 Review
Review

Affiliation:
1Student Research Committee, Tabriz University of Medical Sciences, Tabriz 5166614766, Iran
2Department of Immunology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz 5166614766, Iran
3Immunology Research Center, Tabriz University of Medical Sciences, Tabriz 5166614766, Iran
ORCID: https://orcid.org/0009-0001-2334-9294
Affiliation:
4Department of Clinical Biochemistry, Faculty of Medicine, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
ORCID: https://orcid.org/0000-0003-2589-2191
Affiliation:
2Department of Immunology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz 5166614766, Iran
3Immunology Research Center, Tabriz University of Medical Sciences, Tabriz 5166614766, Iran
ORCID: https://orcid.org/0000-0002-8583-1270
Affiliation:
5Department of Immunology, School of Medicine, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
6Cancer Research Center, Institute of Cancer, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
Email: gh.solgi@umsha.ac.ir
ORCID: https://orcid.org/0000-0001-8929-5658
Explor Immunol. 2025;5:1003229 DOI: https://doi.org/10.37349/ei.2025.1003229
Received: March 16, 2025 Accepted: October 20, 2025 Published: November 23, 2025
Academic Editor: Apostolos P. Georgopoulos, Minneapolis Veterans Affairs Medical Center, USA
The article belongs to the special issue Immunogenetics of Chronic Illnesses
Killer immunoglobulin-like receptors (KIRs) and human leukocyte antigen (HLA) molecules play an essential role in regulating immune responses against hepatitis B virus (HBV) and hepatitis C virus (HCV) infections. HLA-KIRs interactions are crucial for activating and inhibiting the natural killer (NK) cell system through a modulation that shapes these cells to kill infected cells and release cytokines. Regulation underlies the anti-viral function of the NK cell and profoundly affects viral clearance, immune evasion, and the course of disease. Activating KIRs such as KIR2DS1 and KIR3DS1 cooperate with specific HLA ligands in boosting NK cell responses against the virus, thereby facilitating viral elimination. In contrast, inhibitory KIRs like KIR2DL1 and KIR3DL1 bind to HLA-C2 and HLA-Bw4, respectively, imposing a dampening influence on NK cell activation, which allows the virus to persist and progress to chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC). These variations in KIRs and HLA genes will also affect an individual’s susceptibility to infections, disease severity, and their response to antiviral therapies. Observation of the role of KIRs and their interaction with HLA at the immunogenetic level provides valuable insight into host-virus dynamics and opens up many therapeutic avenues. Targeting immunotherapies toward NK cell pathways and developing personalized medicine may boost antiviral immune responses and improve treatment outcomes in chronic viral hepatitis patients. This review recognizes HLA-KIRs interactions as potent biomarkers for disease progression and determining treatment strategies.
Hepatitis has been defined as an inflammatory disease of the liver tissue [1]. Several conditions lead to this disease, including autoimmune liver disease, excessive alcohol consumption, hepatitis B virus (HBV), and hepatitis C virus (HCV) [2]. Among these, HBV and HCV are recognized as the leading causes of viral hepatitis. HBV, which belongs to the Hepadnaviridae family, is a double-stranded DNA virus with an envelope. In contrast, HCV is classified under the Flaviviridae family and is an enveloped single-stranded RNA virus [1]. As much as a very effective vaccination is available for HBV, around 260 million people live with the chronic form of HBV infection, and about 1% of the world population is infected with HCV, confirming an enormous public health dilemma caused by these pathogens [1, 3]. Liver cirrhosis and hepatocellular carcinoma (HCC) are among the most common chronic diseases resulting from the progression of HBV and HCV infections worldwide [1, 4]. About 80% of individuals with HCV infection are predisposed to develop chronic liver disease, of whom up to 40% further progress to cirrhosis, and about 3% to HCC after several decades of chronic infection [1, 4]. HBV infections present symptoms that can range from asymptomatic or subclinical conditions, along with acute or chronic hepatitis. Alarmingly, the yearly mortality resulting from complications associated with chronic HBV infection that progresses to cirrhosis or HCC is approximated to be 900,000 cases [1, 5].
The immunopathogenesis of HBV and HCV infections involves complex interactions between the viruses and the host immune system. In this evolving relationship, immune responses play a critical dual role: they are required to control viral replication, which, alternatively, may mediate liver damage [6–8]. Both HBV and HCV are typically considered non-cytopathic viruses, and liver inflammation, as well as the potential for viral clearance and recovery, result from a cooperation of innate and adaptive immune responses [9, 10]. Within HBV infection, most of the liver damage can be attributed to the immune response itself, with HBV possessing distinct limitations with regard to cytopathic effects on hepatocytes [1, 6]. Chronic activation of inflammatory pathways and sustained infiltration of lymphocytes into the liver can trigger fibrosis, cirrhosis, and ultimately HCC [1, 4, 11]. Following being infected with HCV, persistent inflammatory responses activated by lymphocytic infiltration into the liver and excessive production of pro-inflammatory cytokines further aggravate the progression of fibrosis and cirrhosis [9, 12]. The situation is further complicated by the release of reactive oxygen species (ROS) and inflammatory cytokines from immune cells, which is detrimental to liver injury. Collectively, these create a microenvironment in favor of HCC progression [9, 12].
HBV evades innate immune recognition by inducing minimal activation of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and retinoic acid-inducible gene-I (RIG-I)-like receptors, thereby dampening type I interferon (IFN-I) and proinflammatory cytokine production and facilitating viral persistence [6, 8, 9, 12]. In contrast, HCV actively disrupts innate signaling by encoding proteins, such as NS3/4A, that degrade key mediators of the mitochondrial antiviral-signaling (MAVS) and TIR-domain-containing adapter-inducing IFN-β (TRIF) pathways [10, 13], resulting in the suppression of IFN-I responses [14]. Within this immunological context, natural killer (NK) cells constitute a pivotal component of the host defense, contributing to viral clearance or persistence through the secretion of cytokines, particularly IFN-γ [12, 15] which activate hepatic Kupffer cells and CD4⁺ T lymphocytes, as well as through the direct cytotoxic elimination of hepatocytes in concert with cytotoxic T lymphocytes (CTLs) [15]. The functional outcome of NK cell activity is critically shaped by interactions between killer immunoglobulin-like receptors (KIRs) and their human leukocyte antigen (HLA) ligands (Figure 1), which collectively influence the balance between protective immunity, immunopathogenesis, and therapeutic responsiveness in viral hepatitis; in parallel, natural cytotoxicity receptors (NCRs) further amplify NK cell activation and antiviral defense, while their dysregulation may contribute to hepatic injury [6, 15].
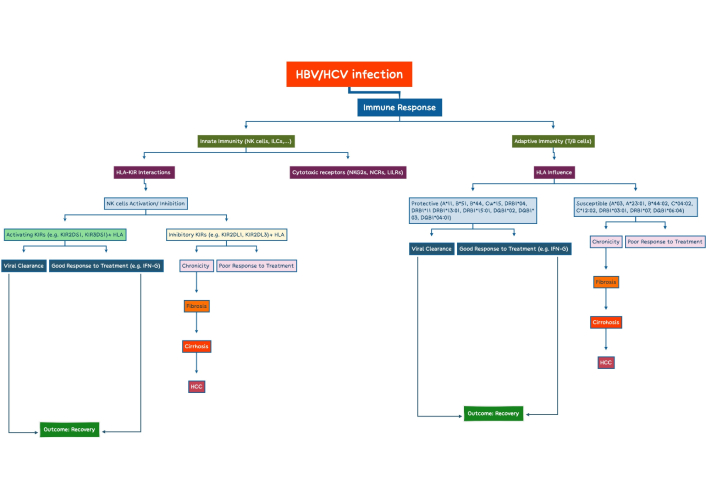
Flowchart of the immune response pathways in HBVs and HCVs: the role of HLA-KIRs interactions and treatment outcomes. HBV: hepatitis B virus; HCV: hepatitis C virus; HLA: human leukocyte antigen; KIRs: killer immunoglobulin-like receptors; NK: natural killer; HCC: hepatocellular carcinoma. This figure was created with XMind.
B cells play a central role in the immune response against HBV, via producing antibodies against hepatitis B surface antigen (HBsAg), hepatitis B core antigen (HBcAg), and hepatitis B e antigen (HBeAg) [12]. HBcAg-specific B cells are more abundant and functional, whereas HBsAg-specific B cells are fewer and less effective, with distinct mRNA profiles linked to antigen presentation and innate immunity [12, 16]. IL-27 can partially restore HBsAg-specific antibody production by promoting plasmablast and plasma cell differentiation via B lymphocyte induced maturation protein-1 (BLIMP-1) induction [12]. Anti-HBc IgG is present in past, active, and occult infections, while anti-HBc IgM indicates acute infection or severe chronic hepatitis B (CHB) flares [12]. Early anti-HBe appearance reflects a better prognosis, but robust HBsAg-specific humoral responses are essential for viral control. In CHB, memory B cells display elevated programmed cell death protein-1 (PD-1) expression; their functional capacity can be restored through PD-1 blockade combined with IL-2, IL-21, and CD40L stimulation, underscoring the potential of targeted immunomodulation strategies [12, 16, 17]. T follicular helper (TFH) cell-derived IL-21 deficiency limits B cell responses, whereas Treg-derived IL-27 can enhance antibody production [12]. Impaired memory B cell maturation into anti-HBs-secreting plasma cells contributes to HBV persistence. Elevated B-cell activating factor (BAFF) in CHB is linked to B cell hyperactivation, cirrhosis, and HCC [12]. Although early studies suggested that antibodies against HCV could protect chimpanzees from homologous viral strains, their significance in human infection was often underestimated. Evidence indicates viral clearance in agammaglobulinemic patients, the presence of non-neutralizing autoantibodies (nnAbs) such as rheumatoid factor, and weak correlations between standard serological antibodies and infection outcomes [7, 18]. Active neutralizing antibodies (nAbs) primarily target hypervariable region 1 (HVR1) of E1, but their effectiveness is often limited by rapid viral mutation and the use of surrogate viral strains in earlier studies [7]. Recent work using autologous viral sequences has highlighted the critical role of early nAb responses in viral clearance, particularly through broadly nAbs (bnAbs) [7]. Unlike strain-specific nAbs, bnAbs can target multiple HCV genotypes and effectively control infection in various models [7, 19]. These bnAbs recognize key structural epitopes on the E1E2 envelope protein, including antigenic regions AR1–AR5 and the CD81 receptor binding site, as defined by alanine-scanning studies [7, 18]. Their importance is supported by both natural infection and vaccination studies [7]. However, the mechanisms underlying bnAb production in most patients remain unclear, possibly reflecting B cell deficiencies or suboptimal TFH support [7, 18].
CD4⁺ T helper cells are central to the immune response, promoting the activation of CD8⁺ T cells and B lymphocytes [9]. Robust, multifunctional CD4⁺ T-cell responses are associated with clearance of HBV and HCV infections, whereas weak responses contribute to chronicity [5, 9]. CD8⁺ T cells play a critical role in HBV control by eliminating infected hepatocytes and secreting antiviral cytokines, including IFN-γ and tumor necrosis factor-α (TNF-α) [6, 15], but their overactivation can induce liver injury and fibrosis [15]. Similarly, HCV-specific CD8⁺ T cells are essential for viral control but often become exhausted during chronic infection, exhibiting upregulation of inhibitory receptors such as PD-1 and reduced effector functions [7, 10, 15].
Antigen-specific CD8⁺ and CD4⁺ T cells are activated upon recognition of viral peptides presented by HLA class I and II molecules (HLA-I, HLA-II), respectively [20]. Polymorphisms within HLA genes critically shape both the magnitude and quality of immune responses to hepatitis infections [21]. Studies have identified specific HLA alleles that influence the outcomes of viral hepatitis (Table 1). For example, DRB1*13:02 and A*03:01 are associated with viral clearance, whereas B*08, B*44, and the DRB1*11:02~DQA1*05:01~DQB1*03:01 haplotype predispose to CHB infection [22–24]. In the Iranian population, DRB1*03:01, DQA1*05:01, and DQB1*06:04 have been linked to increased susceptibility to CHB, while DRB1*15:01, DQB1*04:01, and DRB1*13:01 appear to be protective [25]. The implications of these associations extend beyond genetic predisposition, underscoring the complex interplay between viral infections and autoimmune responses. Notably, DRB1*13:02 has been implicated in protection against persistent HBV infection [26]. At the molecular level, HLA-II molecules contain nine binding pockets (P1–P9) within the peptide-binding groove that interact with antigenic peptides through non-covalent forces, including hydrogen and electrostatic bonds [27]. Importantly, DRB1*13:01 and DRB1*13:02 differ by a single amino acid at position β86 in pocket P9: valine in DRB1*13:01 vs. glycine in DRB1*13:02 [27, 28]. The presence of glycine generates a more open, less hydrophobic pocket, thereby permitting greater flexibility and diversity in peptide accommodation. Conversely, valine introduces bulk and hydrophobicity, narrowing the pocket and tightening peptide binding. These structural differences substantially influence peptide repertoires and the stability of HLA-peptide-T cell receptor (TCR) interactions, ultimately shaping T-cell responses. Specifically, DRB1*13:02 may present a broader and more immunogenic repertoire of HBV-derived peptides, enhance T-cell activation, and facilitate viral clearance [28]. In contrast, the restricted peptide-presenting capacity of DRB1*13:01 may limit effective antiviral responses, thereby predisposing carriers to viral persistence and autoimmune sequelae such as autoimmune hepatitis (AIH) [28, 29].
Additionally, Corghi et al. [30] reported an association between the DRB1*07 allele and the chronicity of HCV infection in Brazilian patients. A study conducted by Ursu et al. [31] on Romanian patients indicated that A*23:01, B*44:02, and C*04:02 alleles are linked to an increased genetic predisposition to chronic hepatitis due to HCV. Furthermore, they showed that the presence of C*12:02, A*03, and A*11 alleles is associated with the induction of fibrosis grading F3–F4 [31]. The patterns of HLA alleles and haplotypes not only influence the quality of the specific immune response to HBV and HCV infections but also significantly affect treatment response and viral clearance (Table 1). The impact of allelic and haplotypic diversity on treatment outcomes in patients with chronic hepatitis C (CHC) has been well documented [32]. The study involving patients in Taiwan, China, found a direct relationship between the presence of A*11, B*51, Cw*15, and DRB1*15 alleles and a favorable response to IFN-α treatment [33]. Also, Romero-Gómez et al. [34] study demonstrated that the presence of the HLA-B*44 was significantly associated with a sustained response to IFN and ribavirin combined therapy in CHC patients.
Impact of HLA alleles and haplotypes on HBV and HCV outcomes: a global view.
| References | Year | Population | Findings |
|---|---|---|---|
| Thio et al. [23] | 1999 | African American cohort | HLA-II alleles → pathogenesis of HBV infection |
| Malhotra et al. [22] | 2001 | American patients | DRB1*13~DQB1*06 haplotype → improved outcomes with early viral treatment |
| Thio et al. [24] | 2003 | Caucasian individuals | HLA-I molecules influence the outcome of HBV infection and may provide insights into HBV pathogenesis |
| Yu et al. [33] | 2003 | Patients in Taiwan, China | A direct relationship between the presence of A*11, B*51, Cw*15, and DRB1*15 alleles and a favorable response to IFN-α therapy DRB1*15~DQB1*05 haplotype was associated with response to IFN-α and A*11~DRB1*15 haplotype was strongly associated with sustained response |
| Romero-Gómez et al. [34] | 2003 | Spanish patients | HLA-B*44—associated with sustained response to IFN-ribavirin in CHC patients |
| Corghi et al. [30] | 2008 | Brazilian patients | An association between the DRB1*07 allele and the chronicity of HCV infection |
| Gauthiez et al. [35] | 2017 | -- | An association between DQB1*02, DQB1*03, DRB1*04 and DRB1*11 with spontaneous HCV clearance |
| Ursu et al. [31] | 2021 | Romanian patients | A*23:01, B*44:02, and C*04:02 alleles are linked to an increased genetic predisposition to chronic hepatitis due to HCVThe presence of C*12:02, A*03, and A*11 alleles is associated with the induction of fibrosis grading F3–F4 |
| Ursu et al. [31] | 2021 | Romanian patients | |
| Naderi et al. [25] | 2023 | Iranian population | DRB1*03:01, DQA1*05:01, DQB1*06:04—associated with increased susceptibility to CHBDRB1*15:01, DQB1*04:01, DRB1*13:01—protective against CHB |
| Tălăngescu et al. [36] | 2024 | Romanian population | Heterozygosity of HLA-DQB1 and HLA-DRB1 was significantly associated with a lower risk of HBV infection |
CHB: chronic hepatitis B; CHC: chronic hepatitis C; HBV: hepatitis B virus; HCV: hepatitis C virus; HLA: human leukocyte antigen; IFN: interferon.
The regulation of HLA expression in viral hepatitis epitomizes the delicate balance between effective antiviral immunity and viral immune evasion. In both HBV and HCV infections, HLA-I molecules are frequently upregulated on hepatocytes, thereby enhancing cytotoxic T cell-mediated surveillance (Table 2), but concurrently increasing susceptibility to immune-mediated hepatic injury [37]. In contrast, viral proteins such as HBV X and the HCV core have been shown to suppress HLA-I surface expression, facilitating immune escape and promoting viral persistence [38]. Aberrant induction of HLA-II molecules on hepatocytes and cholangiocytes, cell types that normally lack such expression, has also been documented and may contribute to chronic inflammation and autoimmune-like manifestations [39] (Table 2). Notably, the extent and direction of HLA modulation appears to differ according to viral genotype, host genetic background, and therapeutic context, underscoring its dual role as both a mediator of protective immunity and a driver of immunopathogenesis [40].
Comparative analysis of HLA expression in HBV and HCV infections.
| Features | HBV | HCV | References |
|---|---|---|---|
| HLA-I expression | Upregulation of hepatocytes enhances CD8+ T cell recognition but promotes immune-mediated liver injury | Consistent upregulation in hepatocytes supports sustained immune surveillance | [41] |
| Viral protein interference | HBV X protein downregulates HLA-I, enabling viral escape and persistence | HCV core, NS3, and NS5A proteins interfere with antigen processing, reducing peptide loading | [38] |
| Regulation pattern | Bidirectional: upregulation vs. suppression depending on stage and immune context | Predominantly upregulation with indirect suppression via antigen-processing disruption | [42] |
| HLA-II expression | Aberrant induction of hepatocytes and cholangiocytes, associated with chronic inflammation and autoimmune-like pathology | Similar aberrant induction fuels intrahepatic inflammation and autoimmune-like injury | [43] |
| Immune evasion strategy | Direct suppression of surface HLA expression by viral proteins | Disruption of antigen processing machinery, broader upstream dysregulation | [44] |
| Clinical implications | Balance between viral clearance and immune-mediated injury; linked to fibrosis progression | Persistent immune activation but dampened cytotoxicity; linked to chronicity and HCC risk | [45] |
HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; HLA: human leukocyte antigen.
NK cells are derived from the lymphoid lineage; however, unlike B and T cells, they have limited specificity, diversity, and memory [46]. These cells are part of the innate immune system and play crucial roles in the initial control of viral infections, enhancing innate immune responses, and guiding and assisting in the activation of acquired immunity [46, 47]. Some NK cells express the very late activation antigen-4 (VLA-4) molecule, allowing them to bind to vascular cell adhesion molecule-1 (VCAM-1) present on the surface of endothelial cells [46, 48]. This interaction plays a crucial role in facilitating the migration of NK cells to various tissues, including the liver, lungs, and the decidua layer of the uterus [49]. In these tissues, NK cells express CD69, an early activation marker expressed on the surface of various immune cells, including NK cells, upon activation. CD69 plays a key role in tissue residency by promoting retention of NK cells within specific organs, such as the liver, lungs, and decidua layer of the uterus. This expression reflects that NK cells are not only present in these tissues but are also functionally engaged in local immune surveillance and antiviral defense [49]. NK cells residing in lymphoid tissues (such as tonsils and lymph nodes), skin, gut, and the decidua primarily consist of NK-CD56bright cells, whereas NK-CD56dim cells are more abundant in the lung and liver [46, 50]. The activation or inhibition of NK cells is dependent on the balance of signals received from receptors on their surface. If target cells, such as HBV- or HCV-infected hepatocytes, express ligands that bind to the activating receptors of NK cells while displaying few or no inhibitory ligands, the NK cell becomes activated and triggers the death of the target cells [46, 51, 52]. This cytotoxic effect is mediated either through the release of perforin and granzymes, where perforin forms transmembrane pores allowing granzymes to enter and initiate caspase-dependent apoptosis, or via receptor-ligand interactions such as Fas-Fas ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL)-TRAIL receptor (TRAILR) signaling pathways, which also induce PD-1 in the infected hepatocytes [46, 51–53]. Conversely, if the target cells have inhibitory ligands and exhibit minimal or absent activating ligands, the NK cell recognizes them as healthy and refrains from mounting an attack [52]. In an effort to escape death by CD8+ T cells, virus-infected and cancerous hepatocytes reduce the expression of HLA-I molecules on their surface [47]. In such situations, these cells become more susceptible to killing by NK cells, as the presence of HLA-I molecules serves as an indicator of the target cell’s health [52]. The differentiation between healthy and abnormal cells (recognition of missing self-cells, RMSCs) by NK cells is recognized as immune surveillance, which plays a critical role in the immune response to virus-infected and neoplastic hepatocytes [54, 55]. In addition to signaling through NK cell surface receptors, cytokines such as IL-12, IL-15, IL-18, and IFN-I produced by dendritic cells and macrophages bind to their receptors on NK cells, thereby promoting their growth and activation [55]. Upon activation, NK cells induce the death of virus-infected and cancerous hepatocytes through the exocytosis of granules containing perforin and granzymes [56]. Furthermore, NK cells contribute to the immune response by secreting IFN-γ, which activates macrophages and aids in the differentiation of Th cells and naive CTLs into Th1 cells and effector CTLs.
There are two groups of receptors on the surface of NK cells; some provide activating signals [NKG2C/E/D, NKP30/44/46, leukocyte immunoglobulin-like receptor (LILR) A1/2/4/5/6, KIR2DS1/2, KIR3DS1], while others deliver inhibitory signals (NKG2A/B, LILRA3, LILRB1–5, KIR2DL1/2/3, KIR3DL1) [48, 57]. The net outcome of signals received from these receptors determines the activation fate of NK cells.
NKG2 receptors, members of the C-type lectin family, are expressed on NK cells and CD8⁺ T cells and mediate both activating and inhibitory signals depending on the receptor subtype [57–62]. NKG2A/CD94 and NKG2B/CD94 heterodimers are inhibitory, whereas other NKG2 receptors, including the NKG2D homodimer, are activating [57–62]. NKG2A–C and NKG2E recognize HLA-E, whose binding affinity varies by receptor, allowing it to function as both an inhibitory and activating ligand [57, 63]. HLA-E presents hydrophobic leader peptides from classical HLA-I molecules, and its two major alleles, HLA-E*01:01 and HLA-E*01:03, differ at codon 107, affecting membrane expression and peptide presentation [64–66]. HLA-E*01:01 has been linked to increased HBV susceptibility [67], while the HLA-E*01:03 G/G genotype may confer protection by presenting virus-derived peptides to NK cells, enhancing lysis of infected hepatocytes [66, 68–70]. Elevated soluble HLA-E (sHLA-E) in CHB patients contributes to immune tolerance, inversely correlating with HBV DNA levels and modulating the timing of antiviral responses [66]. NKG2D recognizes stress-inducible HLA-I-like molecules, such as MHC class I polypeptide-related sequence A/B (MICA, MICB) and UL16-binding protein (ULBP). In CHB, exogenous HBsAg upregulates NKG2D on NK cells, promoting antiviral activity against HCV in co-infected patients [60]. MICA polymorphisms further modulate viral outcomes; MICA*015 is associated with enhanced HCV clearance but increased CHB susceptibility [71], while the promoter single nucleotide polymorphism (SNP) rs2596542 (TT genotype) confers higher HCC risk in HBV/HCV infection, likely via altered NKG2D binding and immune activation [71]. Collectively, these findings underscore the crucial role of NKG2-HLA interactions in shaping antiviral immunity, viral persistence, and disease progression in chronic hepatitis.
NCRs, including NKp30, NKp44, and NKp46, contribute to the cytotoxicity of NK cells [72]. NCRs mediate direct recognition of pathogen-associated ligands or stress-induced cellular alterations. Beyond this receptor-ligand interaction, the repertoire of peptides derived from viruses or infected cells [73], together with polymorphism in HLA-I molecules, most notably HLA-C, and the inherited distribution of activating and inhibitory KIR genes, critically determine the threshold and magnitude of NK cell responses [74]. NKp30 and NKp46 are constitutively expressed, whereas NKp44 is induced following cytokine stimulation [75]. In viral hepatitis, NKp44 demonstrates a dual functionality; upon engagement with stress-induced or viral ligands on hepatocytes, it amplifies NK cell cytotoxicity and cytokine secretion, yet in certain contexts it transmits inhibitory signals that dampen NK activity [75–77]. This bidirectional capacity positions NKp44 as a pivotal immunoregulatory checkpoint, orchestrating the balance between effective antiviral defense and the risk of immune-mediated liver injury, thereby influencing disease persistence and progression. NKp46, together with NKG2D, drives cytotoxicity in resting NK cells and is highly expressed on both NK cells and HBV-infected hepatocytes, facilitating viral clearance [62, 77]. Notably, an NKp46highNKG2Ahigh subset exhibits strong cytotoxicity but reduced IFN-γ production, correlating with liver injury, HBV replication, and elevated serum alanine transaminase (ALT) and HBV DNA [62]. In the context of CHB infection, NKp30 expression is markedly diminished and exhibits an inverse correlation with viral load, underscoring its contribution to antiviral immune surveillance and the containment of viral replication. In contrast, studies in HCV infection have paradoxically associated reduced NKp30 expression with spontaneous viral clearance, suggesting that the immunoregulatory consequences of NCR downregulation are pathogen-specific and context-dependent [78–80]. Furthermore, studies indicate that sex hormones exert additional modulatory effects on NKp30 expression, with consistently higher levels observed in males, cyclical fluctuations reported across the female menstrual cycle, and significant therapy-induced downregulation documented in female patients receiving antiviral treatment [81, 82]. Collectively, these observations highlight the intricate interplay between viral context, host sex-specific biology, and therapeutic interventions in shaping the functional impact of NCR expression on disease trajectory.
LILRs are NK cell receptors that primarily interact with non-classical HLA-I molecules [83]. Encoded within the leukocyte receptor cluster (LRC) on chromosome 19, this family comprises 11 members, each containing two to four extracellular immunoglobulin-like domains with either inhibitory or activating functions [84, 85]. The inhibitory receptors LILRB1–5 and activating receptors LILRA1–6 (except LILRA3) play distinct roles in modulating NK cell activity [84, 85]. LILRB1, with four immunoglobulin-like domains, engages a broad spectrum of ligands, including HLA molecules and UL18, a cytomegalovirus HLA-I homolog [86], whereas LILRB2 selectively binds HLA-G alongside classical and non-classical HLA-I molecules [83, 86]. Functionally, Zhang et al. [87] reported that LILRB1 expression is markedly elevated in circulating CD56dimCD16bright NK cells from patients with active hepatitis compared to inactive carriers and healthy controls. This upregulation impairs NK cell functionality and promotes apoptosis, suggesting a critical role for LILRB1-mediated inhibition in the pathogenesis of CHB [87].
The most important receptors on NK cells are the KIRs, which can deliver either activating or inhibitory signals [52] (Figure 2). These receptors can be broadly categorized as either inhibitory or activating, depending on the structural configuration of their intracellular domains. Inhibitory KIRs contain long cytoplasmic tails (KIR**L*) harboring immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which recruit phosphatases to dampen NK cell activation. In contrast, activating KIRs are defined by short cytoplasmic tails (KIR**S*) that lack signaling capacity on their own but associate with adaptor molecules bearing immunoreceptor tyrosine-based activation motifs (ITAMs) to initiate downstream activating pathways [88]. KIRs are encoded by the LRC region on chromosome 19 [89, 90]. Similar to HLA genes, KIRs are highly polymorphic, with allele distributions varying across populations and ethnic groups, and are inherited as two haplotypes, A and B [89, 90]. Haplotype A primarily contains genes encoding inhibitory receptors, and haplotype B predominantly consists of genes encoding activating receptors [90] (Figure 2). Polymorphism in KIRs, such as KIR2DL2 variants, influences the binding affinities of the receptors to their ligands, which affects the activation or inhibition of NK cells. Hence, inheritance of different KIR genes could contribute to an increased or decreased susceptibility to autoimmune diseases and assist in the clearance of hepatitis viruses [89, 91, 92]. KIRs interact with classical and non-classical HLA-I molecules through their extracellular immunoglobulin-like domains, thus regulating the activation or inhibition of NK cells [93, 94]. The most principal ligands for KIR receptors are classified into four groups according to the amino acid sequence of the KIR-binding epitopes in HLA molecules, as curated in the IPD-IMGT/HLA database (https://www.ebi.ac.uk/ipd/imgt/hla); 1. C1 alleles group (C*01, C*03, C*07, C*08, C*12, C*14 and C*16), 2. C2 alleles group (C*02, C*04, C*05, C*06, C*15, C*17 and C*18). 3. Bw4 alleles group (B*13, B*27, B*37, B*38, B*39, B*40, B*41, B*42, B*44, B*45, B*47, B*48, B*49, B*51, B*52, B*53, B*57, B*58, B*59). 4. Bw6 alleles group (B*07, B*08, B*15, B*18, B*35, B*40, B*42, B*44, B*46, B*47, B*48, B*50, B*54, B*55, B*56, B*62, B*63, B*67, B*73, B*78, B*81).
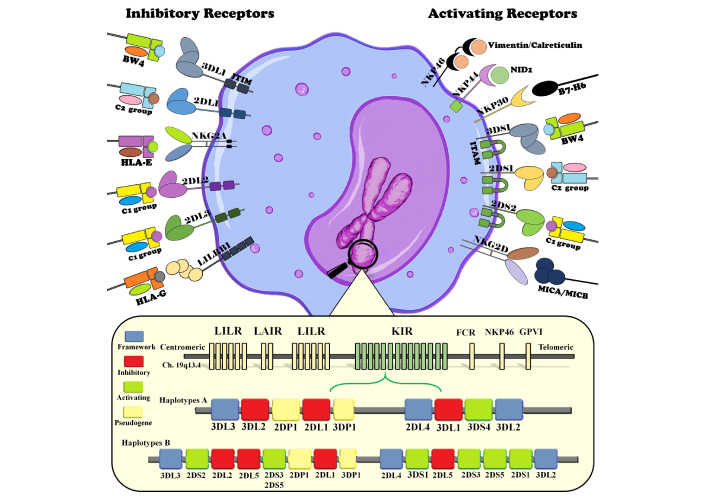
Composition of inhibitory and activating receptors on the surface of NK cells and their specific ligands on the surface of target cells. Also, the bottom of the figure illustrates the chromosomal arrangement of the leukocyte receptor cluster (LRC) on chromosome 19, with centromeric and telomeric regions labeled. Key polymorphic sites and gene content variations are highlighted, demonstrating how haplotype inheritance influences NK cell activation thresholds, susceptibility to viral hepatitis, and disease outcomes. NK: natural killer.
The genetic organization of KIRs into A and B haplotypes has profound implications for the immune response against viral infections. Lu et al. [95] demonstrated that in patients with HBV infection, the frequency of haplotype A, which is largely inhibitory in composition, is reduced, while haplotype B, which carries a greater number of activating receptors, is increased relative to healthy individuals. This skewing suggests that haplotype B may provide a selective advantage in the context of HBV infection by promoting more robust NK cell activation and antiviral responses, whereas haplotype A may be less effective due to its inhibitory bias. Also, Ursu et al. [31, 96] further implicated that multiple KIR alleles (KIR2DL3, KIR2DL5, KIR3DL3, KIR2DP1, KIR3DP1, KIR2DS4) are associated with CHC susceptibility, and the presence of KIR2DL2 is related to elevated post-treatment aspartate transferase (AST) and bilirubin levels. Such observations underscore the importance of KIR haplotypic and allelic diversity in shaping host susceptibility or resistance to viral pathogens.
Structural studies have identified key amino acid residues that govern HLA-KIR interactions. Residues 80 of the HLA molecule and residue 44 of KIRs are critical contact points, mediating recognition through hydrogen and ionic bonds [94] (Figure 3). Beyond these conserved contacts, fine specificity is determined by subtle variations within KIRs. Yang et al. [97] demonstrated that a single amino acid substitution at position 45 distinguishes activating from inhibitory receptors: tyrosine (Tyr45) in the activating KIR2DS2 vs. phenylalanine (Phe45) in inhibitory KIRs. This structural difference alters binding geometry and affinity, thereby defining distinct interaction models. Moreover, KIR2DS2 recognition extends beyond the HLA-C framework to the bound peptide itself; in particular, interaction with the threonine residue at position P8 of the peptide is essential for stable binding. These findings highlight that KIR2DS2 specificity is shaped not only by the HLA-C allotype but also by the peptide repertoire it presents, underscoring the peptide-dependent nature of HLA-KIR interactions [97].
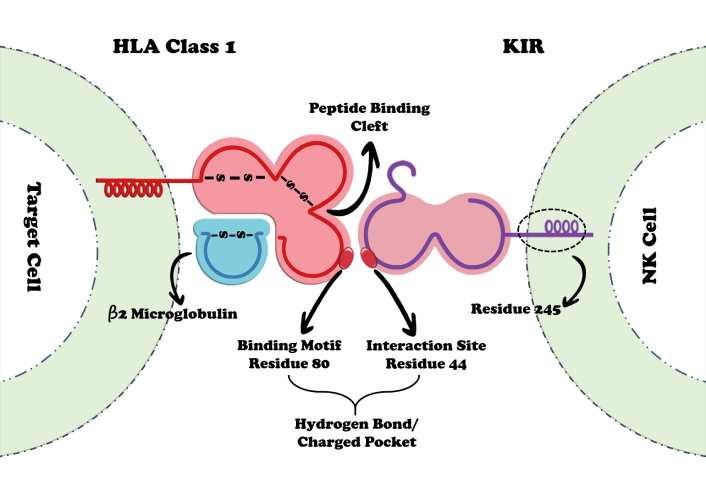
Schematic representation of polymorphic interactions between KIRs and HLA-I molecules. The critical residues identified are KIRs position 44 and HLA-I position 80, which govern specificity, alongside KIRs position 245, which exerts an influence on the strength of inhibitory signaling. HLA: human leukocyte antigen; KIRs: killer immunoglobulin-like receptors.
The peptide dependence of KIR2DS2 recognition illustrates a level of specificity that parallels TCR-peptide-HLA interactions, indicating that NK cell receptors, though innate in nature, can also exhibit highly refined selectivity. Indeed, the preference of KIR2DS2 for peptides containing an “AT” motif at positions P7 and P8 suggests that NK cells may have evolved to recognize conserved viral peptide sequences that are less likely to undergo immune escape mutations. One striking example is the HCV-derived nonstructural peptide LNPSVAATL, presented by HLA-C*01:02, which contains the conserved “AT” motif and has been validated as a ligand for KIR2DS2 [78]. Such findings not only deepen our understanding of peptide-specific KIR recognition but also emphasize the role of NK cells in directly sensing viral antigens in a manner once thought to be exclusive to adaptive immunity. The broader implications of these findings are considerable. First, they suggest that certain activating KIRs, such as KIR2DS2, may contribute to differential outcomes in viral hepatitis through peptide-dependent mechanisms that modulate NK cell activation thresholds. Second, the identification of conserved recognition motifs raises the possibility of harnessing such sequences as biomarkers or therapeutic targets. For example, peptides bearing the “AT” motif could serve as prototypes for the development of NK cell-based vaccines or immunotherapies designed to enhance antiviral responses. Finally, these studies reinforce the concept that the evolutionary balance between activating and inhibitory KIRs reflects selective pressures exerted by pathogens such as HBV and HCV, with haplotypic variation and peptide-level recognition working in concert to shape disease susceptibility and progression.
The interplay between KIRs and HLA molecules is increasingly recognized as a critical determinant of NK cell-mediated immunity in viral hepatitis, influencing not only infection clearance but also disease progression to cirrhosis and HCC. The differential binding affinities, expression patterns, and functional capacities of these receptors shape NK cell activation thresholds and downstream effector functions, including cytotoxicity and cytokine secretion [74].
HLA-KIR interactions critically shape NK cell responses in chronic viral hepatitis, yet their effects diverge across receptor-ligand combinations and disease contexts. In HBV, KIR2DL3/HLA-C1 homozygosity confers protection by enabling NK activation through weak inhibitory signaling [98], whereas KIR2DL1/HLA-C2 combinations deliver stronger inhibition and increase susceptibility. Similarly, HLA-Bw4 allelic subtypes influence KIR3DL1-mediated inhibition: HLA-Bw4 80 isoleucine (80I) binds with higher affinity than HLA-Bw4 80 threonine (80T), intensifying NK suppression and elevating HCC risk [99]. Genetic profiles including HLA-C1 homozygosity, HLA-Bw4 (80I), and KIR2DS4/1D expression have all been associated with heightened HCC risk post-hepatitis [100] (Table 3).
Population-specific studies further highlight this heterogeneity. In Spanish men with alcoholic cirrhosis, KIR2DL2/HLA-C2C2 modulated susceptibility to viral hepatitis [101]. In the Japanese HBV cohort, KIR2DS3 was associated with HBV-related HCC despite no overall HLA-KIR link to cirrhosis [102]. Other studies showed HLA-A (Bw4 group) and HLA-C2 increased HBV persistence, whereas KIR2DL3 was protective [103]. In Bulgarian patients, reduced frequencies of KIR2DL5B and HLA-Bw4 (80I) were seen in self-limiting HBV, while KIR3DL1*004 predisposed to chronicity [104]. Iranian cohorts instead revealed enrichment of KIR2DL5A, KIR2DS1, and KIR3DS1, along with protective KIR3DS1/HLA-Bw4 and KIR3DS1/HLA-A-Bw4 combinations in recovered cases, suggesting population-dependent drivers of clearance [105].
In contrast, in HCV, KIR2DL2/HLA-C1 and KIR2DL3/HLA-C1 combinations confer protection while KIR2DS4 favors chronic infection [106]. Protective effect of KIR2DL3/HLA-C1 combination is lost in HIV co-infection, likely due to NK dysfunction [107]. Additionally, the presence of KIR2DS2/KIR2DL2 has been associated with a predisposition to lymphoproliferative disorders, KIR2DS3 carriage with disease progression, and the KIR3DL1/HLA-Bw6 combination with increased lymphoma susceptibility; in contrast, the presence of the KIR3DS1/HLA-Bw4 combination may confer protection against HCC [108]. In Japanese cohorts, KIR3DL1/Bw4 correlated with HCC progression [109], and post direct-acting antivirals (DAAs) treatment, inhibitory KIR2DL1/HLA-C2 and KIR3DL1/HLA-Bw4 combinations signaling predicted higher HCC risk [110]. These contrasting outcomes between HBV and HCV underscore that identical HLA-KIR pairs can exert either protective or pathogenic effects, depending on the viral context. Factors such as the repertoire and binding affinity of viral peptides presented by HLA molecules, which in turn modulate HLA-KIR recognition, critically shape the balance between NK cell activation and inhibition. Within this framework, KIR2DS3 represents a paradoxical activating receptor whose unique functional attributes defy conventional paradigms of NK cell biology, thereby warranting focused consideration in the context of viral persistence.
KIR2DS3, an activating receptor with atypical functional properties, has emerged as a key immunogenetic marker of impaired NK cell-mediated viral control. While classified as activating, KIR2DS3 exhibits low cell surface expression and suboptimal signaling efficiency, potentially retained intracellularly, which limits its ability to elicit effective NK responses [111, 112]. This atypical behavior may create an “immune decoy” effect, where the presence of the receptor genetically does not translate into functional activation, tipping the balance toward inhibitory HLA-KIR interactions and enabling viral persistence.
Clinical studies corroborate this functional paradox. In Chinese HBeAg-positive HBV patients, KIR2DS3 carriage is associated with reduced virological response to entecavir therapy, suggesting that impaired NK activation undermines antiviral treatment efficacy [113]. Studies on Europeans with HCV demonstrated that KIR2DS3 is a major risk allele for failure of spontaneous viral clearance, favoring chronic infection and accelerated disease progression [35, 114]. Argentine cohorts further demonstrated that KIR2DS3-positive patients exhibit altered NK receptor expression and clinical phenotypes consistent with weakened immune control [115]. Mechanistically, KIR2DS3 may compete with other activating or inhibitory KIRs for HLA-C ligands, thereby dampening NK cell cytotoxicity and cytokine secretion (e.g., IFN-γ, TNF-α), reducing antiviral efficacy, and fostering a tolerogenic hepatic environment conducive to chronic inflammation, cirrhosis, and oncogenesis [116].
In HBV, KIR3DS1 carriers in Gambian populations are HBeAg-positive with higher viral loads, whereas homozygosity for telomeric haplotype A KIRs is associated with lower viral load and improved HBsAg clearance [117]. Correlations between KIR3DL1/HLA-Bw4 and nucleot(s)ide therapy response [102], as well as KIR3DS1/HLA-B (Bw4-80Ile group) and favorable IFN-α therapy outcomes in Chinese HBeAg-positive CHB patients, further emphasize the translational relevance of these interactions [116] (Table 3). In HCV, KIR2DL3 and KIR2DS4 predict positive IFN-α responses, whereas KIR2DL5 is associated with suboptimal treatment outcomes [118, 119].
The likelihood of spontaneous viral clearance in HCV infection is significantly influenced by HLA-KIR interactions. KIR2DL3/HLA-C1 is consistently associated with spontaneous clearance in transfusion- or high-risk-acquired HCV infections [118]. In HCV/HIV co-infection, HLA-C2C2 signaling through KIR2DL1 enhances NK-mediated viral clearance [120]. Chronic HCV patients exhibit reduced NK cells expressing KIR2DS1 and KIR2DL2, whereas recovered individuals show higher frequencies of T cells expressing KIR2DL2/L3/S2, indicating a coordinated role of NK and T cell KIR expression in viral resolution [120].
Impact of HLA-KIR combinations on HBV and HCV outcomes: a global perspective.
| References | Year | Population | Findings |
|---|---|---|---|
| Lu et al. [95] | 2008 | CHB Chinese patients | Lower and higher frequencies of A and B haplotypes in patients with HBV, respectively |
| Gao et al. [98] | 2010 | HBV Chinese patients | The homozygosity of KIR2DL3/HLA-C1 has a protective role against HBV |
| Pan et al. [100] | 2011 | HBV Chinese patients | Increased risk of developing HCC following viral hepatitis through homozygous genotype for HLA-C group 1, HLA-Bw480I, and a combined pattern of KIR2DS4/1D |
| Moralès et al. [99] | 2012 | - | HLA-Bw480I allele inhibits NK cells more effectively than HLA-Bw480T via stronger binding affinity for KIR3DL1 |
| De Re et al. [108] | 2015 | CHC Italian patients | The KIR2DS3 gene is related to the progression of HCV-related liver disease |
| Buchanan et al. [114] | 2015 | -- | KIR2DS3 promotes chronic infection and rapid progression |
| Di Bona et al. [103] | 2017 | CHB Italian patients | KIR2DL3 is protective in controlling HBV infection |
| Shah-Hosseini et al. [105] | 2017 | HBV Iranian patients | Recovered individuals had higher frequencies of KIR2DL5A, KIR2DS1, KIR3DS1 alleles, and specific KIR3DS1 genotypes |
| Yindom et al. [117] | 2017 | Gambian HCC and Cirrhosis patients | Patients with HBV carrying the KIR3DS1 allele are HBe antigen-positive and exhibit high viral loads |
| Li et al. [116] | 2017 | CHB Chinese patients | A direct relationship between the KIR3DS1/HLA-BBw4-80Ile gene combination and favorable response to IFN-α therapy |
| Podhorzer et al. [115] | 2017 | Argentine HCV cohorts | NK receptor alterations accompany KIR2DS3 positivity |
| Zhuang et al. [113] | 2018 | Chinese HBeAg-positive cohorts | KIR2DS3 carriage reduced entecavir response; impaired NK activation reduces antiviral efficacy |
| Djigma et al. [121] | 2020 | West African Cohort (Burkina Faso) | A and B KIR haplotypes were associated with protection against HBV chronic infection evolution to cirrhosis and/or HCC |
| Auer et al. [106] | 2020 | HBV Vietnamese patients | KIR2DS4 allele is linked to chronic infection, whereas the combinations KIR2DL2/HLA-C1 and KIR2DL3/HLA-C1 lower the risk of CHB |
| Ursu et al. [96] | 2020 | CHC Romanian patients | Associations between the KIR2DL3, KIR2DL5, KIR3DL3, KIR2DP1, KIR3DP1, and KIR2DS4 norm allele and an increased genetic predisposition to CHC |
| Joshita et al. [102] | 2021 | HBV Japanese patients | A direct association between the presence of the KIR2DS3 allele and HBV-related HCC |
| Varbanova et al. [104] | 2021 | HBV Bulgarian patients | A direct association between the presence of the KIR3DL1*004 allele and the development of CHB |
| Umemura et al. [109] | 2021 | HCV cirrhotic Japanese patients | KIR3DL1/HLA-Bw4 combination correlates with the progression of disease to HCC |
| Ursu et al. [31] | 2021 | CHC Romanian patients | Elevated AST, ALT, and GGT levels in patients with the KIR2DL2/KIR2DL2-C1C1 genotype |
| Legaz et al. [101] | 2024 | Spanish man with alcoholic cirrhosis | The KIR2DL2/C2C2 combination plays a role in determining the genetic susceptibility of patients with alcoholic cirrhosis to viral hepatitis infections |
| Ryan et al. [110] | 2024 | HCV American patients | KIR2DL1/HLA-C2 and KIR3DL1/Bw4 combinations are associated with an increased risk of HCC |
| Martín-Sierra et al. [122] | 2024 | HCV Spanish patients | No association found between HLA-KIR combinations and seroconversion following virus exposure in patients with HCV |
ALT: alanine transaminase; AST: aspartate transferase; CHB: chronic hepatitis B; CHC: chronic hepatitis C; HBeAg: hepatitis B e antigen; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; HLA: human leukocyte antigen; IFN: interferon; KIR: killer immunoglobulin-like receptor; NK: natural killer; GGT: gamma-glutamyl transferase.
In CHB and CHC infections, NK cells play a paradoxical role, contributing both to viral control and to liver pathology (Figure 4). Activated NK cells upregulate cytotoxic receptors, enabling lysis of infected hepatocytes and, importantly, apoptosis of hepatic stellate cells (HSCs), thereby limiting fibrosis progression [9, 12, 54]. However, their effector functions are finely tuned by the balance between activating and inhibitory HLA-KIR combinations [52]. Strong inhibitory signaling, mediated through KIR2DL1/HLA-C2 and KIR3DL1/HLA-Bw4, dampens cytotoxicity and IFN-γ release [48], facilitating viral persistence, NK exhaustion, and fibrogenesis [54]. Over time, persistent engagement of high-affinity inhibitory pathways drives immune tolerance, impaired surveillance, and heightened risk of cirrhosis and HCC [104, 110]. Conversely, weaker inhibitory interactions such as KIR2DL3/HLA-C1 provide only limited suppression, enabling sustained NK activity and favoring spontaneous HCV clearance [104]. Individuals carrying this genotype often exhibit stronger antiviral potential and reduced chronicity compared with those with high-affinity inhibitory HLA-KIR combinations [91]. In parallel, activating KIRs, including KIR2DS1 and KIR3DS1, recognize HLA-C2 and HLA-Bw4 ligands to potentiate cytotoxicity and IFN-γ production [52]. These high-affinity activating interactions promote viral clearance and support adaptive immunity, although excessive NK activation may exacerbate hepatocyte injury and inflammation. Overall, NK cell function in viral hepatitis reflects the delicate balance of HLA-KIR signaling. High-affinity inhibitory interactions foster viral persistence, fibrosis, and HCC risk, whereas low-affinity inhibitory or strong activating combinations favor viral clearance and limit disease progression. Thus, HLA-KIR combinations serve as critical immunogenetic determinants of NK cell functional thresholds, disease outcome, and responsiveness to therapy (Figure 4).
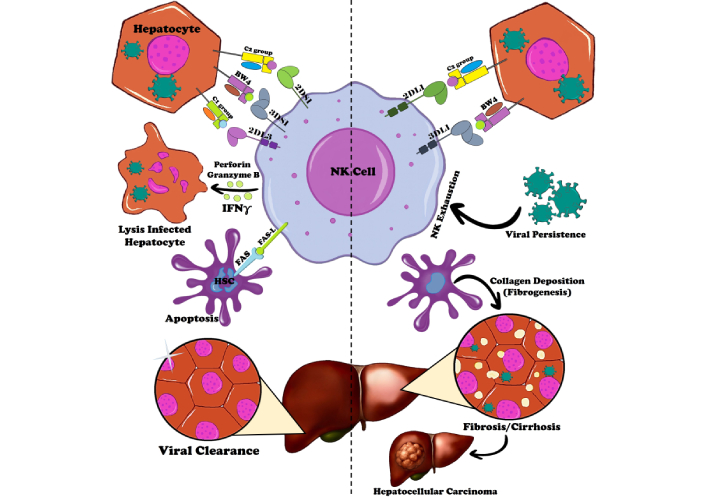
Immune responses in hepatitis B and C virus infections: key mechanisms and HLA-KIR interactions driving viral clearance, chronic infection, and progression to hepatocellular carcinoma. Key HLA-KIR interactions are depicted, with inhibitory pairs (e.g., KIR2DL1/HLA-C2, KIR3DL1/HLA-Bw4) shown in red, promoting viral persistence, exhaustion, and fibrosis, while activating or low-affinity inhibitory pairs (e.g., KIR2DL3/HLA-C1, KIR2DS1/HLA-C2) in green favor NK cell cytotoxicity, IFN-γ production, and clearance. The diagram includes pathways for NK cell-mediated lysis of infected hepatocytes and HSCs, cytokine modulation (e.g., IFN-γ enhancing adaptive immunity), and the paradoxical role of NK cells in both antiviral defense and liver pathology. HLA: human leukocyte antigen; HSCs: hepatic stellate cells; IFN-γ: interferon gamma; KIR: killer immunoglobulin-like receptor; NK: natural killer.
NK cells are central to innate defense against viral hepatitis, exerting antiviral activity through direct lysis of infected hepatocytes and secretion of cytokines such as IFN-γ. Exogenous IFN-γ mediates pleiotropic antiviral effects by upregulating HLA-I for improved antigen presentation to cytotoxic T cells, activating macrophages and dendritic cells, and inducing IFN-stimulated genes (ISG) expression to restrict viral replication [123, 124]. Despite the proven efficacy of nucleos(t)ide analogs and DAAs in suppressing HBV and HCV replication, these agents rarely restore NK cell function in chronic infection [110]. In contrast, IFN-based regimens, particularly pegylated IFN-α (Peg-IFN-α), enhance NK activity by upregulating activating receptors (e.g., NKG2D, NKp30, NKp46), promoting degranulation, and partially reversing NK cell exhaustion [56, 77]. Patients harboring favorable activating HLA-KIR combinations (e.g., KIR2DS1/HLA-C2 or KIR3DS1/HLA-Bw4) demonstrate stronger IFN-γ responses and improved outcomes under IFN-α-based therapies, whereas individuals dominated by inhibitory HLA-KIR profiles exhibit impaired NK function, poor viral clearance, and accelerated fibrosis progression [116]. Notably, IFN-γ also mitigates inhibitory checkpoint pathways such as PD-1 and NKG2A, further amplifying NK effector responses [17, 61].
Building on these insights, novel immunotherapies are being designed to directly reprogram NK cell responses by targeting the HLA-KIR axis [125]. The most advanced strategy involves KIR-blocking antibodies [126]. The fully human anti-KIR2DL1/2DL2/2DL3 antibody lirilumab (IPH2102/BMS-986015) has demonstrated safety and durable disruption of inhibitory signaling in early-phase oncology trials [127]. While primarily tested in hematologic malignancies, preclinical data suggest that KIR blockade can restore NK cytotoxicity against HBV- and HCV-infected hepatocytes, particularly when combined with DAAs or checkpoint inhibitors. Combination approaches offer additional translational promise [127]. KIR blockade synergizes with anti-NKG2A antibodies (e.g., monalizumab), targeting parallel inhibitory pathways, and may be further enhanced by PD-1/PD-L1 inhibitors, already approved for HBV-related HCC, by reversing NK and T cell exhaustion [128].
Adoptive NK cell transfer provides another platform for KIR modulation. Donor NK cells can be selected or engineered for HLA-KIR mismatch to maximize antiviral alloreactivity [129]. Advances in genetic editing, particularly CRISPR/Cas9, enable deletion of inhibitory KIRs or introduction of activating alleles (e.g., KIR2DS1, KIR3DS1), reprogramming NK cells toward sustained antiviral and antitumor activity. Early studies have confirmed the feasibility of multiplex CRISPR editing in primary NK cells, offering proof-of-concept for application in viral hepatitis and related cancers [129]. Additionally, chimeric antigen receptor (CAR)-NK cells represent a rapidly advancing modality in which inhibitory KIRs can be knocked out to prevent host HLA-mediated suppression, while engineered CAR constructs direct NK activity toward viral or tumor targets [130]. Dual-modified CAR-NK cells are already under investigation in hematologic malignancies and may be adapted for HBV- or HCV-driven HCC [125, 130].
Collectively, these emerging approaches underscore that direct modulation of HLA-KIR interactions, via blocking antibodies, adoptive transfer, genetic engineering, or CAR platforms, may overcome one of the central immune bottlenecks in CHB and CHC. By restoring NK effector function, these interventions hold the potential not only to improve viral control but also to reduce progression to HCC. Future directions should emphasize integration of KIR-targeted therapies with antiviral and checkpoint-based regimens, guided by biomarker-driven patient selection, to fully exploit the therapeutic potential of NK cells in viral hepatitis.
In conclusion, this review highlights the essential function of interactions between HLA and KIRs in influencing the immune responses of NK cells against infections caused by HBV and HCV. The presence of activating KIRs, such as KIR2DS1 and KIR3DS1, alongside certain HLA ligands, promotes the cytotoxic activity of NK cells and enhances the production of cytokines, thereby facilitating viral elimination and favorable treatment results. In contrast, inhibitory KIRs, including KIR2DL1 and KIR3DL1, by binding with high affinity to HLA-C2 and HLA-Bw4 epitopes, reduce NK cell activation, which contributes to the persistence of the virus, chronic inflammation, fibrosis, cirrhosis, and the advancement to HCC. Genetic differences in KIR haplotypes and HLA alleles that are specific to populations further affect the likelihood of disease occurrence, severity, and response to treatment, emphasizing the immunogenetic variances that govern the interactions between the host and virus.
These findings present combinations of HLA and KIRs as potential biomarkers for anticipating disease progression, spontaneous viral clearance, and the effectiveness of antiviral treatments. From a therapeutic perspective, focusing on the HLA-KIR interaction through strategies such as KIR-blocking antibodies, adoptive transfers of NK cells, genetic modifications, or combination immunotherapy provides new paths to restore NK cell functionality, counteract immune fatigue, and enhance outcomes in chronic viral hepatitis. Future investigations should emphasize longitudinal studies across varied populations to confirm these relationships, clarify peptide-specific mechanisms, and further develop personalized medical approaches, thereby ultimately alleviating the global impact of liver diseases associated with HBV and HCV.
AIH: autoimmune hepatitis
ALT: alanine transaminase
AST: aspartate transferase
BAFF: B-cell activating factor
BLIMP-1: B lymphocyte induced maturation protein-1
bnAbs: broadly neutralizing antibodies
CAR: chimeric antigen receptor
CHB: chronic hepatitis B
CHC: chronic hepatitis C
CTLs: cytotoxic T lymphocytes
DAAs: direct-acting antivirals
GGT: gamma-glutamyl transferase
HBcAg: hepatitis B core antigen
HBeAg: hepatitis B e antigen
HBsAg: hepatitis B surface antigen
HBV: hepatitis B virus
HCC: hepatocellular carcinoma
HCV: hepatitis C virus
HLA: human leukocyte antigen
HSCs: hepatic stellate cells
IFN-I: type I interferon
ITAMs: immunoreceptor tyrosine-based activation motifs
ITIMs: immunoreceptor tyrosine-based inhibitory motifs
KIRs: killer immunoglobulin-like receptors
LILR: leukocyte immunoglobulin-like receptor
LRC: leukocyte receptor cluster
MAVS: mitochondrial antiviral-signaling
MICA: MHC class I polypeptide-related sequence A
nAbs: neutralizing antibodies
NCRs: natural cytotoxicity receptors
NK: natural killer
NKG2: natural killer group 2
PD-1: programmed cell death protein-1
PRRs: pattern recognition receptors
RIG-I: retinoic acid-inducible gene-I
RMSCs: recognition of missing self-cells
ROS: reactive oxygen species
sHLA-E: soluble human leukocyte antigen-E
SNP: single nucleotide polymorphism
TCR: T cell receptor
TFH: T follicular helper
TLRs: Toll-like receptors
TNF-α: tumor necrosis factor-α
TRAIL: tumor necrosis factor-related apoptosis-inducing ligand
TRAILR: tumor necrosis factor-related apoptosis-inducing ligand receptor
TRIF: TIR-domain-containing adapter-inducing interferon-β
VCAM-1: vascular cell adhesion molecule-1
VLA-4: very late activation antigen-4
AS and AMZ: Investigation, Writing—original draft. AS and TK: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Validation, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The research protocol was approved and supported by the Student Research Committee, Tabriz University of Medical Sciences [Grant No: 76511]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

