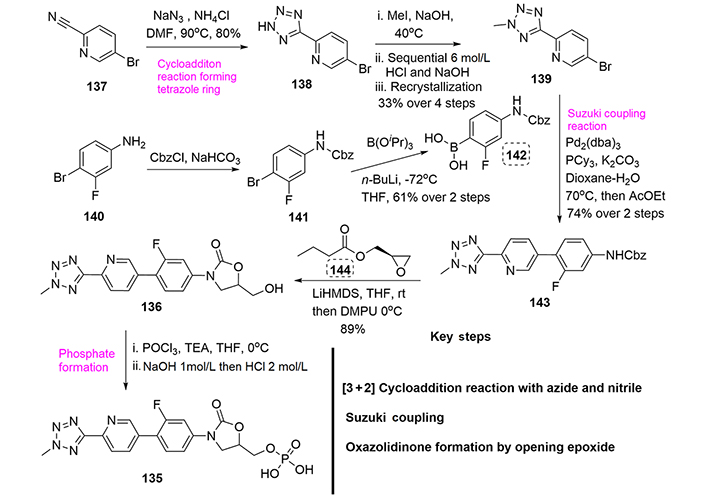
The main route for the preparation of tedizolid was patented by Trius Therapeutics and is depicted in Figure 33 [80]. This synthesis starts from bromocianopyridine (137), which after reaction with sodium azide in the presence of ammonium chloride at 90℃ yields the tetrazole derivative (138) in the form of the ammonium salt. This compound is then methylated using methyl iodide. Sequential treatment with 6 mol/L hydrochloric acid and sodium hydroxide to basic pH and final recrystallization in isopropyl acetate affords the compound (139). Then, again a Suzuki coupling reaction is required as a key step to connect the two advanced precursors, compound (139) and boronic acid (142). Deprotonation of the carbamate within (143) using the strong base lithium bis(trimethylsilyl)amide, followed by reaction with R-(–)-glycidyl butyrate (144) in the presence of DMPU, generates tedizolid (136) in 85% yield through an opening of the oxirane ring of (144) and subsequent cyclization of the ring-opened intermediate. Finally, a phosphate formation reaction using phosphorous(V) oxychloride at 0℃ to provides tedizolide phosphate (135) in a good yield of 76% (Figure 33).
Additionally, Im et al. [81] published in 2011 a similar synthesis of tedizolid, but using a coupling protocol with a tributyltin derivative and Pd(II) utilizing a classical Stille coupling reaction as the key step [82].
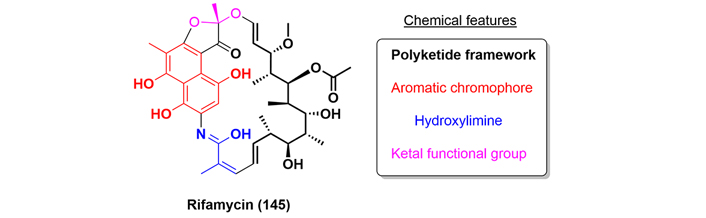
The rifamycin antibiotics are a subclass of the larger family of ansamycins. Rifamycins antibiotics are distinguished by a singular chemical structure, specifically, an aromatic chromophore with an ansa chain, which is a polyketide framework (Figure 34). In addition, rifamycins are featured by a specific antibacterial mode of action, which consists of selective blocking of the β-subunit of the DNA-dependent RNA polymerase (DDRP) enzyme, responsible for controlling the rebuilding process of bacterial peptides [83]. Rifamycin was discovered in the 1950s and received approval from the FDA in 2018 for the treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli.
All patents covering the preparation of rifamycin are based on biosynthesis, by fermentation of different microorganisms, such as Amycolatopsis mediterranei [84, 85] or Amycolatopsis kentuckyensis [86]. For complete synthetic studies of synthetic derivatives, we recommend the primary literature [83, 87, 88].
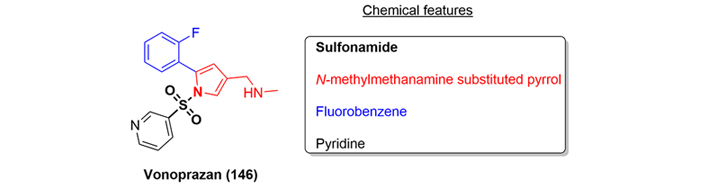
Vonoprazan fumarate (147) is a novel PCAB that was approved in Japan in 2015 and by the FDA in 2022 for the treatment and prevention of acid-related diseases as treatment of Helicobacter pylori infection [89]. Structurally, vonoprazan (146) is a small organic molecule containing a sulfonamide as a key pharmacophore, N-methylmethanamine-substituted pyrrole ring, pyridine, and fluorobenzene moieties (Figure 35).
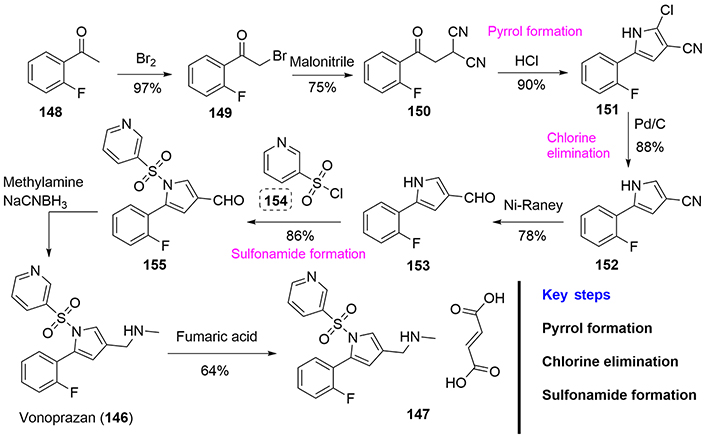
Within the literature one finds the academic publication covering the synthesis of vonoprazan by Yu et al. [89], and several preparations in the patent literature [90, 91]. Among the numerous patents regarding the synthesis of vonoprazan (146), we highlight herein one of the most interesting from the didactic point of view [92]. To this end, ortho-fluoroacetophenone (148) was brominated using bromine to form compound (149) in 97% yield. Subsequent treatment with malonitrile yields the dicyano-compound (150), which contains the desired number and type of required nitrogen atoms for pyrrole ring formation, which is achieved by reaction with hydrochloric acid in high yield. Dechlorination of (151) was achieved using Pd/C catalyst and hydrogen [93]. The cyano compound (152) was reduced using Ni-Raney to afford the aldehyde (153), which was sulfonylated with the corresponding sulfonyl chloride (154), to provide the aldehyde (155), which was used in a reductive amination reaction with methylamine to provide vonoprazan (146). The addition of fumaric acid renders the corresponding fumarate salt (147) as the final form of the drug (Figure 36).
According to the definition of glycopeptide antibiotics provided by the American National Cancer Institute (NCI), they are “originally isolated from plant and soil bacteria with structures containing either a glycosylated cyclic or polycyclic nonribosomal peptide” [94].
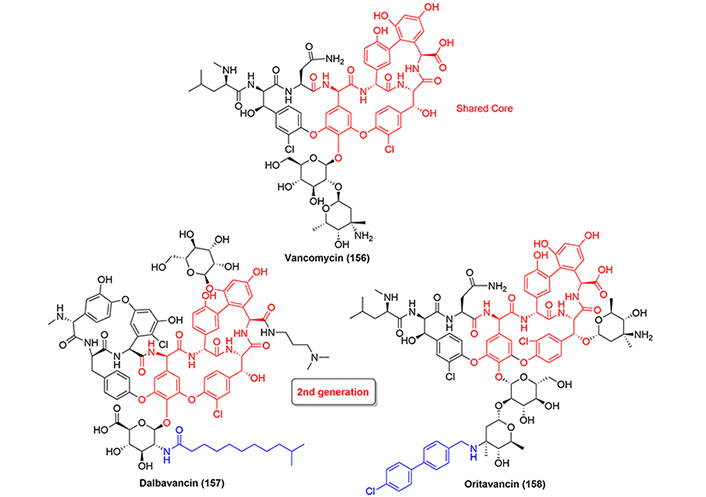
The first drug in this class of antibiotics was vancomycin (156), which represented a new age in the fight against bacterial infections. The antibiotic inhibits the cell wall biosynthesis by non-covalent bonds with the terminal carbohydrates of the peptidoglycans, in a process that ultimately inhibits cross-linking by the transpeptidase enzyme [95].
Both dalbavancin (157) and oritavancin (158) belong to the second-generation of glycopeptides and are semisynthetic derivatives of vancomycin, which have been approved by the FDA in 2014 and 2015 respectively for the treatment of ABSSSI caused or suspected to be caused by susceptible isolates of designated Gram-positive microorganisms (Figure 37). Because of the large amount of literature regarding both drugs and since these drugs are always prepared by biosynthesis and/or semisynthesis, we believe this class of antibiotics deserves special attention and we direct the reader to previously published literature [79, 96–99].
In the present review, we have covered all organic molecules that have been approved by different medicine agencies, mainly the FDA, to be used as antibiotics during the last decade (2012–2022). We have focused our analysis from the organic and synthetic points of view, highlighting the key organic reactions and synthetic methodologies which served as critical tools to successfully prepare the compounds and in some cases, the only way to provide industrial amounts of these drugs. This review not only can be useful for the scientific community to know all names and chemical structures of the most recently approved antibiotics but also to emphasize the importance of learning and knowledge of the most basic and advanced organic chemistry for these molecules.
It has been amply demonstrated that, from the beginning of the era of modern antibiotics, chemical synthesis has played a pivotal role in the discovery and development of new antibacterial agents.
Whereas for small molecule-type antibiotics, their total syntheses have made possible their access and production for the market, for larger molecules, particularly antibiotics such as ramiplanin, oritavaricin, or thiostrepton among others, total synthesis in large-scale production remains elusive, and represents a veritable challenge. In such cases, semisynthesis or fermentation processes provide a solution for cost-effective production at the industrial level. Despite few antibiotics, corresponding to this category, are currently manufactured by total synthesis, their synthesis is of great importance for the understanding of their mechanisms of action through the preparation of analogues and SAR studies. Thus, the knowledge derived from these SAR studies is allowing for the design of improved antibiotics, particularly effective against resistant bacterial strains, which represent a continuous and severe threat to the health of humankind.
We hope this review would assist professional physicians and students of the pharmaceutical sciences to study all methodologies that have been discussed herein with a critical view to point out their drawbacks and limitations. Additionally, we hope that contributing to this pedagogical work could boost the contributions of didactic articles explaining the chemical features and syntheses of small organic molecules which serve as drugs to treat other diseases.
ABSSSI: acute bacterial skin and skin structure infections
BLI: β-lactamase inhibitor
CABP: community-acquired bacterial pneumonia
“CI”: “critically important”
cIAIs: complicated intra-abdominal infections
cUTIs: complicated urinary tract infections
EMA: European Medicines Agency
FDA: Food and Drugs Administration
HABP: hospital acquired bacterial pneumonia
“HI”: “highly important”
“I”: “important”
SAR: structure-activity relationship
TB: tuberculosis
VABP: ventilator-associated bacterial pneumonia
WHO: World Health Organization
MGC: Conceptualization, Formal analysis, Data curation, Methodology, Investigation, Writing—original draft. FS: Writing—review & editing, Supervision. ADM and JMLR: Validation, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The corresponding author MGC is funded by I Plan Propio of University of Malaga (UMA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Jia Li Guo ... Ji Zhong Zhao
Maria G. Ciulla ... Kamal Kumar
Rozita Takjoo ... Norelle L. Daly
Anton Kolodnitsky ... Vladimir Poroikov
Atri Das ... Shantanabha Das
Galana Siro, Atanas Pipite
Maya G. Pillai, Helen Antony