 Review
Review

Affiliation:
1Molecular Oncology Research Center, Barretos Cancer Hospital, Barretos 14784-400, Brazil
ORCID: https://orcid.org/0000-0002-7533-0763
Affiliation:
1Molecular Oncology Research Center, Barretos Cancer Hospital, Barretos 14784-400, Brazil
ORCID: https://orcid.org/0009-0005-4103-8771
Affiliation:
2Human Genetics Laboratory, Programa de Pós-Graduação em Ciências da Saúde, Universidade Federal do Triângulo Mineiro, Uberaba 38025-180, Brazil
Email: jescaferreira@gmail.com
ORCID: https://orcid.org/0000-0003-3556-9849
Explor Immunol. 2026;6:1003245 DOI: https://doi.org/10.37349/ei.2026.1003245
Received: December 19, 2025 Accepted: March 13, 2026 Published: April 13, 2026
Academic Editor: Wantong Song, Chinese Academy of Sciences, China
Breast cancer exhibits substantial clinical and molecular heterogeneity, partly shaped by interactions between tumor biology and the host immune system. Germline variants in immune-related genes may influence inflammatory tone, immune regulation, and tumor-immune interactions. However, evidence linking inherited immune genetic variation to breast cancer risk and clinical behavior remains fragmented and heterogeneous across studies. We conducted a structured integrative review of 33 human genetic associations evaluating germline variants in immune-related genes and their associations with breast cancer risk, prognosis, and clinicopathological features. Data were synthesized using a comparative, pathway-oriented analytic framework. Variants in cytokine genes, particularly TGFB1, IL6, IL1B, and IL10, were the most frequently associated with susceptibility, although effect directions varied across populations and genetic models. In contrast, variants in chemokine pathways (CXCL12) and immune checkpoint regulator genes (B7-H4/VTCN1, PD-1/PDCD1) showed consistent associations with tumor progression, immune evasion, and subtype-specific clinical features, including human epidermal growth factor receptor 2 (HER2)-positive disease and metastatic presentation. Across studies, substantial heterogeneity was observed, driven by differences in ethnic composition, sample size, methodological design, and deviations from the Hardy-Weinberg equilibrium. The findings support a pathway-oriented interpretation in which germline immune variation differentially influences immune regulation and tumor progression rather than uniformly determining disease risk. While inflammatory and immunoregulatory pathways appear to shape basal immune tone, immune effector function is less consistently associated with germline variation. Further progress will require extensive, multiethnic studies integrating genetic, transcriptomic, and functional data to clarify how inherited immune variation contributes to breast cancer biology.
Breast cancer is a multifactorial disease with broad clinical and molecular heterogeneity, reflecting complex interactions between genetic, hormonal, and environmental factors. Advances in molecular subtyping have deepened our understanding of this diversity, revealing that components of the immune response play a decisive role in tumor initiation, progression, and clinical behavior [1].
The antitumor immune response is modulated by both acquired and germline factors that define activation thresholds, inflammatory profiles, and immunoregulatory axes. Polymorphisms in immune-related genes may influence the regulation of cytokines, receptors, and costimulatory molecules, thereby shaping immune tone and responsiveness. Rather than exerting direct, deterministic effects at the protein level, these variants are thought to act as modulators of immune regulation, with clinical relevance arising from their cumulative, context-dependent impact [2, 3].
Several immune pathways mediate host-tumor interactions, including cytokine and chemokine signaling, immune checkpoints, and mechanisms of immune cell recruitment and tolerance. However, studies exploring the germline genetic contribution of these pathways remain sporadic and heterogeneous, with divergent results across populations, tumor subtypes, and clinical endpoints [1, 4, 5]. As a result, individual gene associations are often challenging to interpret in isolation, underscoring the need for pathway-level and integrative analyses.
In this context, this integrative review aimed to gather and organize current evidence on the association between genetic variants in immune response genes and breast cancer risk, prognosis, and clinical characteristics. The focus was restricted to inherited host genetic variation, conceptually distinct from somatic alterations occurring within tumor cells. Rather than focusing on isolated gene effects, we sought to examine recurring patterns across immune-related pathways and to contextualize heterogeneous findings within a pathway-oriented, interpretive perspective. By adopting an integrative and exploratory approach, this review aims to support hypothesis-generating interpretations regarding how germline immune variation may contribute to immune regulation, tumor progression, and clinical heterogeneity in breast cancer.
This study was designed as a structured integrative review to provide a conceptual, hypothesis-generating synthesis of germline immune-related variants in breast cancer. PRISMA 2020 reporting guidance was used to transparently document the search and study selection workflow (identification, screening, eligibility, and inclusion) to enhance methodological transparency and reproducibility. However, the objective was not to conduct quantitative pooling (meta-analysis) or a formal risk-of-bias assessment. The methodological approach was guided by the integrative review framework proposed by Whittemore and Knafl [6], which supports the inclusion of heterogeneous empirical evidence and enables iterative, comparative, and interpretive synthesis across heterogeneous study designs.
Given the heterogeneity of included studies (primarily candidate-gene case-control studies and, in some cases, genomic analyses), quantitative pooling and formal risk-of-bias scoring were not performed. Instead, key methodological domains were systematically extracted to contextualize the evidence’s strengths and limitations.
A comprehensive search was conducted across PubMed, Embase, Scopus, and Web of Science, with no publication date restrictions. Filters were set to include original human research articles (case-control, cohort, and clinical trials) and to exclude reviews and non-primary reports. Although no open-access filter was applied during database searches, a small number of studies (n = 5) were excluded due to the unavailability of full text, which may introduce minimal residual selection bias.
The search strategy was structured around a core conceptual domain: breast cancer, germline genetic variation, immune-related terms, and tumor microenvironment. Controlled vocabulary [medical subject headings (MeSH)/Emtree, when available] and free-text keywords were combined using Boolean operators. The PubMed query served as the reference structure and was adapted to each database’s syntax. Complete database-specific strategies, search dates, and applied filters are available in Supplementary material 1.
Genes were not preselected a priori; rather, all eligible studies evaluating germline variants in immune-related genes retrieved through the predefined search strategy were considered. No gene- or population-based filters were applied at the search stage.
Studies were considered eligible if they were observational (case-control or cohort) or randomized clinical trials, conducted in human populations, published in English, and investigated genetic variants, predominantly single-nucleotide polymorphisms, as well as other variant types, in immune-related genes in the context of antitumor immune responses in breast cancer, and that evaluated associations with breast cancer susceptibility, prognosis, or clinicopathological features. Studies were excluded if they lacked Ethics Committee approval; did not assess genetic variants; failed to explore the relationship between genetic variants and immune system performance in the breast tumor context; were literature reviews, meta-analyses, letters to the editor, editorials, or commentaries; or lacked sufficient methodological description.
Two independent reviewers screened all records in two stages: title and abstract screening, in which articles were assessed against the eligibility criteria; and full-text review, in which potentially relevant studies were examined in full detail, applying the same inclusion and exclusion criteria. In cases of disagreement, a third reviewer was consulted to reach a consensus.
The screening process was conducted using the Rayyan platform, which facilitates blinded, independent review and decision-tracking. Additionally, open-source scripts available at [https://github.com/MuriloPorfirio/Integrative_Review_Tools] were utilized, including a Python-based deduplication utility built with pandas and tqdm, as well as browser-based HTML/JavaScript tools that enable users to include or exclude records based on specified terms in the title, abstract, and keywords.
Data extraction was performed independently by two reviewers, and any discrepancies were resolved through discussion with a third reviewer. Extracted information included: study design and population characteristics, sample size, investigated genes and variants, genotyping methods, Hardy-Weinberg equilibrium assessment, statistical models, and confounder adjustment, and reported associations with breast cancer outcomes. Rather than applying a numerical quality score, methodological characteristics were summarized descriptively to allow contextual interpretation of findings across studies.
Extracted data were synthesized narratively using an integrative, comparative, and iterative analytic approach and examined in relation to functional immune pathways, thereby enabling an integrative and exploratory interpretation of heterogeneous results. A comparative process was applied, in which findings for the same genes, variants, or immune pathways were iteratively compared across studies, populations, and clinical outcomes to identify recurrent patterns and convergent and divergent associations. Data were tabulated to summarize genetic variants, study populations, and associations with breast cancer outcomes, supporting systematic data reduction and cross-study comparison. Figures and tables were generated to illustrate the flow of study selection, the characteristics of included studies, and the most frequently investigated variants.
A comprehensive search of PubMed, Embase, Scopus, and Web of Science identified 411,039 records. After applying the predefined selection filters and removing duplicate entries, 4,481 articles remained for screening. Following the evaluation of the title and abstract, 51 studies were selected for full-text assessment. Of these, 33 studies published between 2004 and 2024, conducted in Asian, Latin American, European, and African populations, with a predominance of case-control designs, were included (Figure 1).
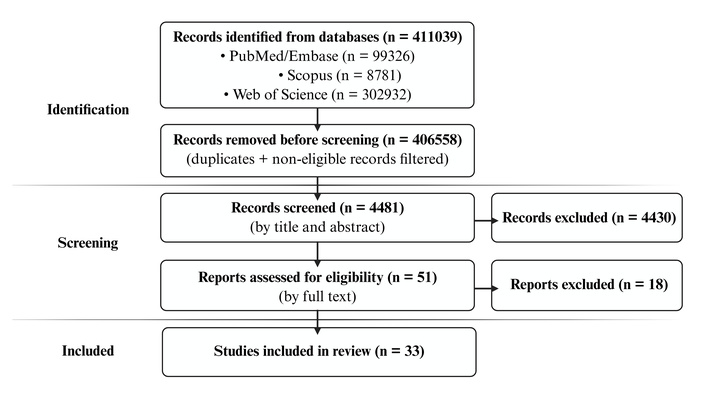
The flowchart illustrates the search strategy used to identify studies on the association between genetic variants and breast cancer. Prepared by the authors, 2025, and adapted from Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA). Distributed under a Creative Commons CC BY 4.0 license.
East Asian study populations, particularly Chinese cohorts, were the most frequently represented in the included studies [7–12]. Brazilian cohorts were the second most frequently represented [3, 13–17]. Other study populations, including USA-based cohorts, were less frequently represented [18, 19].
A structured summary of key methodological domains across all included studies, encompassing study design, sample size, ancestry, Hardy-Weinberg equilibrium assessment, confounder adjustment, and genotyping approach, is presented in Table 1. These domains were considered during narrative interpretation to contextualize the strength and consistency of reported associations.
Methodological domains considered during narrative quality appraisal of the included studies.
| Study (author, year) | Country | Population ancestry | Sample size (cases/controls) | HWE in controls | Confounder adjustment | Genotyping method |
|---|---|---|---|---|---|---|
| Li et al., 2013 [7] | China | Chinese | 1,110/1,060 | Yes | Yes | MassARRAY |
| Padala et al., 2022 [20] | India | South Indian | 300/300 | Yes | No | PCR-RFLP and AS-PCR |
| Alidoust et al., 2021 [21] | Iran | Iranian | 476/524 | Yes | Not reported/unknown | ARMS |
| Chou et al., 2020 [8] | China | People in Taiwan, China | 1,365/0 | Not applicable | Yes | Chip SNP Affymetrix TWB |
| Zhang et al., 2019 [22] | Multicenter | Multi-cohort (TCGA, GTEx; predominantly European ancestry) | Not applicable (secondary genomic datasets) | Not applicable | Yes | Public GWAS datasets + genotype imputation |
| Deligezer and Dalay, 2004 [23] | Türkiye | Turkish | 151/133 | Yes | No | PCR-RFLP |
| Eras et al., 2019 [24] | Türkiye | Turkish | 204/210 | Yes | Yes | TaqMan genotyping assay |
| Sirisena et al., 2018 [25] | Sri Lanka | Sri Lankan | 349 | Yes | Yes | MassARRAY |
| Lee et al., 2017 [26] | South Korea | Korean | 2,027 | Not reported/unknown | Yes | Affymetrix Genome-Wide Human SNP Array 6.0 chip |
| Pal and Dutta, 2023 [27] | India | North Indian | 285/363 | Not reported/unknown | Yes | Sanger sequencing |
| Lin et al., 2022 [9] | China | Chinese | 434/450 | Not reported/unknown | Yes | MassARRAY |
| Hausmann et al., 2021 [17] | Brazil | Brazilian | 243/294 | Yes | Yes | TaqMan genotyping assay |
| Tsai et al., 2020 [28] | China | People in Taiwan, China | 1,232/1,232 | Yes | Yes | PCR-RFLP |
| Fawzy and Toraih, 2020 [29] | Egypt | Egyptian | 200/340 | Yes | Yes | TaqMan genotyping assay |
| Park et al., 2024 [30] | South Korea | Korean | 90/90 | Not reported/unknown | Not reported/unknown | RT-qPCR |
| Shan et al., 2019 [31] | Tunisia | Tunisian | 544/538 | Yes | Yes | TaqMan genotyping assay |
| Vitiello et al., 2019 [15] | Brazil | Brazilian | 388/405 | Not reported/unknown | Yes | PCR-RFLP |
| Vitiello et al., 2018 [16] | Brazil | Brazilian | 323/405 | Not reported/unknown | Yes | PCR-RFLP |
| Vitiello et al., 2018 [14] | Brazil | Brazilian | 338/403 | Yes | Yes | PCR-RFLP |
| Sabet et al., 2017 [32] | Egypt | Egyptian | 105/50 | Yes | No | PCR-RFLP |
| Ren et al., 2016 [10] | China | Chinese | 560/583 | Yes | No | MassARRAY |
| Chen et al., 2015 [11] | China | Chinese | 715/0 | Yes | Yes | 12-plex SNPstream platform |
| Okuyama Kishima et al., 2015 [3] | Brazil | Brazilian | 74/0 | Yes | No | PCR-RFLP |
| de Oliveira et al., 2013 [13] | Brazil | Brazilian | 26/0 | Not reported/unknown | No | PCR-RFLP |
| Resler et al., 2013 [18] | USA | American/Caucasian | 845/807 | Yes | Yes | Illumina GoldenGate multiplex platform |
| Ozgöz et al., 2013 [33] | Türkiye | Turkish | 31/30 | Not reported/unknown | No | PCR-RFLP |
| Joshi et al., 2011 [34] | India | Parsi and Maharashtrian (Indian) | 230/407 | Yes | No | PCR-SSP |
| Madeleine et al., 2011 [19] | USA | Caucasian American | 882/906 | Yes | Yes | Applied Biosystems SNPlex® system |
| Slattery et al., 2014 [5] | Mexico and USA (San Francisco) | USA Hispanic/Native American or MexicanUSA non-Hispanic white | 3,592/4,182 | Yes | Yes | Multiplexed bead array assay format based on GoldenGate chemistry (Illumina, San Diego, CA) |
| Yu et al., 2015 [12] | China | Chinese | 376/366 | Yes | Not reported/unknown | PCR-RFLP |
| Safonov et al., 2017 [1] | Multicenter | TCGA (predominantly European ancestry) | 1,025/0 | Not applicable | Yes (multivariate linear models) | Public GWAS datasets + genotype imputation TCGA germline SNVs |
| Adolf et al., 2019 [35] | Tanzania | Tanzanian | 75/84 | Yes | No | LightSNiP typing assay (TIBMolBiol, Berlin, Germany) |
| Lei et al., 2016 [4] | Consortia-BCAC | European | 42,510/40,577 | Yes | Yes | Illumina iCOGS array + imputation (1000 Genomes) |
GWAS: genome-wide association study; HWE: Hardy-Weinberg equilibrium assessed in control groups when applicable; SNP: single-nucleotide polymorphism; TCGA: The Cancer Genome Atlas. Not applicable refers to secondary analyses of publicly available genomic datasets without an independent control group. Confounder adjustment refers to multivariable models including demographic, clinical, or ancestry-related covariates.
The studies included in this review examined 116 genetic variants across 58 genes related to the immune system. These genes were primarily involved in cytokine and interleukin signaling, immune cell receptor expression, inflammatory pathways, and regulation of immune checkpoints. Across studies, specific variants were found to be associated with an increased risk of breast cancer (susceptibility), often linked to advanced disease stages, metastasis, or poorer prognosis. Conversely, some variants appeared to have a protective effect, correlating with improved survival outcomes or reduced tumor susceptibility. Recurrently investigated variants are summarized in Table 2, and an endpoint-oriented evidence summary for repeatedly investigated variants is presented in Table 3. Complete variant panels for each included study, including both statistically significant and non-significant results, are provided in Table S1.
Recurrently investigated germline variants in breast cancer susceptibility studies included in this integrative review.
| Gene | Genetic variant | Consequence | No. of papers [references] | Hardy-Weinberg equilibrium < 0.05 [reference]* |
|---|---|---|---|---|
| TGFB1 | rs1800470 | Missense variant | 4 [15, 16, 27, 34] | 2 [15, 27] |
| rs1800469 | 2 kb upstream variant | 2 [15, 16] | 1 [15] | |
| rs1800471 | Missense variant | 2 [15, 16] | 1 [15] | |
| IL6 | rs1800795 | Intron variant | 3 [5, 19, 20] | 1 [20] |
| rs1800797 | Non-coding transcript variant | 2 [5, 20] | 1 [20] | |
| IL1B | rs1143627 | 2 kb upstream variant | 2 [5, 24] | 0 |
| IL10 | rs1800871 | Intron variant | 2 [5, 32] | 0 |
| CXCL12 | rs1801157 | 3' UTR variant | 2 [9, 13] | 2 [9, 13] |
| B7H4 | rs10754339 | Non-coding transcript variant | 2 [12, 33] | 1 [33] |
*: Hardy-Weinberg equilibrium in control groups, as reported in the original publications.
Evidence summary for repeatedly investigated variants by outcome category, genetic model, and population.
| Gene | Genetic variant | Reference | Population | Outcome | Genetic model | Effect size(OR, 95% CI) | Direction |
|---|---|---|---|---|---|---|---|
| TGFB1 | rs1800470 | [27] | North Indian | Susceptibility | DominantTT + TC vs. CC | 1.7 (1.1–2.6) | ↑ Risk |
| [15]1 | Brazilian | Susceptibility | Haplotype | Not extractable | — | ||
| [16] | Brazilian | Susceptibility | RecessiveTT vs. CT + CC | 0.39 (0.1–0.8) | ↓ Risk | ||
| [34]2 | Indian | Susceptibility | Dominant TC + CC vs. TT | 0.50 (0.2–0.9) | ↓ Risk | ||
| rs1800469 | [15]1 | Brazilian | Susceptibility | Haplotype | Not extractable | — | |
| [16] | Brazilian | Susceptibility | DominantCT + TT vs. CC | 2.1 (1.1–3.8) | ↑ Risk | ||
| rs1800471 | [15]1 | Brazilian | Susceptibility | Haplotype | Not extractable | — | |
| [16] | Brazilian | Susceptibility | RecessiveCC vs. GG + GC | 11.3 (1.6–78.2) | ↑ Risk | ||
| IL6 | rs1800795 | [20] | South Indian | Susceptibility | DominantGC + CC vs. GG | 2.1 (1.5–2.9) | ↑ Risk |
| [5] | Multiethnic* | Susceptibility | Dominant GC + CC vs. GG | 0.8 (0.8–0.9) | ↓ Risk | ||
| [19] | Caucasian Americans | Susceptibility | Log-additiveper C allele | 1.0 (0.9–1.2) | No effect | ||
| rs1800797 | [20] | South Indian | Susceptibility | Dominant GC + CC vs. GG | 2.11 (1.5–2.9) | ↑ Risk | |
| [5] | Multiethnic* | Susceptibility | Dominant GT + TT vs. GG | 0.8 (0.7–0.9) | ↓ Risk | ||
| IL1B | rs1143627 | [5] | Multiethnic* | Susceptibility | Dominant TC + CC vs. TT | 0.9(0.8–0.9) | ↓ Risk |
| [24] | Turkish | Susceptibility | Codominant/homozygote comparisonTT vs. CC | 2.06 (1.1–3.6) | ↑ Risk | ||
| IL10 | rs1800871 | [32] | Egyptian | Susceptibility | DominantTC + CC vs. TT | 4.02 (1.4–10.9) | ↑ Risk |
| [5] | Multiethnic* | Susceptibility | RecessiveTT vs. CC + CT | 0.79 (0.6–0.9) | ↓ Risk | ||
| CXCL12 | rs1801157 | [9] | Chinese | Susceptibility | DominantGA + AA vs. GG | 1.35 (1.0–1.7) | ↑ Risk |
| [13] | Brazilian | Gene expression | DominantGA vs. GG | Not applicable | ↓ CXCL12 expression | ||
| B7H4 | rs10754339 | [33] | Turkish | Susceptibility | CodominantAG + GG vs. AA | Not reported | No effect |
| [12] | Chinese | Susceptibility | 1-locus model3 | 1.67 (1.2–2.2) | ↑ Risk |
1: Ref. [15] focused on TGFBR2 and TGFB1 haplotypes and did not report extractable OR (95% CI) for the individual variants listed in this table; 2: this study reports multiple genetic models; the value shown here corresponds to the dominant model in Maharashtrian women aged < 40 years and is age-adjusted; 3: high-risk genotype combination vs. low-risk genotype combination; *: non-Hispanic White and Hispanic/Native American; ↑: increased; ↓: decreased. OR: odds ratio; CI: confidence interval.
Among the 116 genetic variants identified in this study, nine were investigated in more than one study (Table 2). A predominance of intronic and promoter-region variants was observed, suggesting a potential regulatory effect on gene expression rather than direct protein-coding alterations. Cytokine-related genes, particularly IL6 and TGFB1, accounted for the most recurrently studied variants, with the rs1800470 (TGFB1) variant investigated in four studies [15, 16, 27, 34] and the rs1800795 (IL6) variant in three studies [5, 19, 20]; VTCN1 (B7H4) appeared in two studies, whereas CTLA4 polymorphism was identified in only one eligible study and therefore did not meet the threshold for inclusion in Table 2.
All genetic variants in Table 2 were tested for Hardy-Weinberg equilibrium in at least one study. Regarding the consequences of these variants, the analysis revealed missense variants, such as those in the TGFB1 gene (rs1800470 and rs1800471), as well as non-coding transcript variants, including rs1800797 in the IL6 gene, rs10754339 in the B7H4 gene, and rs1801157 in the CXCL12 gene. Additionally, there were 2 kb upstream variants, such as rs1143627 in the IL1B gene and rs1800469 in the TGFB1 gene, as well as intron variants, including rs1800795 in the IL6 gene, and rs1800871 in the IL10 gene.
To enhance cross-study comparability, recurrent variants were additionally categorized according to clinical endpoint (susceptibility, prognosis, and clinicopathological features), genetic model, and direction of effect across populations, using standardized effect-size measures when available, reported as odds ratios (OR) with 95% confidence intervals (CI) (Table 3).
Of the 33 included studies, 21 (64%) reported at least one statistically significant association between at least one immune-related germline variant and an increased risk or worse prognosis for breast cancer. Six studies (18%) described protective associations, and six (18%) reported no significant association. Variants in cytokine and receptor genes (IL6, IL1B, IL10, TGFB1, and TNFRSF1A) were most frequently associated with risk and tumor progression (susceptibility). In contrast, immune checkpoint-related genes (PDCD1, CTLA4, and VTCN1/B7H4) showed a predominant association with prognosis and immune response modulation. Variants in chemokine-related genes (CXCL12, IL8, and CCL5) were commonly associated with advanced stages and aggressive subtypes, and variants in PDCD1 and autoimmune regulator (AIRE) showed protective effects across different populations.
These findings indicate that inherited variations in inflammatory and immunoregulatory pathways may influence both breast cancer susceptibility and disease progression. The summary of the findings highlights an interconnection between immunoregulatory variants and tumor progression mechanisms. Most of the variants affect cytokine and checkpoint genes, suggesting that the balance between inflammation and immunosuppression may be genetically modulated in breast cancer.
After examining the literature included in this review, it becomes evident that germline genetic variants in immune-related genes are associated with breast cancer risk, prognosis, and clinical phenotype in a context-dependent manner. The available evidence suggests that these variants may act as genetic markers of altered tumor-immune interactions, influencing inflammatory tone, immune tolerance, and immune cell dynamics throughout breast cancer development. When considered collectively, the heterogeneous findings across studies point to recurrent immune pathways, supporting an integrative, hypothesis-generating interpretation that extends beyond isolated gene associations toward functional immune axes relevant to breast cancer biology.
Importantly, the associations discussed herein do not imply direct functional effects of individual variants at the protein level. Most studies included in this review evaluated germline single-nucleotide polymorphisms as genetic markers associated with breast cancer phenotypes, without functional validation of downstream transcriptional or proteomic consequences. Thus, the variants summarized here should be interpreted as proxies of altered immune regulation rather than deterministic drivers of protein expression or activity. Their biological relevance likely arises from cumulative, context-dependent effects within immune pathways, influenced by genetic background, epigenetic regulation, and tumor microenvironmental factors.
Variants in genes involved in immune activation and inflammatory regulation were the most frequently investigated across the included studies and were predominantly associated with breast cancer susceptibility. These genes encode cytokines and related signaling molecules that regulate basal inflammatory tone and immune surveillance, thereby influencing early tumor-immune interactions. Although individual associations were heterogeneous and strongly population-dependent, the recurrent involvement of cytokine-related pathways suggests that inherited modulation of inflammatory balance may shape differential susceptibility to breast carcinogenesis, rather than acting as independent causal determinants [36, 37].
Within this context, variants in cytokine genes and their receptors emerged as the most consistently investigated genetic factors associated with breast cancer risk. Key genes repeatedly examined across studies include TGFB1, IL6, IL1B, and IL10, which collectively regulate pro- and anti-inflammatory signaling and contribute to the dynamic balance between immune activation and immunosuppression during early stages of tumor development [38–40]. The TGFB1 gene, which encodes the immunoregulatory cytokine TGF-β1, was among the most frequently investigated immune-related genes in the included studies. Three variants (rs1800470, rs1800469, and rs1800471) were recurrently analyzed and showed context-dependent associations with breast cancer susceptibility and molecular subtype.
Among them, rs1800470 was the most extensively studied. While the C allele was associated with a reduced overall risk in some populations, particularly in CC homozygotes [27, 34], other studies reported an association with the human epidermal growth factor receptor 2 (HER2)-positive subtype [16]. These divergent findings suggest that the effect of rs1800470 is heterogeneous and may depend on population-specific haplotypic backgrounds and tumor molecular contexts. Supporting this notion, rs1800469 and rs1800471 showed no consistent association with overall breast cancer risk but were repeatedly linked to the HER2-positive subtype under dominant and recessive models, respectively [16].
Functionally, TGF-β1 plays a central role in immune regulation by limiting immune activation and promoting tolerance [41]. Experimental studies indicate that HER2 overexpression can alter canonical TGF-β signaling, shifting its effects from growth inhibition toward pro-migratory and pro-invasive programs through HER2/EGFR/AKT-dependent mechanisms and Smad3 linker phosphorylation [42, 43]. This biological interaction provides a plausible framework for the subtype-specific genetic associations observed in TGFB1.
Taken together, the evidence suggests that inherited variation in TGFB1 does not act as a general susceptibility factor but may preferentially influence immune regulation in specific tumor contexts. In particular, TGFB1 variants appear to be associated with HER2-positive breast cancer, supporting a model in which altered immune tolerance and immunosuppressive signaling may contribute to subtype-specific disease susceptibility rather than heightened inflammatory activation.
In contrast to the predominantly immunoregulatory profile observed for TGFB1, variants in the IL6 gene, particularly rs1800795 and rs1800797, were associated with breast cancer risk and progression in a highly context-dependent manner, reflecting the pleiotropic role of IL-6 in inflammation and tumor-host interactions.
The rs1800795 (–174G > C) variant was the most frequently investigated and showed divergent associations across populations. In a multiethnic cohort, the C allele was associated with reduced breast cancer risk [5], whereas in a Caucasian-American cohort, no significant overall effect was observed [19]. In contrast, in a South Indian population, it was consistently associated with increased risk under both dominant and recessive models, as well as with metastatic disease [20]. However, deviation from the Hardy-Weinberg equilibrium in the control group was reported in this study, which may reflect potential genotyping inconsistencies or underlying population stratification and therefore warrants cautious interpretation of the reported associations. These opposing effects suggest that the functional impact of rs1800795 is modulated by population-specific genetic backgrounds and environmental or inflammatory contexts, rather than by a universal susceptibility allele.
A similar pattern was observed for rs1800797. In the same South Indian cohort, the A allele was associated with increased breast cancer risk and advanced metastatic disease under both dominant and recessive models [20]. In contrast, this allele was linked to reduced risk in a multiethnic cohort including non-Hispanic White, Hispanic, and Native American women [5]. Such discrepancies reinforce the notion that IL6 variants alone are insufficient to predict disease behavior without considering broader biological context.
Biologically, these heterogeneous associations are consistent with IL-6’s central role as a pleiotropic cytokine involved in acute inflammation, immune cell differentiation, and tumor-promoting signaling. IL-6 can exert both pro- and anti-tumor effects depending on signaling intensity, duration, and cellular context [44–46]. Genetic variants influencing IL-6 expression or signaling thresholds may therefore shift the inflammatory milieu toward tumor-promoting or tumor-restraining states in a context-dependent manner, shaped by ancestry, environmental exposures, and tumor microenvironmental cues. Collectively, the IL6 findings support a model in which inherited variation in inflammatory cytokine pathways contributes to breast cancer susceptibility by modulating basal inflammatory tone rather than through fixed, directionally consistent effects across populations.
Variants in IL1B and IL10 further reinforce the role of inflammatory balance in shaping breast cancer susceptibility. These genes encode cytokines with opposing immunological functions, and their genetic variants appear to modulate the equilibrium between pro- and anti-inflammatory signaling during early tumor-immune interactions [47].
The IL1B rs1143627 polymorphism has been associated with breast cancer risk in two studies, although with divergent effect directions. One investigation reported that the T allele was associated with increased risk, consistent with the pro-inflammatory role of IL-1β [24]. In contrast, another study observed a protective effect related to the C allele in a multiethnic cohort [5]. These findings suggest that genetic variation in IL1B may influence susceptibility by modulating the intensity or persistence of inflammatory signaling rather than acting as a fixed risk determinant.
In contrast, variants in IL10, a key anti-inflammatory cytokine [48], were more consistently associated with protective or less aggressive disease phenotypes. The IL10 rs1800871 variant was linked to inflammatory breast cancer in an Egyptian cohort, in which the TC and CC genotypes were overrepresented among affected individuals [32]. Conversely, the TT genotype was associated with protection against non-inflammatory breast cancer in an Egyptian cohort [32], and the TT genotype was also associated with reduced overall risk in a multiethnic population [5]. These observations align with IL-10’s immunoregulatory role in dampening excessive inflammatory responses.
Taking everything into consideration, the opposing associations observed for IL1B and IL10 variants support a model in which breast cancer susceptibility is influenced not by inflammation per se, but by the balance between pro- and anti-inflammatory immune signaling. Inherited variation in these cytokine pathways may shape basal immune tone and inflammatory set points, thereby modulating the host environment in which early tumor-immune interactions occur.
Across studies, associations between inflammatory cytokine variants and specific clinical and molecular features, including HER2-positive disease, metastatic presentation, and inflammatory versus non-inflammatory phenotypes, suggest that these genetic effects are subtype-dependent. Collectively, these findings reinforce the idea that immune-related germline variants are associated with susceptibility and may be linked to early disease characteristics by modulating the inflammatory equilibrium.
A second interpretative axis emerging from the reviewed studies concerns germline variants in genes regulating immune suppression, peripheral tolerance, and tumor-immune interactions. Although less frequently investigated than inflammatory cytokine genes, variants in immune checkpoint regulators and chemokine pathways showed more consistent associations with tumor progression, aggressive phenotypes, and clinical features related to immune evasion. Collectively, these findings suggest that inherited differences in inhibitory immune signaling and immune cell trafficking may modulate breast tumors’ ability to escape immune control during later stages of disease, rather than influencing initial susceptibility.
The CXCL12 gene, which encodes a chemokine essential for the migration and spatial organization of immune cells within tissues, harbors the rs1801157 polymorphism, investigated in two independent cohorts. In a Han Chinese population, Lin et al. [9] reported that the A allele was associated with an increased risk of breast cancer (p = 0.014 in the codominant model; p = 0.032 in the dominant model), with stronger associations observed in advanced-stage disease (p = 0.012) and among estrogen receptor-positive patients (p = 0.023). In a Brazilian cohort, de Oliveira et al. [13] observed that the same A allele was associated with reduced CXCL12 expression in tumor tissues (p = 0.012), suggesting a potential impairment of immune cell recruitment to the tumor microenvironment.
Although direct functional studies linking rs1801157 to breast cancer progression are currently lacking, the convergence of genetic association with expression data is consistent with a model that may be associated with tumor progression, potentially through effects on immune cell trafficking and local immune surveillance, rather than being primarily involved in tumor initiation [49, 50].
Among genes involved in the negative regulation of immune responses, VTCN1 (also known as B7H4) emerged as a recurrent signal across studies. B7H4 encodes an immunoinhibitory molecule that suppresses T-cell activation and contributes to peripheral immune tolerance within the tumor microenvironment [51, 52].
Association studies conducted in Turkish [33] and Han Chinese [12] populations independently reported that the G allele of the rs10754339 variant was associated with increased breast cancer risk and/or poorer clinical outcomes, with statistical significance observed in the latter cohort (p < 0.05). Importantly, functional evidence supports a mechanistic basis for these associations. El Din et al. [53] demonstrated, through combined in vitro and in silico analyses, that the A→G substitution at rs10754339 disrupts the binding site of the tumor-suppressive microRNA miR-506-3p, resulting in increased B7-H4 expression.
These findings may support a model in which germline variation in VTCN1 may be associated with enhanced tumor immune evasion, potentially reflecting reinforced inhibitory immune signaling, thereby contributing to disease progression rather than tumor initiation. This convergence of genetic association and functional validation highlights B7H4 as a relevant immunoregulatory axis in breast cancer.
In contrast to the immunosuppressive pattern observed for VTCN1, the PDCD1 rs2227982 variant has been associated with a reduced risk of breast cancer in a Chinese cohort, particularly among HER2-positive patients [10], suggesting a protective effect. PD-1 is a central immune checkpoint receptor that regulates T-cell activation and exhaustion, and germline variation in this pathway may influence the balance between immune surveillance and tolerance [54, 55].
Although evidence on the impact of rs2227982 on survival or treatment response remains limited, these findings indicate that inherited modulation of PD-1 signaling may differentially affect tumor-immune interactions across molecular subtypes. Rather than uniformly promoting immune escape, variation in checkpoint pathways may exert bidirectional effects, reinforcing the complexity of immune regulation in breast cancer and underscoring the relevance of checkpoint genes in shaping tumor behavior. Although only one eligible study on CTLA4 polymorphism met the predefined inclusion criteria, its findings align with the broader concept that immune checkpoint variation may influence breast cancer susceptibility and immune modulation.
Collectively, the evidence summarized in this axis suggests that germline variation in immune checkpoint and chemokine pathways is more frequently associated with progression-related features than with initial disease susceptibility. Variants affecting immune suppression and immune cell trafficking appear to modulate tumor-immune interactions by shaping immune evasion, local immune exclusion, and subtype-specific clinical behavior. Significantly, the relatively consistent direction of associations observed for these pathways contrasts with the heterogeneity seen in inflammatory cytokine genes, supporting a model in which inherited differences in immune inhibitory signaling contribute to later stages of tumor evolution and aggressiveness rather than tumor initiation.
A third interpretative axis emerging from the reviewed literature concerns genes directly involved in immune effector mechanisms, including antigen presentation, innate immune sensing, and cytotoxic lymphocyte function. Despite their central biological role in antitumor immunity, most variants investigated in these pathways did not show significant or consistent associations with breast cancer risk, progression, or clinical outcomes across the included studies.
Notably, several studies evaluated polymorphisms in genes related to immune activation and effector responses, including pathways involved in antigen processing, lymphocyte activation, and innate immune signaling; however, these variants largely failed to demonstrate reproducible associations across populations or clinical subgroups (Table S1). This pattern contrasts with the more consistent associations observed for genes involved in inflammatory regulation and immune suppression, suggesting a differential contribution of germline variation across immune functional layers.
Rather than representing inconclusive or negative findings, this recurrent lack of association supports a functional distinction between immune regulation and immune execution. While germline variation appears to be associated with differences in basal immune tone and regulatory thresholds, effective immune effector responses are highly plastic and context-dependent, relying on dynamic signals derived from cytokine exposure, metabolic constraints, and chronic antigenic stimulation within the tumor microenvironment [56, 57]. In this context, Fcγ receptor (FCGR) genes constitute a biologically plausible effector layer because they mediate IgG-dependent mechanisms, including antibody-dependent cellular cytotoxicity (ADCC) and immune complex handling. However, FCGR variants were not represented among the observational germline association studies included in our synthesis of breast cancer risk/prognosis and clinicopathological features; thus, they could not be evaluated within our evidence base. Notably, FCGR polymorphisms have been more commonly examined in treatment-specific (predictive) settings, particularly in the context of anti-HER2 monoclonal antibody therapy, where IgG-Fc gamma receptor interactions contribute to immune effector engagement [58].
This interpretation aligns with current models of cancer immunoediting, which propose that immune escape and effector dysfunction arise predominantly through acquired mechanisms, including somatic alterations, epigenetic reprogramming, and tumor-driven immune remodeling, rather than inherited defects in immune effector machinery [59]. Consequently, the limited contribution of germline variation in immune effector genes observed in this review supports a hierarchical model in which inherited genetic variation preferentially shapes immune regulation and tolerance. In contrast, immune effector function is governed mainly by somatic and microenvironmental factors during breast cancer evolution [60, 61].
Collectively, the findings of this integrative review suggest a possible conceptual framework in which germline immune variation may differentially contribute to breast cancer susceptibility, immune regulation, and clinical behavior across functional immune pathways. Rather than exerting uniform effects, immune-related genetic variants appear to cluster into interpretative axes that modulate basal immune tone, immune suppression and tolerance, and tumor-immune interactions. This framework integrates heterogeneous genetic associations into a pathway-oriented model that emphasizes immune regulation as a dynamic continuum, as summarized in Figure 2.

Conceptual framework integrating germline immune variation across functional immune pathways in breast cancer. Germline immune-related variants identified in the reviewed studies cluster into three interpretative axes: modulation of basal immune tone (primarily cytokine-related genes), immune suppression and tolerance (including checkpoint and chemokine-related pathways), and tumor-immune interaction. The framework highlights the differential contribution of germline immune variation to breast cancer susceptibility and tumor progression, while suggesting a limited influence of germline factors on immune effector function.
A global evaluation of the included studies reveals substantial heterogeneity in methods and populations. Most investigations were conducted in Asian and Latin American cohorts, with limited representation of African and European populations, restricting the generalizability of the findings. Divergent findings across populations may reflect differences in minor allele frequencies, linkage disequilibrium structures, and haplotypic backgrounds, which can modify the functional context of specific variants. In addition, variability in environmental exposures, inflammatory burden, lifestyle factors, and control of population stratification may further influence observed associations. Methodological heterogeneity, including genotyping platforms, sample size, and statistical modeling approaches, likely also contributes to inconsistent results across cohorts. Additional immune-related genes (including CTLA-4) may have been investigated in breast cancer; however, the set of genes included in this review reflects studies retrieved through the predefined search strategy, and that met the eligibility criteria. In addition, small sample sizes and recurrent deviations from the Hardy-Weinberg equilibrium reduce the robustness of several reported associations. Collectively, these factors warrant caution in interpreting individual results and preclude broad generalizations.
Although several associations identified in this review are biologically plausible within known immune pathways, direct functional validation remains limited for many of the investigated variants. Evidence such as expression quantitative trait locus analyses, experimental assays, or consistent replication across independent populations is not uniformly available. Therefore, the mechanistic interpretations discussed herein should be regarded as hypothesis-generating rather than confirmatory, underscoring the need for integrative genomic, transcriptomic, and functional studies in large, multiethnic cohorts.
Despite these limitations, an integrative analysis of the available evidence supports a coherent conceptual framework in which germline immune variation exerts differential effects across immune pathways. Variants in inflammatory and immunoregulatory cytokine genes were more frequently associated with breast cancer susceptibility, suggesting a role in shaping basal immune tones during early tumor-immune interactions. In contrast, variants in immune checkpoint and chemokine pathways were more consistently associated with tumor progression, immune evasion, and aggressive clinical features, suggesting a greater association with later disease stages. Conversely, genes directly involved in immune effector execution rarely showed significant germline associations, suggesting a limited contribution of inherited variation to these pathways.
This hierarchical pattern suggests a potential functional distinction between germline modulation of immune regulation and acquired control of immune effector function in breast cancer. Rather than acting as isolated determinants, immune-related germline variants appear to contribute cumulatively and context-dependently by shaping regulatory thresholds and immune permissiveness. Future studies integrating germline genetics with tumor genomics, transcriptomics, and functional immune profiling in large, multiethnic cohorts will be essential to validate these observations and to clarify how inherited immune variation influences disease trajectory and therapeutic responsiveness. In addition, although treatment response and resistance were not primary foci of this review, germline immune variants may modulate outcomes in antibody-driven settings through FCGR-mediated ADCC; however, available pharmacogenetic evidence for FCGR polymorphisms remains inconsistent and context-dependent [62].
Importantly, the associations summarized in this review reflect statistical correlations observed in heterogeneous populations and should not be interpreted as evidence of direct biological causation. Rather, they provide a conceptual framework for future integrative and functional investigations.
In summary, current evidence suggests that germline variation in immune-related genes may contribute to breast cancer susceptibility and disease progression through differential effects across immune pathways. Variants affecting inflammatory and immunoregulatory signaling appear more frequently associated with susceptibility. In contrast, polymorphisms in immune checkpoint and chemokine pathways are more often linked to tumor progression and aggressive clinical features. Although the available evidence remains heterogeneous and largely associative, these findings support a conceptual framework in which inherited immune variation modulates the regulatory landscape of tumor-immune interactions. Future large-scale, multiethnic studies integrating germline genetics with tumor genomics, transcriptomics, and functional immune profiling will be essential to clarify the biological relevance and potential clinical implications of these associations.
ADCC: antibody-dependent cellular cytotoxicity
FCGR: Fcγ receptor
HER2: human epidermal growth factor receptor 2
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/1003245_sup_1.pdf. The supplementary table for this article is available at: https://www.explorationpub.com/uploads/Article/file/1003245_sup_2.xlsx.
JFV: Conceptualization, Formal analysis, Methodology, Resources, Writing—original draft, Writing—review & editing. MPA: Formal analysis, Methodology, Resources, Writing—original draft, Writing—review & editing. JHV: Formal analysis, Methodology, Resources, Writing—review & editing. All authors read and approved the submitted version.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The datasets that support the findings of this study are available from the corresponding author upon reasonable request.
Not applicable.
© The Author(s) 2026.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2026. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 860
Download: 20
Times Cited: 0
