 Original Article
Original Article

Affiliation:
1Department of Medical Biotechnology, Aarupadai Veedu Medical College and Hospital, Vinayaka Mission’s Research Foundation (Deemed to be University), Pondicherry 607402, Kirumampakkam, India
2Department of Pathology, Aarupadai Veedu Medical College and Hospital, Vinayaka Mission’s Research Foundation (Deemed to be University), Pondicherry 607402, Kirumampakkam, India
ORCID: https://orcid.org/0000-0003-4890-516X
Affiliation:
2Department of Pathology, Aarupadai Veedu Medical College and Hospital, Vinayaka Mission’s Research Foundation (Deemed to be University), Pondicherry 607402, Kirumampakkam, India
ORCID: https://orcid.org/0000-0002-6867-1746
Affiliation:
3Department of Medical Gastroenterology, Aarupadai Veedu Medical College and Hospital, Vinayaka Mission’s Research Foundation (Deemed to be University), Pondicherry 607402, Kirumampakkam, India
ORCID: https://orcid.org/0000-0002-4633-7739
Affiliation:
2Department of Pathology, Aarupadai Veedu Medical College and Hospital, Vinayaka Mission’s Research Foundation (Deemed to be University), Pondicherry 607402, Kirumampakkam, India
ORCID: https://orcid.org/0000-0003-3149-2557
Affiliation:
1Department of Medical Biotechnology, Aarupadai Veedu Medical College and Hospital, Vinayaka Mission’s Research Foundation (Deemed to be University), Pondicherry 607402, Kirumampakkam, India
Email: ravikumar.sambandam@avmc.edu.in
ORCID: https://orcid.org/0000-0001-7351-0421
Explor Immunol. 2026;6:1003252 DOI: https://doi.org/10.37349/ei.2026.1003252
Received: April 17, 2025 Accepted: March 19, 2026 Published: May 07, 2026
Academic Editor: Calogero Caruso, University of Palermo, Italy
Aim: Colorectal cancer (CRC) remains the second leading cause of cancer-related mortality worldwide. While antibody-drug conjugates (ADCs) offer targeted therapeutic options, they are often limited by toxicity, immunogenicity, complex pharmacokinetics, and high production costs. Polyclonal antibodies—capable of recognizing multiple epitopes—present a promising, yet underexplored, alternative for targeted drug delivery. The stage-specific presence of secreted, stable immunoglobulins (IGs) in CRC and their potential utility in drug conjugation strategies remain largely uncharacterized.
Methods: This study utilized electrospray ionization nano-liquid chromatography tandem mass spectrometry (ESI-nanoLC-MS/MS) proteomic analysis on pre-treatment plasma samples across CRC stages to identify stage-specific IGs. Venn diagram comparisons refined IG candidates, while protein stability was assessed using ProtParam. Molecular docking simulations (via CB-Dock2), epitope mapping (via CABS-dock), and cell-penetrating peptide (CPP) prediction were integrated to explore epitope pairing between IGs and Kirsten rat sarcoma viral oncogene homolog (K-Ras) neoantigen, evaluating their potential for polyclonal drug conjugates (pPDCs).
Results: A total of 325 secreted IGs were initially identified, with 46 found to be stage-specific. Protein stability analysis shortlisted 5 IGs—3 for early-stage and 2 for advanced-stage CRC. Molecular docking revealed that IG heavy variable 3-64 (IGHV3-64) exhibited high-affinity binding with Irinotecan [binding free energy (ΔG) = −10.0 kcal/mol] and showed epitope-level pairing with K-Ras at residues 2–17 and 106–114. Additional CPP motif analysis supported the potential of IGHV3-64-derived peptides for intracellular delivery, reinforcing their promise in pPDC development.
Conclusions: IGHV3-64 emerges as a strong candidate biomarker for advanced-stage CRC, demonstrating consistent binding affinity to Irinotecan and epitope pairing with K-Ras. Its inherent CPP features further support its potential for targeted, intracellular delivery in pPDCs design. These findings highlight a novel direction in personalized cancer immunotherapy, warranting further in vitro and in vivo validation to confirm clinical utility.
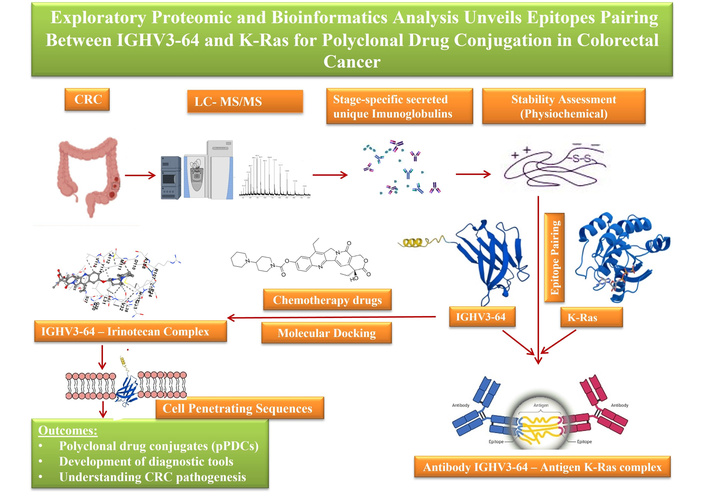
Colorectal cancer (CRC) remains a major global health burden, with both incidence and mortality rates projected to rise significantly. According to the GLOBOCAN report, new CRC cases are expected to increase from 1.88 million in 2020 to 2.94 million by 2040, while related deaths are anticipated to grow from 0.91 million to 1.55 million over the same period. These statistics underscore that CRC remains a leading non-communicable disease with high mortality, carrying a lifetime risk of approximately 5% [1]. Early-onset CRC (EOCRC) adds further complexity, as patients are often diagnosed at advanced stages, requiring aggressive treatments that impose heavy financial and healthcare burdens [2]. This highlights the urgent need for affordable early diagnostic strategies and more effective therapeutic interventions.
Immunotherapy-based approaches have emerged as promising tools in cancer treatment. Among them, monoclonal antibody-drug conjugates (ADCs) are gaining increasing attention in CRC management. However, clinical trials reveal several limitations, including payload-related toxicities, immunogenicity, unpredictable pharmacokinetics, limited efficacy at the maximum tolerated dose, off-target effects, and high treatment costs [1, 2]. Additionally, a recent report describing a patient with multiple myeloma who developed T cell lymphoma following chimeric antigen receptor (CAR)-T cell therapy [3] emphasizes the importance of exploring safer, naturally derived alternatives. This calls attention to polyclonal antibodies, which, due to their ability to bind multiple epitopes, may offer a more adaptable and less immunogenic platform for targeted drug delivery. Leveraging such naturally occurring immunoglobulins (IGs) could improve therapeutic outcomes, mitigate resistance, and enhance both survival and quality of life.
Despite their potential, the role of polyclonal antibodies or IGs in drug delivery remains underexplored. Specifically, there is limited knowledge about stage-specific secretion patterns of unique IGs, their stability, and how they interact with conventional chemotherapy agents or CRC-associated neoantigens. Moreover, most current biomarker research is centered on small molecules, often overlooking valuable IG-related data with translational potential.
The primary objective of this study is to identify stage-specific, unique IGs in CRC. The secondary goal is to evaluate their structural and functional stability using physicochemical parameters. Third, the study aims to identify high-affinity interactions between these IGs and FDA-approved CRC chemotherapies, highlighting their potential as precision tools for personalized diagnostics and therapeutics. The fourth objective is to determine whether these IGs contain antigen-specific epitopes capable of interacting with CRC-related neoantigens. Finally, the fifth aim is to investigate whether the IGs and Kirsten rat sarcoma viral oncogene homolog (K-Ras) harbor cell-penetrating peptide (CPP) sequences.
All methods were conducted in accordance with relevant guidelines and regulations, with full compliance with the Indian Council of Medical Research (ICMR) ethical guidelines. The study was approved by the Aarupadai Veedu Medical College and Hospital (AVMC) Institutional Human Ethics Committee (IHEC) under protocol No. AVMC-2021-04-14. The study was conducted in accordance with the principles of the Declaration of Helsinki (2024 revision). Written informed consent was obtained from all participants prior to sample collection, and all procedures adhered to established ethical standards.
Protein concentrations from twelve CRC plasma samples and three control plasma samples (Table 1) were quantified using a Nanodrop spectrophotometer (Digital Eppendorf D30 BioPhotometer) at A280 nm. An equal amount of 50 µg total protein from each sample was reserved for downstream proteomic analysis. Samples were appropriately diluted to achieve pre-quantification and normalization prior to tryptic digestion.
Baseline characteristics of colorectal cancer (CRC) patients and healthy controls included in the discovery-phase proteomics study.
| Group | N | Age, mean (range) | Sex (male/female) | Comorbidities/Medications | Ethnicity | Treatment status | Clinical tumor, nodes, and metastasis (cTNM), mean (SD), ranges | Pathological staging, ranges | Final anatomic stage, ranges |
|---|---|---|---|---|---|---|---|---|---|
| Stage I CRC | 1 | 70 (–) | 1/0 | No major comorbidities reported | Asian | Pre-treatment | cT1N0M0–cT4N2M1 | pT1bN0–pT4aN2b | I–IV |
| Stage II CRC | 4 | 67 (55–80) | 2/2 | Limited records; no major systemic illness | |||||
| Stage III CRC | 4 | 58 (52–67) | 4/0 | No comorbidities recorded | |||||
| Stage IV CRC | 3 | 46 (35–60) | 1/2 | Limited records | |||||
| All CRC patients | 12 | 59.6 (35–80) | 8/4 | Overall, no major systemic illness was reported | |||||
| Healthy controls | 3 | 66.7 (62–70) | 1/2 | No comorbidities |
Statistical comparison: age difference between CRC patients and controls was not significant (t-test, p = 0.075). Sex distribution did not differ significantly between groups (chi-square test, p = 0.41). Comorbidity information was limited; available records did not indicate major systemic illness or medications affecting metabolic/immune function.
Sample preparation and in-gel digestion were performed following the protocol described by Keeratichamroen et al. (2020) [4], with minor modifications.
Protein samples were denatured and briefly resolved by 12% SDS-PAGE to assess integrity and molecular weight distribution. SDS-PAGE was employed to achieve preliminary protein separation before in-gel digestion, facilitating simplification of complex samples and enabling excision of individual protein bands for targeted enzymatic digestion. Briefly, 10 μL of protein sample from each group (normal control, CRC stages I–IV) was loaded onto a 12% SDS-polyacrylamide gel and electrophoresed using a Bio-Rad electrophoresis system (Bio-Rad Inc.) at 100 mA for 45 min. Following electrophoresis, gels were stained with 0.1% Coomassie Brilliant Blue R-250 (Bio-Rad Inc.) prepared in 40% methanol and 10% acetic acid. Each gel lane was excised into five equal slices, which were subsequently destained using 0.1 M ammonium bicarbonate (NH4HCO3) in 50% acetonitrile (ACN).
Gel slices were completely dried using a SpeedVac evaporator (Labconco Corporation). Reduction was carried out with 50 μL of 0.1 M NH4HCO3 containing 10 mM dithiothreitol (DTT) and 1 mM EDTA, followed by incubation at 60°C for 45 min. Alkylation was performed using 100 mM iodoacetamide in 0.1 M NH4HCO3, incubated for 30 min at room temperature in the dark. The solution was then removed, and the gel slices were re-dried before enzymatic digestion. Proteolytic digestion was achieved by adding sequencing-grade trypsin (Promega Corporation) and incubating the samples overnight at 37°C. The digestion was quenched with 1% trifluoroacetic acid (TFA), and peptides were extracted using 5% formic acid in 50% ACN. All peptide extracts were pooled and concentrated to dryness using a SpeedVac. The resulting peptide mixtures were purified using C18 ZipTip® (EMD Millipore) and stored at −20°C until further analysis.
Following sample preparation, ESI-nanoLC-MS/MS was performed to profile plasma-derived peptides. A detailed overview of instrument parameters, gradient conditions, and acquisition settings is provided in the section below.
A nano ACQUITY UPLC System (Waters) chromatography system coupled to a quadrupole time-of-flight (Q-TOF) mass spectrometer (SYNAPT-G2, Waters) was used for the ESI-nanoLC-MS/MS analysis. All analyses were carried out in positive mode electrospray ionization with a NanoLockSpray™ source. Each sample was analyzed three times (three technical replicates). The MassLynx 4.1 software was used for the control and operation of both instruments. Three microliters of peptides were injected for the measurement. A trap column (Symmetry® 180 μm × 20 mm C18, 5 μm, Waters) was used to remove the salts from the sample. A 75 μm × 100 mm BEH C18 column (Waters), with a particle size of 1.7 μm, was employed for the peptide separation. The chromatography column was maintained at 40°C. The mobile phase-A was water, and the mobile phase-B was ACN; both contained 0.1% formic acid. Gradient elution of mobile phase-B (1–40%) was employed for 55.5 min. The column was washed with 80% mobile phase-B for 7.5 min. One percent mobile phase-B was used for the re-equilibration of the column.
Total run time: 60 min, flow 0.300 µL/min. Initial 0.00–42.00 min, 99.0% phase-A (water), 1.0% phase-B (ACN); 43.00–45.00 min, 60.0% phase-A, 40.0% phase-B; 46.00–50.00 min, 20.0% phase-A, 80.0% phase-B; 51.00–60.00 min, 99.0% phase-A, 1.0% phase-B.
The autosampler was maintained at 4°C throughout the analysis to preserve sample integrity, with an injection volume of 3 µL for each run. A constant flow rate of 300 nL/min was applied during chromatographic separation. Mass spectrometric analysis was conducted with online mass correction using leucine enkephalin (m/z 556.2766) as a reference compound in positive ion mode. The nano-electrospray ionization (nanoESI) source was operated with a capillary voltage of 3.4 kV, sample cone voltage of 40 V, and extraction cone voltage of 4 V. Ion mobility spectrometry (IMS) was performed using nitrogen gas at a flow rate of 90 mL/min, with a T-Wave™ pulse height of 40 V and a T-Wave™ velocity of 800 m/s. The IMS voltages were maintained at 8 V and 20 V. Data were acquired in continuum format using the elevated collision energy (MSE) acquisition mode with ion mobility. The mass spectrometer operated at a resolving power of 18,000 full width at half maximum (FWHM) under resolution mode. Data were collected with an acquisition time of 0.9 s per scan and an interscan delay of 0.024 s. Collision energies were dynamically ramped from 20 eV to 45 eV to achieve efficient peptide fragmentation and improve sequence identification.
Raw ESI-nanoLC-MS/MS spectra were processed using Progenesis QI for Proteomics v4.2 (Non-Linear Dynamics, Waters). Protein identification was carried out against the reviewed Homo sapiens entries in the UniProt database (https://www.uniprot.org/). Search parameters were set with a 1% false discovery rate (FDR) and allowed for one missed tryptic cleavage.
Minimum identification thresholds were defined as one fragment ion per peptide, three fragment ions per protein, and one unique peptide per protein. Oxidation of methionine was specified as a variable modification, while carbamidomethylation of cysteine residues was designated as a fixed modification.
Quantitative proteomic data were analyzed using one-way ANOVA integrated within Progenesis QI for Proteomics v4.2 to determine statistically significant differences between sample groups. A p-value < 0.05 was considered statistically significant for all analyses.
To identify proteins and peptides uniquely associated with specific CRC stages, Venn diagram analysis was performed to visualize and compare overlapping and stage-specific features. The Multiple List Comparator tool from Molbiotools (https://molbiotools.com/listcompare.php) was used to narrow down candidate lists and generate visual representations of stage-specific peptide distributions.
The ProtParam tool (https://web.expasy.org/protparam) [5] was employed to assess the physicochemical properties of unique IGs, especially their molecular stability. All 46 IGs identified through Venn diagram analysis were subjected to ProtParam screening to refine the selection. This systematic approach enabled the identification of the most stable IGs, highlighting promising candidates for future therapeutic development and biomarker discovery.
The three-dimensional structure of K-Ras was retrieved from the AlphaFold Protein Structure Database (EMBL-EBI, UK). Protein entries were filtered by organism (Homo sapiens) and annotation status to ensure the use of a validated, manually reviewed structure (Swiss-Prot). Based on these criteria, the final structure selected for downstream analyses was GTPase K-Ras (AlphaFold ID: AF-P01116-2-F1-v6). The structure was accessed from the AlphaFold database (https://alphafold.ebi.ac.uk/entry/AF-P01116-2-F1).
To evaluate the therapeutic potential of the unique IGs, blind molecular docking was performed using CB-Dock2 (https://cadd.labshare.cn/cb-dock2/php/blinddock.php) [6]. Structural data files (SDF) for six FDA-approved CRC drugs—Irinotecan (IRN), oxaliplatin (OXA), 5-fluorouracil (5-FU), lonsurf (trifluridine-tipiracil) (FTD-TPI), capecitabine (CAP), and leucovorin (LEU)—were downloaded from PubChem and used as ligands for docking. CB-Dock2 facilitated the structure-based evaluation of IG-drug interactions, while AutoDock Vina refined the binding conformations, offering insights into the strength and stability of IG-drug complexes.
Epitope prediction for both IG heavy variable 3-64 (IGHV3-64) and K-Ras was performed using the Immune Epitope Database (IEDB; https://www.iedb.org/) [7]. For modeling epitope-epitope interactions, we used CABS-dock (https://biocomp.chem.uw.edu.pl/CABSdock/) [8] under standard simulation settings, allowing flexible peptide-protein docking to simulate native-like interactions and assess the immunological and structural compatibility between IGHV3-64-derived epitope peptides and K-Ras neoantigenic epitopes. As four IGHV3-64 epitopes were identified, each was individually screened in separate docking simulations.
Molecular simulation setup:
K-Ras protein sequence(s):
MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHHYREQIKRVKDSEDVPMVLVGNKCDLPSRTVDTKQAQDLARSYGIPFIETSAKTRQRVEDAFYTLVREIRQYRLKKISKEEKTPGCVKIKKCIIM
IGHV3-64 peptide sequence: EVQLVESGEGLVQPGG
Simulation mc cycles: 50
Peptide secondary structure: CCEEEECCCCCCCCCC
As the objective is to find epitope-epitope pairing between K-Ras and IGHV3-64-derived epitope candidates (short peptides), intending to reveal naturally compatible or selective interactions that could be harnessed for precise delivery of polyclonal drug conjugates (pPDCs). We investigated the binding interactions between the K-Ras protein and the IGHV3-64 epitope, Monte Carlo (MC) simulations were performed. A total of 50 MC cycles were conducted to assess the structural stability and conformational dynamics of the complex.
To determine whether the unique IGs possess cell-penetrating capabilities, we analyzed their corresponding full-length protein sequences—sourced from UniProt—using the CellPPD server (http://crdd.osdd.net/raghava/cellppd/) [9]. This analysis identified putative CPP regions, indicating peptides with potential for enhanced intracellular delivery.
The identification of IGs specific to different CRC stages requires high-resolution analysis of human plasma samples. IGs are membrane-bound or secreted glycoproteins produced by B lymphocytes. During the recognition phase of humoral immunity, membrane-bound IGs function as antigen receptors. Upon antigen binding, they initiate clonal expansion and differentiation of B cells into plasma cells, which subsequently secrete IGs. These secreted IGs mediate the effector phase of humoral immunity by neutralizing and eliminating bound antigens. The variable (V) region of the IG heavy chain plays a key role in antigen recognition. Motivated by this functional specificity, we initiated a discovery-phase proteomics study to identify circulating, stage-specific IGs. A total of 12 pre-treatment plasma samples, representing clinical tumor, nodes, and metastasis (TNM) stages I–IV, were analyzed using high-resolution ESI-nanoLC-MS/MS. An overview of the experimental workflow is presented in Figure 1.
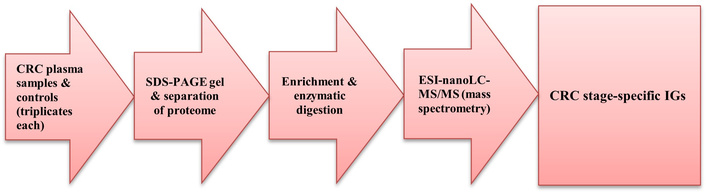
Proteomics ESI-nanoLC-MS/MS. CRC: colorectal cancer; ESI-nanoLC-MS/MS: electrospray ionization nano-liquid chromatography tandem mass spectrometry; IGs: immunoglobulins; SDS-PAGE: sodium dodecyl-sulfate polyacrylamide gel electrophoresis.
Mass spectrometry analysis identified a total of 138 IGs across the four CRC stages, with 64, 25, 25, and 24 IGs detected in stages I, II, III, and IV, respectively. To clarify the distribution of IGs across disease stages, Venn diagram analysis was performed to distinguish stage-specific IGs—defined as IGs uniquely detected in only one CRC stage—from overlapping IGs shared between two or more stages (Figure 2).
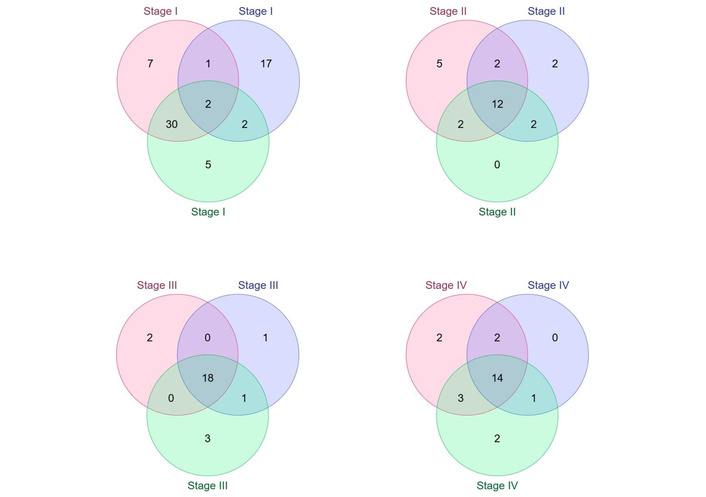
Venn diagram illustrating the distribution of immunoglobulins (IGs) identified across colorectal cancer (CRC) samples and stages. The Venn diagram summarizes IGs identified by electrospray ionization nano-liquid chromatography tandem mass spectrometry (ESI-nanoLC-MS/MS) in plasma samples from CRC stages I–IV. Each colored circle represents an individual biological sample (triplicate samples) analyzed within the same CRC stage; colors are used solely for visual distinction between replicate samples and do not indicate different disease stages or biological conditions. Overlapping regions denote IGs shared between two or more samples or stages, whereas non-overlapping regions indicate IGs uniquely detected in a single CRC stage. The numbers within each segment represent the total number of IGs identified in that category. Using this approach, 29 IGs were identified as unique to stage I, 7 to stage II, 6 to stage III, and 4 to stage IV. Stage-specific IGs were defined as those detected exclusively in one stage and absent in all others and were prioritized for downstream stability filtering and computational analyses.
Physicochemical stability analysis assesses a protein’s physical and chemical properties to evaluate its suitability for therapeutic or diagnostic use—key for ensuring efficacy, safety, and manufacturability. Parameters such as amino acid (AA) composition, molecular weight, isoelectric point (pI), and hydrophobicity were evaluated using ProtParam. All 46 stage-specific IGs were analyzed, and 5 were identified as stable—3 from early-stage CRC (ESCRC; 2 from stage I and 1 from stage II) and 2 from advanced-stage CRC (1 each from stages III and IV), as summarized in Table 2. These 5 IGs met the defined stability criteria, highlighting their potential for drug conjugation and further clinical development.
Stage-specific immunoglobulin (IG) proteins identified across CRC progression.
| Component | Proteins | ||||
|---|---|---|---|---|---|
| Early stages | Advanced stages | ||||
| Stage I | Stage II | Stage III | Stage IV | ||
| Sequence | MAWTPLFLFLLTCCPGSNSQTVVTQEPSLTVSPGGTVTLTCASSTGAVTSGYYPNWFQQKPGQAPRALIYSTSNKHSWTPARFSGSLLGGKAALTLSGVQPEDEAEYYCLLYYGGAQ | MAWAPLLLTLLSLLTGSLSQPVLTQPPSASASLGASVTLTCTLSSGYSNYKVDWYQQRPGKGPRFVMRVGTGGIVGSKGDGIPDRFSVLGSGLNRYLTIKNIQEEDESDYHCGADHGSGSNFV | MDLMCKKMKHLWFFLLLVAAPRWVLSQLQLQESGPGLVKPSETLSLTCTVSGGSISSSSYYWGWIRQPPGKGLEWIGSIYYSGSTYYNPSLKSRVTISVDTSKNQFSLKLSSVTAADTAVYYCAR | MMEFGLSWVFLVAIFKGVQCEVQLVESGEGLVQPGGSLRLSCAASGFTFSSYAMHWVRQAPGKGLEYVSAISSNGGSTYYADSVKGRFTISRDNSKNTLYLQMGSLRAEDMAVYYCAR | MEFGLSWLFLVAILKGVQCEVQLVESGGGLVQPGGSLRLSCAASGFTFSSYAMSWVRQAPGKGLEWVSAISGSGGSTYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAK |
| No. of AA | 117 | 123 | 125 | 118 | 117 |
| Formula | C560H847N141O171S5 | C575H903N157O180S4 | C634H979N159O181S6 | C573H880N152O171S8 | C561H868N148O169S6 |
| Molecular weight (mol.wt) | 12,451.03 | 13,023.68 | 13,917.06 | 12,890.69 | 12,582.31 |
| IIx/stability | 32.73/stable | 21.31/stable | 39.81/stable | 37.58/stable | 39.20/stable |
| Uniprot accession, protein & gene ID | P04211, IG lambda variable 7-43 (IGLV7-43) | A0A0B4J1Y8 (IGLV9-49) | P01824, IG heavy variable 4-39 (IGHV4-39) | A0A075B6Q5 ( IGHV3-64) | P01764 (IGHV3-23) |
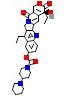
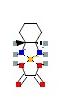
| Structure |  |  |  |  | 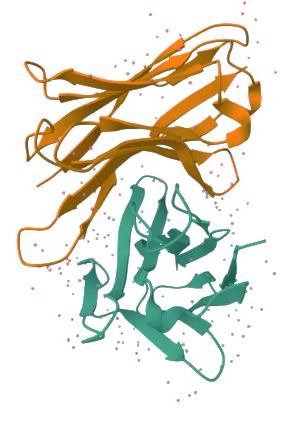 |
| Functions | -Prognostic biomarker in esophageal squamous cell carcinoma (ESCC)-Involved in immune tolerance and suppression | -Participates in antigen recognition-Triggers clonal expansion and differentiation of B lymphocytes; mediates effector phase of humoral immunity-Facilitates antigen-based affinity maturation | -Implicated in aggressive forms of chronic lymphocytic leukemia (CLL)-Strongly associated with Richter transformation-Exhibits polyreactive antigen binding; cooperates with NOTCH1 mutations and trisomy 12-Driver of disease aggressiveness and transformation | -Prognostic biomarker in head and neck squamous cell carcinoma (HNSCC)-Contributes to B cell malignancy pathogenesis [e.g., Waldenström’s macroglobulinemia (WM), IgM monoclonal gammopathy of undetermined significance (IgM-MGUS)] | -Interacts with defensin alpha 3 (DEFA3) via SNPs; involved in gene regulation in CLL-Serves as a scaffold for PD-1-blocking antibodies-Linked to site-specific genetic mutations in mucosa-associated lymphoid tissue (MALT) lymphoma-Defines a distinct subtype in splenic lymphomas-Highly frequent and mostly mutated; essential for B cell clone development in WM |
This table summarizes representative IG variable region proteins identified by electrospray ionization nano-liquid chromatography tandem mass spectrometry (ESI-nanoLC-MS/MS) in plasma samples from patients with early-stage (stages I–II) and advanced-stage (stages III–IV) CRC. For each IG, the amino acid (AA) sequence, length, molecular formula, mol.wt, instability index (IIx), UniProt accession number, protein and gene identifiers, and reported biological functions are shown. Stage-specific classification was based on Venn diagram analysis, where proteins were uniquely detected in a single CRC stage and absent in other stages. These IGs were subsequently prioritized for stability assessment and downstream computational analyses.
The 5 prioritized IGs underwent extensive molecular docking analysis against 6 FDA-approved CRC chemotherapies: IRN, OXA, 5-FU, FTD-TPI, CAP, and LEU. This analysis aimed to determine potential drug-specific binding affinities and ultimately identify promising pPDCs. These FDA-approved drugs form the backbone of standard chemotherapy regimens and are widely used across many countries, earning their place as core chemotherapy agents in the WHO Essential Medicines List (WHO-EML). They play critical roles in treatment protocols—either as monotherapy or in combination—depending on cancer stage, patient-specific factors, and therapeutic intent (curative vs. palliative) (Table 3).
Overview of FDA-approved chemotherapeutic agents used in colorectal cancer (CRC).
| Chemotherapies | Mechanisms and clinical use in CRC |
|---|---|
| Irinotecan (IRN) |
|
| Oxaliplatin (OXA) |
|
| 5-Fluorouracil (5-FU) |
|
| Leucovorin (LEU) (folinic acid) |
|
| Lonsurf (trifluridine-tipiracil) (FTD-TPI) |
|
| Capecitabine (CAP) |
|
This table summarizes commonly used FDA-approved chemotherapies for CRC, highlighting their primary mechanisms of action and established clinical roles. These agents form the backbone of standard treatment regimens across different disease stages and are frequently used alone or in combination to improve therapeutic efficacy, overcome resistance, and extend patient survival.
Binding free energy (ΔG) were calculated to evaluate interaction strength and specificity. Among the heavy chain variable regions (IGHVs), IGHV3-64 showed the strongest affinity for IRN (ΔG = –10.0 kcal/mol) (Figure 3), followed by IGHV3-23 (ΔG = –9.8 kcal/mol) and IGHV4-39 (ΔG = –9.6 kcal/mol).
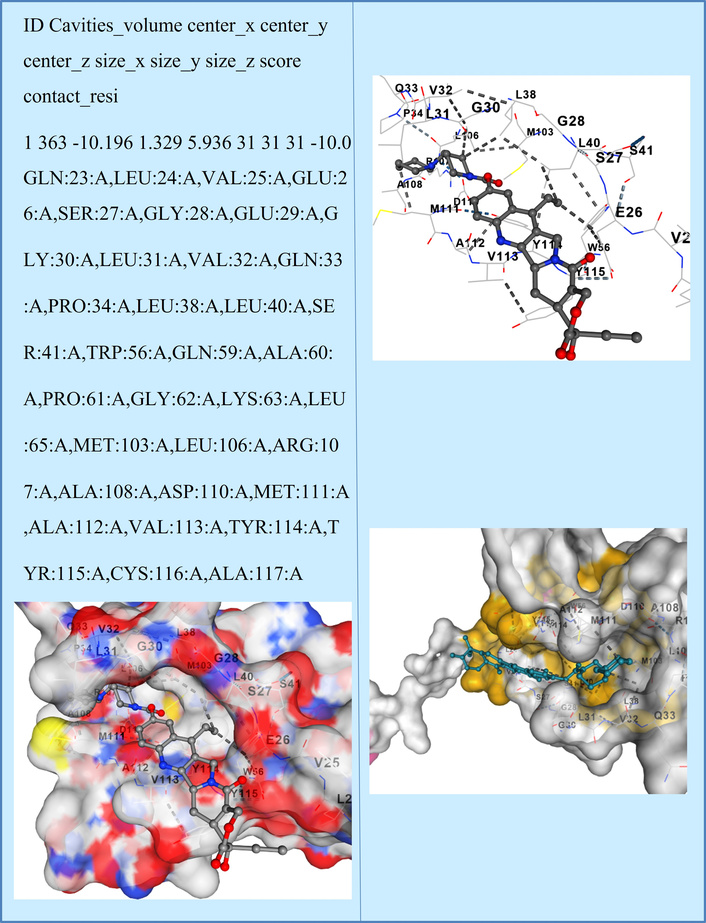
Molecular docking pose of immunoglobulin heavy variable 3-64 (IGHV3-64) with Irinotecan. The figure illustrates the predicted binding orientation of Irinotecan within the variable region of IGHV3-64, as determined by molecular docking analysis. The interaction occurs within a defined binding cavity and involves multiple residues at the IGHV3-64 variable domain interface, resulting in a predicted binding free energy of −10.0 kcal/mol. Residues contributing to ligand contact are listed in the table and reflect spatial compatibility for stable drug association. Docking results represent computational predictions and do not imply pharmacological inhibition or biological activity.
Similarly, light chain variable regions IG lambda variable 9-49 (IGLV9-49) and IGLV7-43 exhibited notable binding to IRN (ΔG = –9.3 and –8.8 kcal/mol, respectively), indicating their potential suitability for IRN-conjugated payload systems. A summary of the docking results is provided in Table 4.
Molecular docking analysis of shortlisted immunoglobulins (IGs) with FDA-approved colorectal cancer (CRC) chemotherapeutic agents.
| Molecular docking analysis IGs protein vs. chemotherapies | IRN | OXA | 5-FU | FTD-TPI | CAP | LEU |
|---|---|---|---|---|---|---|
 |  |  | 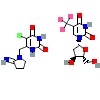 | 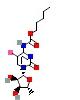 | 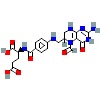 | |
| P04211 | –8.8 | –4.5 | –4.6 | –7.2 | –7.2 | –7.8 |
| A0A0B4J1Y8 | –9.3 | –4.4 | –4.8 | –8.1 | –8.0 | –8.4 |
| P01824 | –9.6 | –4.3 | –4.7 | –6.7 | –6.6 | –8.4 |
| A0A075B6Q5 | –10.0 | –4.0 | –4.7 | –6.6 | –6.7 | –7.5 |
| P01764 | –9.8 | –4.4 | –4.8 | –6.8 | –6.9 | –8.4 |
This table presents the predicted binding free energy (ΔG, kcal/mol) obtained from molecular docking simulations between five shortlisted IGs proteins and six commonly used CRC chemotherapies, including Irinotecan (IRN), oxaliplatin (OXA), 5-fluorouracil (5-FU), lonsurf (trifluridine-tipiracil) (FTD-TPI), capecitabine (CAP), and leucovorin (LEU). More negative ΔG values indicate stronger predicted binding affinity. Docking results were used to comparatively rank IGs for their feasibility as potential drug carrier scaffolds and do not imply pharmacological activity or therapeutic efficacy.
In contrast, all IGs showed significantly weaker interactions with OXA and 5-FU, with binding energies ranging from –4.0 to –4.8 kcal/mol, indicating a limited potential for stable complex formation with these agents. Moderate binding was observed with FTD-TPI and CAP, particularly by IGLV9-49 (ΔG = –8.1 and –8.0 kcal/mol, respectively). Notably, LEU showed consistent moderate binding across multiple IGs—IGHV3-23, IGHV4-39, and IGLV9-49—all with a ΔG = –8.4 kcal/mol. These findings support the hypothesis that IGs, especially IGHV3-64, IGHV3-23, and IGLV9-49, can serve as high-affinity carriers in pPDC strategies targeting CRC. The polyclonal nature of these IGs may provide broader epitope coverage and increased drug loading capacity, which are advantageous for enhancing therapeutic efficacy and specificity. The data further validate the rational selection of IG scaffolds based on binding energetics, laying the groundwork for future conjugation experiments and in vivo functional validation.
Based on the top binding scores, IGHV3-64 was selected for further analysis due to its strong binding affinity not only to IRN but also to several other CRC chemotherapeutic agents. This finding prompted us to investigate whether IGHV3-64 shares any molecular epitopes with known CRC oncogenes, and whether it contains any CPP sequences—two critical factors that could support its development as a pPDC candidate. To explore potential pathways for pPDC design and enable downstream functional analysis, we focused on K-Ras, one of the most frequently mutated oncogenes in CRC (Figure 4).
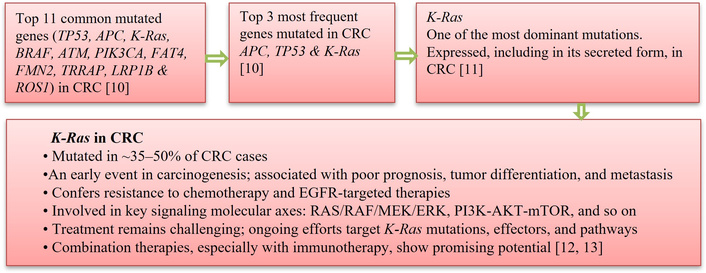
Finalization of Kirsten rat sarcoma viral oncogene homolog (K-Ras) oncogene selection and downstream computational analysis in colorectal cancer (CRC). This workflow outlines the rationale and stepwise process used to prioritize K-Ras as a key oncogenic target in CRC, based on its high mutation frequency, early involvement in carcinogenesis, association with poor prognosis, and resistance to standard chemotherapies and EGFR-targeted treatments. The workflow summarizes the integration of literature evidence, mutational prevalence data, and biological relevance to guide subsequent epitope mapping, molecular docking, and proximity-guided pPDC design.
K-Ras, a pivotal oncogene implicated in a range of malignancies such as colorectal, pancreatic, and non-small cell lung cancer, continues to be a notoriously difficult protein to drug due to its high structural flexibility and absence of deep, druggable pockets. Despite the approval of covalent G12C inhibitors, many K-Ras mutations remain untargetable, necessitating new therapeutic strategies [14–16]. Here, we explore a novel approach based on epitope-epitope molecular recognition, specifically between short peptide epitopes derived from the IGHV3-64 variable domain and functional epitope regions of K-Ras. These interactions are investigated as a platform to guide the rational design of peptide drug conjugates (PDCs) for intracellular precision delivery.
To identify relevant epitopes, we utilized the IEDB. The resulting epitopes are summarized in Table 4. Four selected epitopes from IGHV3-64 were then cross-analyzed against the full K-Ras protein sequence using the CABS-dock server for flexible protein–peptide docking. In this analysis, the K-Ras protein served as the target, using the AA sequence from IGHV3-64 (Table 5).
Predicted and curated epitope regions identified in IGHV3-64 and K-Ras.
| IEDB ID | Epitopes | Peptide sequence position | Antigen | Organism |
|---|---|---|---|---|
| IGHV3-64 | ||||
| 722846 | RQAPGKGLEY | 58–67 AA | IGHV 3-64 (UniProt: A0A075B6Q5) | Homo sapiens |
| 2260399 | EVQLVESGEG | 21–30 AA | ||
| 2260400 | EVQLVESGEGLVQPG | 21–35 AA | ||
| 2260401 | EVQLVESGEGLVQPGG | 21–36 AA | ||
| K-Ras | ||||
| 1275428 | SEDVPMVLV | 106–114 AA | GTPase K-Ras (UniProt: P01116) | Homo sapiens |
| 2226767 | TEYKLVVVGAGGV | 2–14 AA | ||
| 2268524 | KLVVVGAGGVGKS | 5–17 AA | ||
This table summarizes epitope sequences identified within IGHV3-64 and K-Ras proteins, including epitope IDs, peptide sequences, AA positions, antigen names, and source organisms. Epitope information was retrieved from the IEDB and used to support epitope-level pairing analysis between circulating immunoglobulin variable regions and oncogenic K-Ras. These epitopes were evaluated for spatial compatibility and proximity-guided targeting potential and do not imply direct inhibition of K-Ras signaling or immune activation. AA: amino acid; IEDB: Immune Epitope Database; IGHV3-64: immunoglobulin heavy variable 3-64; K-Ras: Kirsten rat sarcoma viral oncogene homolog.
Amongst the 10 models that were from the results, we opted for model 1, because model 1 is typically the most reliable as it’s the top-ranked by clustering and has the biggest cluster (suggesting most sampled structures converged) with better root mean square deviation (RMSD)/contact scores. The steps of formation and the epitope interaction were presented in Figure 5.
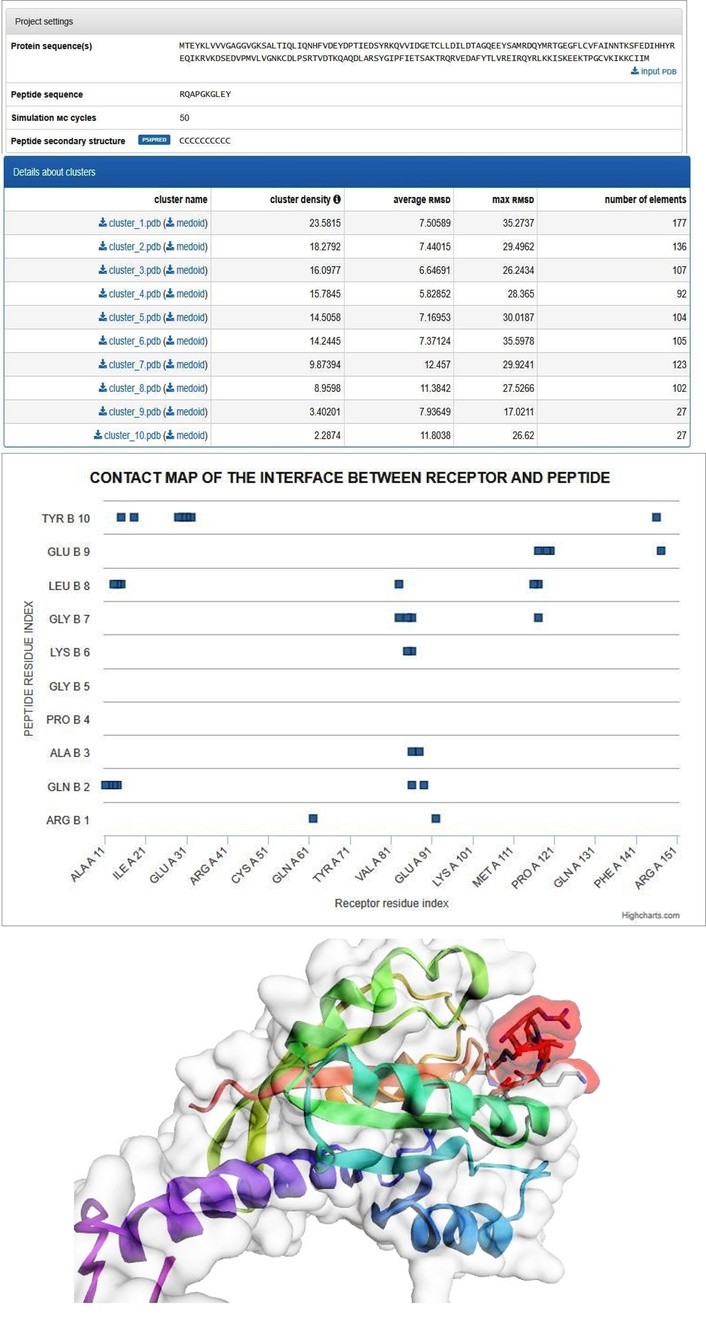
Epitope pairing and structural simulation of IGHV3-64 with K-Ras. The figure illustrates the computational epitope-pairing analysis between IGHV3-64-derived peptide regions and K-Ras, integrating epitope mapping and structure-based docking simulations. Predicted epitope regions on IGHV3-64 and K-Ras are shown along with their spatial alignment, highlighting proximity at functionally relevant K-Ras domains. Molecular simulation outputs demonstrate structural compatibility and feasible interaction interfaces rather than direct enzymatic inhibition. This analysis was used to assess proximity-guided targeting potential for polyclonal drug conjugate (pPDC) design. IGHV3-64: immunoglobulin heavy variable 3-64; K-Ras: Kirsten rat sarcoma viral oncogene homolog.
Using CABS-dock simulations, 4 candidate peptides from IGHV3-64 were modeled for their binding affinity and specificity to K-Ras across two functional epitope regions: the N-terminal segment (residues 2–17), encompassing the P-loop and GTP-binding motif, and the C-terminal segment (residues 106–114), part of the switch II region critical for effector interactions. The peptide RQAPGKGLEY demonstrated exclusive binding within the N-terminal epitope, establishing contacts with residues Ala11 to Gly15 (Figure 5). Notably, this region aligns with the highly dynamic GTP-binding site, suggesting a natural conformational compatibility between this IGHV3-64-derived epitope and a critical functional domain of K-Ras. The ability of this short peptide epitope to interact so selectively suggests potential molecular mimicry or immunological memory-driven recognition between the IG domain and the Ras protein—an insight that could be leveraged for epitope-guided targeting. Likewise, EVQLVESGEG, another peptide of similar origin, consistently bound to the same N-terminal region, suggesting a recurring binding motif or “hotspot” preference. In contrast, EVQLVESGEGLVQPG exhibited dual-site engagement, bridging both the N-terminal and C-terminal epitopes. This dual epitope pairing implies a capacity for bivalent binding, which could increase payload stability and intracellular retention when used in drug conjugation strategies. Finally, the extended peptide EVQLVESGEGLVQPGG localized exclusively to the C-terminal epitope (106–114), forming strong interactions with residues Glu107, Asp108, Val109, and Met111—components of the switch II domain involved in effector modulation.
Following epitope pairing analysis, we investigated the presence of CPP sequences within IGHV3-64 and K-Ras to evaluate their theoretical potential for intracellular delivery. Identification of CPPs within CRC-associated proteins provides a bioinformatic strategy to assess whether endogenous protein fragments possess physicochemical properties compatible with membrane translocation.
CPP prediction was performed using the CellPPD server, which applies a support vector machine (SVM)-based classification model. In accordance with the tool’s recommendations, peptides with SVM scores greater than 0.0 were considered CPP-positive, while negative scores indicated non-CPP sequences. This threshold was applied uniformly across all analyzed peptides and is now explicitly reported to improve methodological transparency. CPP profiling revealed that both IGHV3-64 and K-Ras harbor multiple peptide segments predicted to possess cell-penetrating properties (Table 6). These peptides demonstrated moderate-to-high SVM scores, balanced hydrophobic-hydrophilic profiles, and compact structural features—attributes commonly associated with membrane interaction and intracellular access. Within IGHV3-64, two CPP-positive peptides were identified: MHWVRQAPGK (AA 54–63) and KGRFTISRDN (AA 85–94). These peptides exhibit relatively low molecular weights (~1.2 kDa), basic pIs (~pI 11), and modest amphipathicity, suggesting theoretical compatibility with membrane interaction and endosomal escape under acidic intracellular conditions. Their physicochemical profiles also indicate solubility and chemical accessibility suitable for conjugation with small-molecule payloads such as IRN or OXA. K-Ras contained multiple overlapping CPP-positive regions, particularly within its C-terminal sequence. The peptide RQYRLKKISK (AA 164–173) exhibited the highest SVM score (0.68), along with strong amphipathicity (1.72), a high net positive charge (+5), and a basic pI (11.17). These characteristics are consistent with electrostatic interactions with negatively charged cancer cell membranes. Additional CPP-positive peptides, including REIRQYRLKK and VREIRQYRLK, displayed similar cationic and amphipathic profiles, further supporting their predicted membrane-interactive capacity.
Cell-penetrating peptide (CPP) prediction from IGHV3-64 and K-Ras protein sequences.
| Category | SVM score | Prediction | Hydrophobicity | Steric hindrance | Side bulk | Hydropathicity | Amphipathicity | Hydrophilicity | Net hydrogen | Charge | pI | Mol wt |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IGHV3-64 | ||||||||||||
| MHWVRQAPGK (CPP starts @ 54 AA) | 0.11 | CPP | –0.24 | 0.56 | 0.56 | –1.01 | 0.88 | –0.10 | 1.00 | 2.50 | 11.01 | 1209.58 |
| KGRFTISRDN (CPP @ 85 AA) | 0.00 | CPP | –0.49 | 0.67 | 0.67 | –1.45 | 0.86 | 0.78 | 1.50 | 2.00 | 10.84 | 1193.46 |
| K-Ras | ||||||||||||
| HYREQIKRVK (CPP @ 95–104 AA) | 0.09 | CPP | –0.61 | 0.62 | 0.62 | –1.96 | 1.62 | 0.91 | 1.70 | 3.50 | 10.29 | 1356.74 |
| VREIRQYRLK (CPP @ 160 AA) | 0.15 | CPP | –0.59 | 0.67 | 0.67 | –1.32 | 1.35 | 0.78 | 1.80 | 3.00 | 10.91 | 1360.77 |
| REIRQYRLKK (CPP @ 161 AA) | 0.52 | CPP | –0.75 | 0.67 | 0.67 | –2.13 | 1.72 | 1.23 | 2.00 | 4.00 | 11.01 | 1389.81 |
| EIRQYRLKKI (CPP @ 162 AA) | 0.32 | CPP | –0.50 | 0.67 | 0.67 | –1.23 | 1.48 | 0.75 | 1.60 | 3.00 | 10.29 | 1346.79 |
| IRQYRLKKIS (CPP @ 163 AA) | 0.04 | CPP | –0.47 | 0.66 | 0.66 | –0.96 | 1.35 | 0.48 | 1.60 | 4.00 | 11.10 | 1304.75 |
| RQYRLKKISK (CPP @ 164 AA) | 0.68 | CPP | –0.65 | 0.65 | 0.65 | –1.80 | 1.72 | 0.96 | 1.80 | 5.00 | 11.17 | 1319.76 |
| RLKKISKEEK (CPP @ 167 AA) | 0.12 | CPP | –0.64 | 0.65 | 0.65 | –1.96 | 1.97 | 1.77 | 1.50 | 3.00 | 10.01 | 1258.67 |
| KKISKEEKTP (CPP @ 168 AA) | 0.11 | CPP | –0.54 | 0.62 | 0.62 | –2.12 | 1.72 | 1.61 | 1.20 | 2.00 | 9.55 | 1187.54 |
| KTPGCVKIKK (CPP @ 176 AA) | 0.21 | CPP | –0.32 | 0.63 | 0.63 | –0.71 | 1.47 | 0.73 | 0.90 | 4.00 | 10.05 | 1101.56 |
This table summarizes peptide fragments scanned from IGHV3-64 and K-Ras using the CellPPD prediction server. For each peptide, the AA sequence, positional range, support vector machine (SVM) score, and physicochemical properties are reported. Peptides with SVM scores > 0.0 were classified as CPP-positive according to the tool’s recommended threshold. These predictions were used to assess the theoretical feasibility of intracellular delivery and do not represent experimental confirmation of cell penetration. AA: amino acid; pI: isoelectric point; IGHV3-64: immunoglobulin heavy variable 3-64; K-Ras: Kirsten rat sarcoma viral oncogene homolog.
From a pPDC design perspective, the presence of CPP-positive regions in both IGHV3-64 and K-Ras supports the theoretical feasibility of constructing conjugates capable of intracellular access without reliance on synthetic delivery vectors. However, it is important to emphasize that CPP prediction represents a computational feasibility assessment, not experimental validation. Comparative cytotoxicity assays (e.g., free IRN vs. IGHV3-64-conjugated formulations), cellular uptake studies, and intracellular localization analyses will be essential in future work to confirm functional delivery.
This study introduces a novel multi-omics framework for identifying circulating, stage-specific IGs in CRC, with the ultimate aim of enabling precision-guided pPDC design. High-resolution ESI-nanoLC-MS/MS plasma proteomics across TNM stages I–IV identified 138 IGs, of which 46 were uniquely stage-specific. Through physicochemical filtering based on molecular weight, pI, and stability indices, five stable IGs representative of ESCRC and advanced-stage CRC (ASCRC) were prioritized for downstream analyses. The stage-specific enrichment of circulating IGs is unlikely to be stochastic and instead reflects dynamic immune remodeling within the CRC TME during disease progression. CRC evolution is accompanied by marked shifts in immune cell composition, cytokine gradients, and antigenic burden. While ESCRC is often associated with effective immune surveillance, ASCRC is characterized by a chronically antigen-exposed yet immunosuppressive TME, marked by sustained inflammation, immune exhaustion, and altered lymphocyte functionality.
Within this evolving TME, B cells and humoral immune responses play increasingly recognized and context-dependent roles. ASCRC has been associated with expanded B cell infiltration, tertiary lymphoid structure formation, and clonal expansion of antibody-producing plasma cells. Persistent exposure to tumor-associated antigens and neoantigens—particularly mutant K-Ras—can drive selective B cell activation and biased IGHV gene usage, resulting in repertoire skewing toward specific IGHV families. Accordingly, the detection of stage-specific circulating IGs, including IGHV3-64 in advanced CRC, likely reflects antigen-driven humoral immune adaptation rather than a passive by-product of tumor burden.
From a translational perspective, this stage-associated IG pattern also suggests potential diagnostic and stratification value. Although receiver operating characteristic (ROC) and area-under-the-curve (AUC) analyses were not performed due to limited cohort size, the consistent enrichment of IGHV3-64 in ASCRC supports its candidacy for future validation as a circulating biomarker to distinguish early vs. advanced disease in larger, independent cohorts.
The concept of employing endogenous IGs as precision-targeting agents aligns with the established clinical success of ADCs in solid tumors, including HER2-positive malignancies [17]. Our findings extend this paradigm by identifying tumor stage-associated circulating IGs shaped by the CRC immune microenvironment and proposing their repurposing as endogenous scaffolds for pPDC development—potentially offering a personalized and immunologically compatible alternative to monoclonal ADCs, particularly in heterogeneous and immune-evasive advanced CRC.
Molecular docking analysis revealed that several prioritized IGs—particularly IGHV3-64, IGHV3-23, and IGLV9-49—exhibited selective binding affinity toward IRN. Among these, IGHV3-64 demonstrated the strongest predicted interaction (ΔG = −10.0 kcal/mol), highlighting its feasibility as a drug carrier framework. This observation is consistent with prior studies demonstrating that antibody-based delivery systems can enhance the bioavailability and therapeutic efficacy of chemotherapeutic agents in CRC xenograft models [18]. The preferential association of IGHV3-64 with IRN, relative to other shortlisted IGs, likely reflects distinct structural and physicochemical features of its variable region rather than a generic property of IGs. Specifically, favorable charge distribution, hydrophobic pocket architecture, and conformational flexibility within the complementarity-determining regions (CDRs) may facilitate stable non-covalent interactions with small-molecule chemotherapeutics. Such features may arise through antigen-driven clonal selection in advanced CRC, further supporting the functional relevance of IGHV repertoire skewing.
Importantly, this differential binding does not suggest that other candidate IGs lack utility, but rather positions IGHV3-64 as a higher-priority carrier candidate within the screened pool. Moreover, the analysis does not imply direct modulation of IRN pharmacodynamics by IGHV3-64; instead, it supports its feasibility as a molecular carrier capable of stable drug association—an essential prerequisite for rational PDC design.
Epitope-pairing analysis revealed that IGHV3-64-derived peptides exhibit spatial compatibility with functionally relevant regions of K-Ras, including residues within the P-loop and switch II domains—structural hotspots implicated in CRC oncogenesis. These in-silico findings are biologically plausible in advanced CRC, where chronic exposure to mutant K-Ras-derived neoantigens may promote B cell clonal selection toward K-Ras-reactive IGHV configurations. Crucially, the present study does not propose that IGHV3-64 binding inhibits K-Ras GTPase activity or suppresses downstream RAS/RAF signaling. Instead, the biological significance of this interaction is best understood as a tumor-immune interface phenomenon, reflecting endogenous humoral responses shaped by oncogenic K-Ras signaling. Epitope pairing was therefore intentionally evaluated to assess spatial compatibility and proximity potential, rather than enzymatic inhibition.
With respect to specificity, epitope pairing was assessed at the level of sequence and structural compatibility rather than competitive binding or mutational discrimination. While the observed pairing suggests preferential interaction, cross-reactivity with structurally related epitopes cannot be excluded. Accordingly, epitope pairing is framed here as candidate neoantigen recognition, not definitive specificity. Future studies should incorporate mutant vs. wild-type K-Ras comparisons and competitive binding assays [e.g., surface plasmon resonance (SPR), co-immunoprecipitation (CoIP), Förster resonance energy transfer (FRET)] to rigorously evaluate selectivity. Within this proximity-guided delivery model, IGHV3-64-derived peptides may act as molecular guides, facilitating localized accumulation of conjugated therapeutic payloads in K-Ras-driven tumor cells. This approach aligns with emerging antibody-guided and peptide-based strategies designed to address the long-standing challenge of targeting “undruggable” oncogenes without requiring direct catalytic inhibition [19].
Additionally, the identification of intrinsic CPP motifs within IGHV3-64 strengthens the biological plausibility of this model by suggesting enhanced intracellular accessibility. Collectively, drug association, epitope proximity, and CPP features define a multifunctional but non-inhibitory role for IGHV3-64 in PDC systems, consistent with engineered antibody-peptide platforms targeting intracellular oncogenic or immune signaling pathways [20].
This study represents the first systematic exploration of stage-specific, stable, secreted circulating IGs shaped by the CRC immune microenvironment and their feasibility as endogenous carriers for drug conjugation, including their association with K-Ras. From a translational perspective, several practical considerations are relevant for IGHV3-64-based pPDC development, including conjugation strategy, stability, and pharmacokinetics. Conceptually, pPDCs may be generated through direct chemical conjugation, linker-based attachment, or emerging linker-free approaches such as fusion tags or site-specific antibody modification. While endogenous IG frameworks may offer intrinsic stability and immunological compatibility, factors such as drug-to-antibody ratio heterogeneity, batch variability, and pharmacokinetic behavior will require systematic optimization and validation. These aspects are acknowledged here as developmental challenges rather than resolved advantages, reinforcing the exploratory nature of the present work.
In comparison with conventional monoclonal ADCs, pPDCs are not proposed as replacements but as complementary modalities. While ADCs benefit from precise epitope targeting and established clinical pipelines, they are limited by single-epitope dependence, antigen loss, immunogenicity, manufacturing cost, and reduced efficacy in antigen-heterogeneous tumors. pPDCs, by contrast, offer multi-epitope coverage and immune-context-adapted targeting, which may be particularly relevant in advanced-stage and K-Ras-mutant CRC, where effective ADC targets and anti-EGFR therapies are limited [21]. Clinically, IRN remains a cornerstone of metastatic CRC therapy, particularly in folinic acid, FU, and IRN (FOLFIRI)-based regimens. However, systemic toxicity, therapeutic resistance, and limited selectivity remain significant challenges. In K-Ras-mutant CRC, where EGFR-targeted antibodies are ineffective, IGHV3-64-based pPDCs may offer a conceptually distinct and complementary strategy, integrating chemotherapeutic delivery with tumor- and immune-context-specific molecular guidance rather than competing with existing regimens [22].
Beyond therapeutic considerations, circulating biomarkers are increasingly explored for prognostic and perioperative risk stratification in CRC. Recent studies highlighting serum butyrylcholinesterase (BuChE) as a predictor of postoperative complications further underscore the clinical relevance of blood-based biomarkers. In this context, stage-specific IG profiles identified in the present study may represent an additional layer of biologically informed biomarkers with potential relevance across diagnostic, prognostic, and therapeutic domains [23].
Collectively, our findings propose a biologically grounded, hypothesis-driven framework for a new class of pPDCs leveraging tumor-associated circulating IGs as polyclonal, multi-epitope carriers. While preliminary and computational in nature, this framework is particularly relevant for ASCRC, where immune remodeling, antigenic heterogeneity, and therapeutic resistance converge. Future studies will focus on experimental validation in vitro and in vivo, including CRC organoid-based pharmacokinetics, tumor selectivity, immune interactions, diagnostic performance (ROC/AUC), and targeted validation using SPR, CoIP, and FRET assays. To clarify the conceptual positioning of pPDCs relative to established ADCs, a comparative overview of key features, advantages, and limitations is provided in Table 7.
Conceptual comparison between monoclonal antibody-drug conjugates (ADCs) and polyclonal drug conjugates (pPDCs).
| Feature | ADCs | pPDCs |
|---|---|---|
| Antibody composition | Single, defined monoclonal antibody recognizing one epitope | Mixture of immunoglobulins (IGs) or IG-derived frameworks recognizing multiple epitopes |
| Epitope coverage | Single-epitope targeting; vulnerable to antigen loss or mutation | Multi-epitope recognition; potentially more resilient to tumor heterogeneity |
| Target specificity | High specificity for a predefined surface antigen | Broader targeting spectrum shaped by endogenous immune selection |
| Tumor heterogeneity handling | Limited; reduced efficacy if antigen expression is heterogeneous | Conceptually better suited to heterogeneous tumors due to polyclonality |
| Immunogenicity risk | May require humanization; risk of anti-drug antibody formation | Potentially lower if derived from endogenous or patient-matched IGs |
| Manufacturing complexity | Highly controlled but expensive and time-intensive | Conceptually simpler frameworks, but standardization remains challenging |
| Drug-antibody ratio (DAR) | Precisely controlled and well-characterized | More variable; requires optimization and validation |
| Pharmacokinetics | Well-characterized and predictable | Less well-defined; requires systematic investigation |
| Clinical maturity | Multiple FDA-approved ADCs across solid tumors | Exploratory and early-stage; limited clinical validation |
| Applicability in K-Ras-mutant CRC | Limited, as effective surface targets are scarce | Potential relevance through intracellular targeting or neoantigen association |
| Primary limitations | Antigen escape, toxicity, cost, resistance | Variability, regulatory complexity, need for robust validation |
| Current role | Established therapeutic modality | Emerging therapeutic modality |
| Positioning of current study | — | Computational and proteomic feasibility assessment for CRC-specific pPDC design |
This study presents a first-of-its-kind, integrative proteomic and computational framework for identifying circulating, stage-specific IGs in CRC and evaluating their feasibility as endogenous carriers for pPDCs. Selected IGs, particularly IGHV3-64, demonstrated stable association with IRN and epitope-level spatial compatibility with K-Ras, supported by the presence of intrinsic CPP motifs that may facilitate intracellular access. Rather than establishing therapeutic efficacy, these findings identify IGHV3-64 as a candidate IG scaffold with properties suitable for proximity-guided drug delivery in advanced and treatment-resistant CRC. The results support the concept that circulating, immune-selected IGs—shaped by the TME—can be systematically mined and repurposed as immunologically compatible, precision-guided delivery frameworks, pending rigorous experimental validation.
Although primarily exploratory and computational in nature, this work provides a biologically grounded rationale for future in vitro and in vivo studies, including organoid-based models, pharmacokinetic analyses, and functional validation assays. Collectively, the study establishes a conceptual foundation for immune-context-aware precision therapeutic design, offering a new avenue for integrating tumor immunobiology with next-generation drug conjugation strategies in CRC.
A key strength of this study is its integrative, multi-omics approach—combining high-resolution mass spectrometry, computational docking, and epitope-level biophysical analysis. This strategy enabled the identification of stage-specific IG subtypes in CRC and their evaluation as endogenous drug carriers. Unlike conventional ADCs, the use of naturally circulating IGs offers a personalized and immunocompatible platform, potentially minimizing off-target toxicity and improving bioavailability. Notably, the identification of dual-function peptides—with both drug-binding and cell-penetrating capabilities—presents an innovative solution for intracellular delivery, especially for targeting traditionally “undruggable” protein K-Ras.
However, several limitations warrant consideration. First, the findings are based primarily on in-silico and in vitro analyses, necessitating further validation in cellular and animal models to confirm therapeutic relevance. Second, although IG expression patterns were derived from plasma proteomics, it remains unclear whether these signatures are consistent across diverse patient populations or influenced by variables such as immune status, comorbidities, or the gut microbiome. Moreover, the broader functional role of circulating IGs in tumor progression and immune modulation was not investigated, which may impact their utility as therapeutic vectors.
Future work will focus on validating IGs-drug interactions using SPR and assessing cellular uptake. In vivo studies in CRC mouse models will be crucial to evaluate pharmacokinetics, biodistribution, tumor specificity, and therapeutic efficacy. Additional efforts will explore engineering IG-derived peptides with cleavable linkers and payloads such as siRNAs or immunomodulators, paving the way for next-generation pPDCs tailored to heterogeneous and treatment-resistant CRC subtypes.
5-FU: 5-fluorouracil
AA: amino acid
ACN: acetonitrile
ADCs: antibody-drug conjugates
ASCRC: advanced-stage colorectal cancer
AUC: area-under-the-curve
AVMC: Aarupadai Veedu Medical College and Hospital
CAP: capecitabine
CAR: chimeric antigen receptor
CLL: chronic lymphocytic leukemia
CoIP: co-immunoprecipitation
CPP: cell-penetrating peptide
CRC: colorectal cancer
EOCRC: early-onset colorectal cancer
ESCC: esophageal squamous cell carcinoma
ESCRC: early-stage colorectal cancer
ESI-nanoLC-MS/MS: electrospray ionization nano-liquid chromatography tandem mass spectrometry
FDR: false discovery rate
FOLFIRI: folinic acid, fluorouracil, and Irinotecan
FRET: Förster resonance energy transfer
HNSCC: head and neck squamous cell carcinoma
IEDB: Immune Epitope Database
IGHV3-64: immunoglobulin heavy variable 3-64
IGLV9-49: immunoglobulin lambda variable 9-49
IgM-MGUS: IgM monoclonal gammopathy of undetermined significance
IGs: immunoglobulins
IIx: instability index
IMS: ion mobility spectrometry
IRN: Irinotecan
K-Ras: Kirsten rat sarcoma viral oncogene homolog
LEU: leucovorin
MALT: mucosa-associated lymphoid tissue
MC: Monte Carlo
mol.wt: molecular weight
NH4HCO3: ammonium bicarbonate
OXA: oxaliplatin
PDCs: peptide drug conjugates
pI: isoelectric point
pPDCs: polyclonal drug conjugates
Q-TOF: quadrupole time-of-flight
RMSD: root mean square deviation
ROC: receiver operating characteristic
SDS-PAGE: sodium dodecyl-sulfate polyacrylamide gel electrophoresis
SPR: surface plasmon resonance
SVM: support vector machine
TME: tumor microenvironment
TNM: tumor, nodes, and metastasis
WHO-EML: WHO Essential Medicines List
WM: Waldenström’s macroglobulinemia
ΔG: binding free energy
Other supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/1003252_sup_1.pdf.
We would like to thank Mrs. J.S.Sakkthi Prabha for formatting and proofreading the manuscript.
RA: Conceptualization, Investigation, Methodology, Writing—original draft. VB, SK, JR, and RS: Writing—review & editing, Funding acquisition, Resources. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
The study was approved by the AVMC Institutional Human Ethics Committee (IHEC) under protocol No. AVMC-2021-04-14. The study was conducted in accordance with the principles of the Declaration of Helsinki (2024 revision).
Written informed consent was obtained from all participants prior to sample collection.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Seed funding support to conduct this research (VMRF/Research/Seed Money/30th April, 2021) was provided by VMRF. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 801
Download: 26
Times Cited: 0
