 Review
Review

Affiliation:
1Institute for Clinical Immunotherapy and Advanced Biological Treatments, 65121 Pescara, Italy
†MDG and AP contributed equally to this work.
Email: digioacchino@me.com
ORCID: https://orcid.org/0000-0002-9224-5886
Affiliation:
2Division of Hematology, Department of Human Pathology in Adulthood and Childhood “Gaetano Barresi”, University of Messina, 98125 Messina, Italy
†MDG and AP contributed equally to this work.
Affiliation:
1Institute for Clinical Immunotherapy and Advanced Biological Treatments, 65121 Pescara, Italy
ORCID: https://orcid.org/0009-0002-2075-5112
Affiliation:
3Pneumology Division, University Hospital, 66100 Chieti, Italy
Affiliation:
1Institute for Clinical Immunotherapy and Advanced Biological Treatments, 65121 Pescara, Italy
ORCID: https://orcid.org/0000-0002-7947-158X
Affiliation:
4Allergy and Clinical Immunology Unit, Department of Clinical and Experimental Medicine, University of Messina, 98125 Messina, Italy
#SG and AA contributed equally to this work.
ORCID: https://orcid.org/0000-0001-7001-6532
Affiliation:
2Division of Hematology, Department of Human Pathology in Adulthood and Childhood “Gaetano Barresi”, University of Messina, 98125 Messina, Italy
#SG and AA contributed equally to this work.
ORCID: https://orcid.org/0000-0001-6156-8239
Explor Immunol. 2025;5:1003226 DOI: https://doi.org/10.37349/ei.2025.1003226
Received: May 27, 2025 Accepted: September 25, 2025 Published: November 17, 2025
Academic Editor: Jean Amiral, HYPHEN BioMed, France
Innate lymphoid cells are lymphocytes that are neither T cells nor B cells. They are relatively rare in lymphoid tissues and peripheral blood and are distinguished by their absence of an adaptive antigen receptor. In the present study, we describe the mechanisms underlying the generation of the various cell populations and highlight the functional importance of their plasticity. These cells are indeed capable of transdifferentiating from one type to another. This adds complexity to their functional program, and this feature appears to be crucial for adapting and modulating immune responses under different conditions. These lymphoid cells are of great hematological interest due to their pathophysiological and therapeutic role in many onco-hematological pathologies such as acute myeloid leukemia, multiple myeloma, and several types of lymphomas. In hematological disorders, innate lymphoid cells may exert differential effects on the pathogenesis of hematologic malignancies. Furthermore, within the same disease, certain cell populations have been shown to play a protective role in antitumor immune responses, whereas others appear to suppress these responses. This review aims to provide an integrated description of innate lymphoid cells, their alterations in hematological malignancies, and potential preventive strategies, by proposing new specific targets for correcting anomalies. We also discuss the use of innate lymphoid cells as new therapies by applying chimeric antigen receptor-modified natural killer cells. We examine the current knowledge and outline future perspectives.
Innate lymphoid cells (ILCs) are lymphocytes that do not belong to the T or B cell lineages and are relatively rare in lymphoid tissues and peripheral blood. They are defined by the absence of adaptive antigen receptors [1]. One of their principal functions is to initiate an early immune response upon pathogen invasion, preceding the activation of antigen-specific lymphocytes [2]. ILCs are predominantly located at mucosal surfaces of non-lymphoid organs [3]. Remarkably, even in the absence of recombination-activating genes (Rag-1 and Rag-2) and without the expression of conventional T and B cell antigen receptors, ILCs are still capable of maturing. Although they do not possess immunological memory, they are classified as components of the innate immune system [4, 5]. The mechanisms underlying their activation, however, remain largely undefined [6, 7].
ILCs are categorized into four major subgroups. Natural killer (NK) cells and type 1 ILCs represent two subsets of ILCs that express the T-box transcription factor TBX21 (T-bet). Although NK cells and type 1 ILC1s both belong to the family of ILCs, they exhibit distinct functional, phenotypic, and developmental characteristics.
These cells exert their functions primarily through the production of interferon-gamma (IFN-γ). NK cells are involved in type 1 immune responses and are classified as cytotoxic ILCs. In contrast, ILC1s are widely distributed across various tissues, including the liver, adipose tissue, intestine, and salivary glands. They are activated by soluble cytokines such as interleukin (IL)-15, IL-12, and IL-18. ILC1s contribute to host defense against viral and intracellular bacterial infections by producing effector cytokines and initiating a rapid, first-line immune response [8].
The second group of ILCs (ILC2s) is defined by the expression of the GATA-binding protein 3 (GATA3) and is highly prevalent in mucosal tissues such as the gastrointestinal (GI) tract, lungs, tonsils, and skin. These cells mediate type 2 immune responses by producing IL-5, IL-13, IL-4, and members of the epidermal growth factor family. They also secrete cytokines such as amphiregulin, which play a role in combating helminth infections and regulating tissue repair [9].
ILC3s are characterized by the presence of RAR-related orphan receptor gamma T (RORγt). ILC3s are further subdivided into NKp46+ and NKp46– subsets based on surface marker expression. These cells secrete a range of cytokines and growth factors, including IFN-γ, tumor necrosis factor-alpha (TNF-α), IL-22, IL-17, granulocyte-macrophage colony-stimulating factor (GM-CSF), and heparin-binding epidermal growth factor-like growth factor (HB-EGF) [10, 11]. ILC3s are abundant in the skin, lungs, intestinal mucosa, and mesenteric lymph nodes, where they play a key role in initiating rapid immune responses against extracellular microorganisms and maintaining tissue homeostasis [12].
The final group of ILCs, known as lymphoid tissue inducer (LTi) cells, comprises lymphoid tissue-derived cells that are also dependent on RORγt and originate from fetal liver progenitor cells. These cells contribute to the development of secondary lymphoid organs by promoting lymphoid tissue proliferation, a process mediated by lymphotoxin, a member of the TNF superfamily. LTis are present in various organs and tissues during early embryonic development. By regulating adaptive immune responses and supporting the formation of secondary lymphoid structures, they play a critical role in the establishment of both primary and secondary lymphoid tissues [13, 14].
The differentiation of ILCs from a common progenitor cell is illustrated in Figure 1.
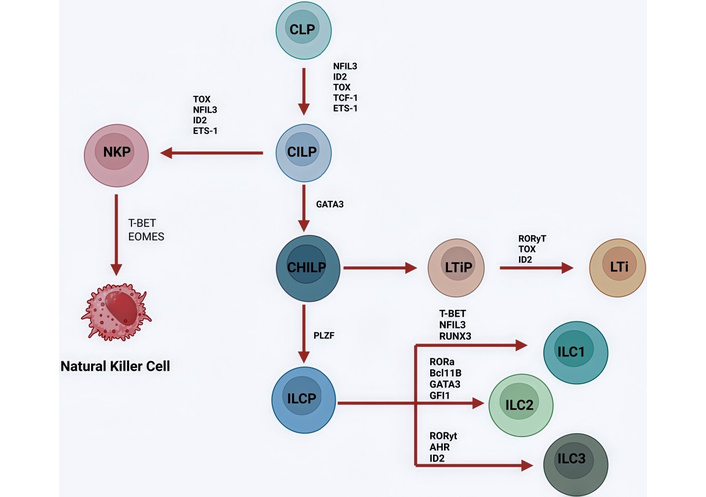
The development of ILCs starts with CLPs (common lymphoid progenitors). CLPs can differentiate into NK cell precursors. These cells will differentiate into CILPs, which themselves differentiate into natural killer progenitors (NKP) cells or into common helper innate lymphoid progenitors (CHILPs), which give rise to lymphoid tissue inducer progenitors (LTiPs) and innate lymphoid cell precursors (ILCPs). LTiPs differentiate into LTis and ILCPs into ILC1, ILC2, or ILC3. Each differentiation step is correlated with the expression of the following transcription factors: NFIL3, ID2, TOX, TCF-1, ETS-1, GATA3, PLZF, T-bet, EOMES, RUNX3, RORα, Bcl11B, GFI1, RORγt, and AHR. The human ILC1 group might have spread from a different ancestor, the identity of which is still unknown. Transcription factors and proteins in ILC development: NFIL3: nuclear factor IL-3 induced; ID2: inhibitor of DNA binding 2; TOX: thymocyte selection-associated high mobility group box protein; TCF-1: T cell factor 1; ETS-1: avian erythroblastosis virus E26 homolog-1; GATA3: GATA binding protein 3; PLZF: promyelocytic leukemia zinc finger; T-bet: T-box transcription factor TBX21; Eomes: eomesodermin; RUNX3: runt-related transcription factor 3; RORα: RAR-related orphan receptor α; Bcl11B: B cell lymphoma/leukemia 11B; GFI1: growth factor independent 1; RORγt: RAR-related orphan receptor gamma T; AHR: aryl hydrocarbon receptor. Created in BioRender. Mirabile, G. (2025) https://BioRender.com/rtss29h.
Although the classification of ILCs provides a valuable theoretical framework for understanding their diversity, immune responses can introduce additional complexity into their functional programming. Evidence suggests that certain ILC subsets exhibit functional plasticity—a feature well-documented in T cells [15, 16]—which may be critical for adapting and modulating immune responses to diverse pathogenic stimuli.
In vitro, human RORγt+ ILC3s stimulated with IL-2 or IL-15 can differentiate into ILC1-like cells, characterized by upregulation of the transcription factor T-bet and the interleukin-12 receptor β2 (IL-12Rβ2) [17] (Figure 2). These cells subsequently produce IFN-γ in response to IL-12 stimulation.
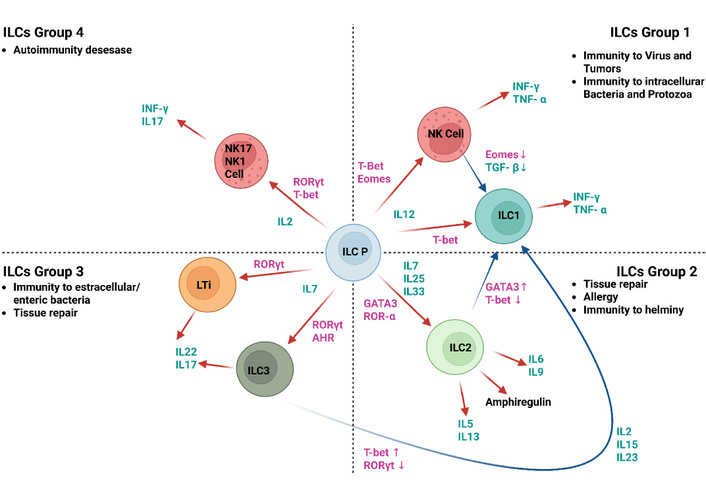
Tissue ICL1s. T-bet+ ILC1s present within tissues might comprise: ILC1s derived from innate lymphoid cell precursors (ILCPs); ILC2s that transition upon exposure to IL-12 and IL-1β, leading to a reduction in GATA3 and an increase in T-bet; ILC3s that undergo conversion when exposed to IL-2, IL-15, and IL-23, resulting in decreased RORγt and elevated T-bet levels; and NK cells that minimize EOMES expression in environments abundant in TGF-β. T-bet: T-box transcription factor TBX21; RORγt: RAR-related orphan receptor gamma T; GATA3: GATA binding protein 3; EOMES: eomesodermin; AHR: aryl hydrocarbon receptor; TGF-β: transforming growth factor-beta. Created in BioRender. Mirabile, G. (2025) https://BioRender.com/huv8ftc.
Furthermore, culturing ILC3s with IL-23 promotes their conversion into ILC1s. Interestingly, IL-23 is also the primary stimulus that induces IL-22 secretion by ILC3s. This seemingly paradoxical effect is enabled by the constitutively high expression of the transcription factor signal transducer and activator of transcription 4 (STAT4) in ILC3s [18, 19]. As a result, sustained exposure to IL-23 activates STAT4 and drives the polarization of ILC3s toward a type 1 phenotype. Some studies also suggest that IL-23 may facilitate the reverse transition—from ILC1s back to ILC3s [17]—although the molecular mechanisms underlying this bidirectional plasticity remain to be elucidated.
Acute myeloid leukemia (AML) is a hematological disease characterized by the growth and proliferation of immature cells. AML utilizes unique immune evasion strategies like those of solid cancers. This section provides an update on recent advances in understanding how AML affects each group of ILCs [20].
The antitumor activity of NK cells has been documented in several malignancies, including AML [21, 22]. In the early 2000s, NK cells emerged as promising candidates for immunotherapy, as those derived from haploidentical donors were shown to enhance alloreactive responses and improve patient survival [23].
Recent findings have provided new insights into NK cell development and their role in AML progression. NK cells originate in the bone marrow and reach full maturation in secondary lymphoid organs such as the tonsils and lymph nodes. The majority of circulating NK cells are mature CD56 dim cells, while a smaller subset consists of CD56 bright cells. Nevertheless, immature NK cell precursors can also be detected in peripheral blood, albeit at lower frequencies [24]. Several studies have reported a developmental block in NK cell maturation in AML patients. In a murine model of AML, splenic NK cells exhibited impaired progression from stage 2 (CD27+CD11b–) to stage 3 (CD27+CD11b+) [25]. Similar findings were observed in human ILC precursors, which failed to differentiate into NK cells in the presence of AML cells in an ex vivo co-culture system [26].
Moreover, AML patients typically exhibit a significantly reduced proportion of circulating NK cells compared to healthy individuals, a condition associated with poorer clinical outcomes [27]. Some studies have also reported a less mature NK cell phenotype in AML, characterized by reduced expression of CD57 and killer cell immunoglobulin-like receptors (KIRs) [28]. However, conflicting data exist, with other reports indicating a more mature NK phenotype in AML patients, marked by increased CD57 and KIR expression and decreased CD56 expression [29]. Notably, these latter studies did not directly assess NK cell functionality. It is possible that both immature and hyper-mature NK phenotypes share similar functional impairments, with hyper-mature cells representing an exhausted state with diminished antitumor activity.
A further reduction in NK cell function has been associated with increased expression of the inhibitory receptor NKG2A at diagnosis [30]. Similarly, decreased expression of activating receptors such as NKp46 and NKp30 correlates with unfavorable clinical outcomes [31, 32], whereas higher expression levels of these receptors are linked to improved prognosis [33, 34].
In patients with myelodysplastic syndrome (MDS)—a condition that frequently progresses to AML—reduced NK cell numbers and impaired function have also been observed, largely due to decreased expression of activating receptors such as NKG2D and NKp30 [35, 36]. In these individuals, the extent of NK cell dysfunction appears to be intermediate between that of healthy controls and AML patients. Furthermore, MDS patients with pronounced NK cell impairment exhibit a higher risk of progression to AML [35]. A genetic predisposition to MDS/AML involving dysfunctional NK cells has also been identified. Mutations in the transcription factor GATA2 predispose individuals to MDS/AML [37, 38] and result in severe NK cell defects, characterized by the preservation of the CD56 dim subset and the loss of CD56 bright cells [35]. This NK cell abnormality has also been observed in individuals with GATA2 loss-of-function mutations who do not present with MDS or AML.
Although NK cell function appears to be profoundly suppressed at AML diagnosis, these cells play a significant role in preventing or delaying relapse during post-remission phases [39–41]. Following chemotherapy, the NK cell compartment is rapidly reconstituted—typically within four months to one year after remission—with a predominance of immature CD56 bright cells [42]. Enhanced surface expression of NKp46 and increased CD107a expression upon exposure to K562 target cells indicate that NK cell activity is at least partially restored in remission, even in patients who exhibited impaired NK function at diagnosis [30]. Therapeutic strategies that promote the expression of additional activating receptors have also been shown to enhance NK cell-mediated cytotoxicity against leukemic targets [43].
Collectively, these findings suggest that NK cell dysfunction in AML may be at least partially reversible, and further research is warranted to better understand the endogenous role of NK cells in AML pathophysiology.
Although the anti-tumor effects of NK cells are well established, the functional roles of other ILC subsets in cancer remain incompletely understood [44]. In particular, the role of ILC1s in AML is still unclear, and ongoing research is focused on elucidating their contribution. One of the major challenges in studying ILC1s is the lack of unique, definitive surface markers for their identification.
Early investigations of peripheral blood and bone marrow from AML patients revealed an enrichment of functionally impaired, lineage-negative ILC1s compared to healthy donors (HDs) [45]. More recently, a study identified a phenotypically distinct ILC1-like subset (Lin–CD56+CD94+CD16–CD127+) with reduced cytotoxic potential at diagnosis, which appeared to be restored in patients who achieved remission [46]. In this study, only surface markers were used to differentiate conventional NK cells from ILC1 or ILC1-like cells, and the phenotypic profile of the latter overlapped with that of circulating CD56 bright NK cells [46, 47].
A key distinction between human and murine NK cells and ILC1s lies in their transcription factor expression: NK cells typically co-express EOMES and T-bet, whereas ILC1s express only T-bet. In mice, the inhibitory receptor CD200R1 has been proposed as a selective surface marker for liver-resident ILC1s and is notably absent in NK cells [44, 48]. Interestingly, CD200 expression on human AML blasts has been shown to suppress IFN-γ production and reduce cytotoxic activity by engaging CD200R1 on human NK cells [49]. Although the expression pattern of CD200R1 in human ILCs is still under investigation, this interaction may reflect an increased presence of ILC1s in CD200 Hi AML cases [20].
Finally, in murine models, NKp46 expressed on ILC1s mediates direct interactions with tumor cells, enhancing cytotoxicity and promoting the production of TNF and IFN-γ. Deletion of NKp46 results in reduced survival and impaired ILC1-mediated tumor control in a mouse model of AML. This phenotype can be reversed by adoptive transfer of NKp46+ ILC1s into NKp46-deficient mice. In humans, NKp46+ ILC1s produce higher levels of cytokines and exhibit greater cytotoxicity compared to their NKp46– counterparts, suggesting that NKp46 plays a similarly critical role in human ILC1 function [50].
Recent studies have revealed a tumor-promoting role for ILC2s. While ILC2s are primarily known for their involvement in allergic responses and anti-helminthic immunity, emerging evidence suggests they may also contribute to leukemogenesis. A study focusing on AML demonstrated that prostaglandin D2 (PGD2), secreted by mesenchymal stem cells (MSCs), activates ILC2s via the chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) [51]. This activation induces the secretion of IL-5, which in turn promotes the expansion of regulatory T cells (Tregs) and the proliferation of hematopoietic stem and progenitor cells (HSPCs). In an AML mouse model, this Treg expansion was associated with reduced survival and accelerated leukemia progression.
In addition, tumor-derived PGD2 and the NKp30 ligand B7-H6 have been shown to activate ILC2s, leading to the secretion of IL-13. This cytokine stimulates the activity of myeloid-derived suppressor cells (MDSCs), whose immunosuppressive functions are well-documented in promoting tumor progression [52]. These findings suggest that therapeutic strategies targeting the PGD2-ILC2-Treg or PGD2-ILC2-MDSC axes may hold promise in the treatment of AML.
However, contrasting evidence has emerged from another study, which found no significant differences in ILC2 frequency or IL-5 and IL-13 levels in the peripheral blood of untreated AML patients compared to healthy controls [53]. These discrepancies may be attributed to differences in the tissue microenvironment, as PGD2-producing mesenchymal cells are primarily located in the bone marrow rather than in peripheral blood. This highlights the importance of investigating the primary tissue microenvironment in systemic diseases such as AML, where distinct tissues may exhibit divergent immunological profiles.
Taken together, these investigations highlight the intricate and situation-specific function of ILC2 cells in the development of AML. Clinical data from peripheral blood studies do not support the tumor-promoting axis suggested by preclinical models, which include PGD2-mediated ILC2 activation and the downstream proliferation of immunosuppressive Tregs and MDSCs. This disparity highlights the crucial role that the bone marrow microenvironment (BMME) plays in determining ILC2 function and most likely reflects the compartmentalized nature of immune regulation. To resolve these conflicting findings and improve therapeutic targeting of the PGD2-ILC2 axis in AML, future research should give priority to tissue-specific analyses.
Despite growing evidence that ILC3s play a significant role in post-chemotherapy prognosis and in the pathogenesis of graft-versus-host disease (GVHD), studies specifically investigating ILC3s in AML remain limited. One study reported a marked reduction in natural cytotoxic receptor-positive (NCR+) ILC3s—but not NCR– ILC3s—in the peripheral blood of treatment-naïve AML patients [54]. In this study, ILC3s were defined as Lin–CD127+CRTH2–CD117+NKp46+/– cells, a phenotype that overlaps with that of immature NK cells and ILC precursors circulating in the bloodstream. Interestingly, no significant differences in IL-17A or IL-22 levels were observed between the ILC compartments of AML patients and HDs [55].
Emerging evidence suggests that NKp44 may be a more reliable marker than CD117 for identifying human ILC3 populations, as NKp44+ cells are typically absent from the circulation of healthy individuals. In AML patients who responded to standard chemotherapy, the frequency of Lin–CD127+CRTH2–CD117+NKp46+ cells was comparable to that of healthy controls. In contrast, non-responders exhibited a reduced percentage of these cells, suggesting a potential prognostic value for this ILC3 subset.
Beyond their role in AML pathophysiology, ILC3s have also been detected during post-induction chemotherapy and hematopoietic stem cell transplantation (HSCT). A study evaluating the reconstitution of ILC subsets following induction chemotherapy and allogeneic HSCT (allo-HSCT) found that donor-derived ILC1s, ILC2s, and NKp44– ILC3s reconstituted more rapidly and at higher levels than other ILC subsets. NKp44+ ILC3s were also observed in the peripheral blood of AML patients undergoing these treatments. These ILC populations expressed activation markers such as CD69 and homing receptors for the gut and skin, including α4β7 integrin, CCR6, CCR10, and cutaneous lymphocyte-associated antigen (CLA). Their presence following chemotherapy or allo-HSCT was associated with a reduced incidence of GVHD [56].
In murine models, NCR+ ILC3s have been shown to promote intestinal tissue regeneration and prevent bacterial translocation—mechanisms that contribute to the mitigation of GVHD [57]. These effects are mediated through IL-22-dependent pathways. Indeed, IL-22 has been shown to confer protection against GVHD in mouse models of allo-HSCT [58]. Further research is needed to clarify the role of ILC3s in AML biology and to assess their potential as biomarkers for treatment response.
An overview of innate lymphoid cell abnormalities in AML is provided in Table 1.
Anomalies of innate lymphoid cell populations in AML.
| Population | Findings in AML |
|---|---|
| NK | CD57+ KIR+ NK cells are elevated in a subset of AML patients [29]. |
| NK | The inhibition of AHR increases the cytotoxic activity of NK cells on AML blasts and returns normal NK maturation. |
| NK | A less mature peripheral NK cell phenotype characterized by the absence of CD57 and KIR is associated with a reduced overall survival rate [27]. |
| NK | IFN-γ secretion is reduced, and NK cytotoxic activity is compromised in AML patients with CD200 HI. |
| NK | A lower level of NK cell function in AML patients is linked to decreased expression of factors that activate the receptors NKp30 and NKp46 and increased production of those that block the receptor NKG2A [31–34]. |
| NK | There have been no observed changes in overall survival in relation to the CMV+ serum status, which is linked to enhanced memory-like NK cell production and extended relapse-free life. |
| ILC1 | In AML patients, we have an enrichment and reduced function of null-deficient ILC1 [45], with reduced cytotoxic capabilities [59], and a lower incidence of GVHD [56]. |
| ILC2 | It was observed that a Treg expansion and HSPC proliferation [51] increased IL-13 secretion [52] and a lower incidence of GVHD [56]. |
| ILC3 | We have a reduction in spontaneous natural cytotoxic receptor-positive (NCR+) ILC360 normal rates are associated with a favorable prognosis [45] and lower incidence of GVHD [56]. |
AML: acute myeloid leukemia; AHR: aryl hydrocarbon receptor; KIR: killer cell immunoglobulin-like receptor; IFN-γ: interferon-gamma; NKp30: natural killer cell protein 30; NKp46: natural killer cell protein 46; CMV: cytomegalovirus; GVHD: graft-versus-host disease; Treg: regulatory T cell; HSPC: hematopoietic stem and progenitor cell.
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia in adults and is characterized by profound immune dysregulation, which contributes to increased morbidity and mortality [60].
A comparative analysis of NK cells and T cells in CLL reveals important distinctions. Higher NK cell counts are associated with improved prognosis in CLL patients, as NK cells are capable of targeting leukemic cells, highlighting their potential therapeutic relevance [61]. However, autologous NK cells often fail to mount an effective response against CLL cells, suggesting that leukemic cells have evolved mechanisms to evade NK cell-mediated surveillance [62].
Unlike T cells, which rely on antigen-specific receptors for target recognition, NK cells integrate signals from a repertoire of activating and inhibitory receptors to regulate their effector functions [63, 64]. Key activating receptors involved in anti-tumor responses include KIRs, NKp46, NKp30, CD16, and DNAX accessory molecule-1 (DNAM-1). However, the expression of these receptors on NK cells in CLL patients has been inconsistently reported [62, 65, 66]. CLL cells can impair NK cell recognition by downregulating ligands for activating receptors and releasing soluble factors that interfere with receptor-ligand interactions [60, 62, 67–69]. Additionally, CLL cells upregulate immunosuppressive molecules such as human leukocyte antigen (HLA)-E, HLA-G, and transforming growth factor-beta (TGF-β), which further inhibit NK cell activity [70, 71].
As a result, NK cell cytotoxic responses are diminished in the presence of CLL cells [62, 65, 66]. Nevertheless, therapeutic strategies such as anti-CD20 monoclonal antibodies can redirect NK cells to eliminate CLL cells via antibody-dependent cellular cytotoxicity (ADCC) [62, 67, 72]. Moreover, NK cell function can be at least partially restored when these cells are removed from the leukemic microenvironment, indicating that CLL cells create an imbalance between activating and inhibitory signals that impairs NK cell recognition and facilitates immune evasion [72, 73].
Recent advances in the understanding of multiple myeloma (MM) have highlighted the pivotal role of the immune system in disease progression. The BMME consists of both cellular components—including bone marrow stromal cells (BMSCs), endothelial cells, osteoclasts, osteoblasts, fibroblasts, T lymphocytes, and dendritic cells—and non-cellular components, such as the extracellular matrix (ECM) and soluble factors including chemokines, cytokines, and growth factors.
The primary function of the bone marrow stroma is to regulate and support the proliferation and differentiation of hematopoietic cells. During MM progression, interactions between microenvironmental cells—particularly endothelial cells and MSCs—and tumor clones are mediated by surface adhesion molecules, receptors, and soluble mediators secreted by these cells. These interactions promote MM cell survival, proliferation, and differentiation [74, 75].
Initial studies of tumor-infiltrating immune cells in MM suggested a dynamic relationship between immune system components and the tumor [76]. While these immune cells were initially believed to exert anti-tumor effects, more recent evidence indicates that they may also contribute to tumor progression. Interactions between ILCs and microenvironmental cells are critical for tumor development and dissemination, as both immune and tumor cells are influenced by cytokines, adhesion molecules, and metalloproteinases [77].
The early immune response to tumor formation involves the activation of cytotoxic mechanisms, recruitment of immune cells, and secretion of cytokines that induce tumor cell apoptosis [78]. However, as neoplastic cells proliferate and dominate the microenvironment, ILCs begin to produce factors that support tumor growth [3, 79]. The ECM plays a dual role in adaptive immunity: it facilitates T-cell migration into tissues while also exerting inhibitory effects on T-cell proliferation [80–82]. Interactions between ILCs and stromal cells are also evident, with ILCs potentially contributing to tumor tissue formation and expansion [83].
As previously discussed, a defining feature of ILCs is their plasticity—the ability to differentiate into various subtypes in response to environmental cues [81]. In MM, this plasticity can modulate ILC function, resulting in either anti-tumor or tumor-promoting activities [84] (Figure 3).
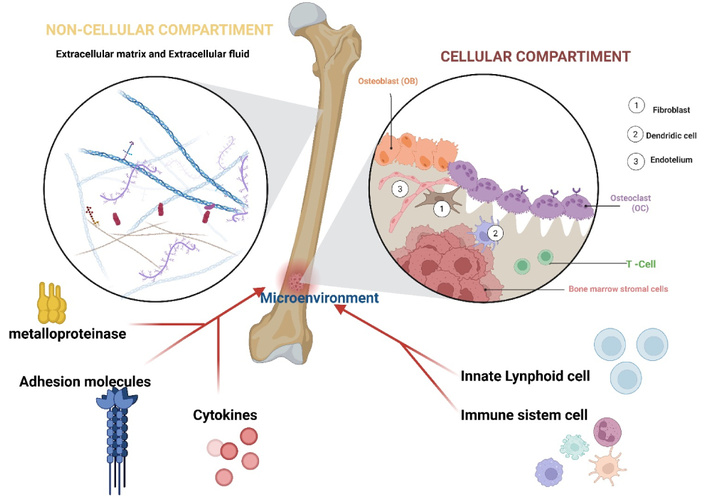
ILCs in the MM microenvironment. The BMME (bone marrow microenvironment) is composed of a cellular and non-cellular component (ECM: extracellular matrix). Through interactions with the BMME, ILCs produce inflammatory mediators, adhesion molecules, and metalloproteinases, which play a crucial role in controlling tissue homeostasis. BMSCs: bone marrow stromal cells. Created in BioRender. Mirabile, G. (2025) https://BioRender.com/zy2uoib.
Recent research has shed light on how ILCs may play a role in preventing MM. These insights could lead to novel therapeutic strategies aimed at leveraging the immune system to combat MM.
NK cells play a critical role in cancer immunosurveillance through both direct cytotoxic mechanisms and indirect immunomodulatory functions [85].
In MM, malignant plasma cells frequently express CD1d, a member of the CD1 family of antigen-presenting molecules. Structurally similar to major histocompatibility complex (MHC) class I molecules, CD1d consists of α1, α2, and α3 domains associated with β2-microglobulin. Although CD1d is monomorphic, its surface expression on MM cells renders them more susceptible to NK cell-mediated cytotoxicity [86, 87].
Interestingly, as MM progresses, a gradual decline in CD1d surface expression has been observed. While CD1d remains detectable within the cytoplasm, its surface expression diminishes significantly in advanced disease stages. This loss of surface CD1d correlates with reduced transcriptional activity, suggesting a mechanism by which MM cells may evade NK cell recognition and immune-mediated clearance [88].
ILC1s contribute to cancer prevention primarily through the production of IFN-γ [89–92]. IFN-γ induces apoptosis in malignant cells by upregulating the expression of Fas and Fas ligand (FasL) on their surface. Additionally, IFN-γ enhances tumor antigen presentation by increasing the expression of MHC molecules, thereby improving immune recognition and targeting of cancer cells [93]. It also promotes a shift toward Th1-type immune responses while suppressing Th2 cell activity [94].
In the context of MM, IFN-γ has been shown to modulate oncogenic transcription factors within malignant cells. Notably, several studies have demonstrated that IFN-γ can inhibit MM progression with efficacy comparable to that of the corticosteroid dexamethasone [94–101].
Therefore, although early research focused on IFN-γ’s anti-proliferative and immunostimulatory actions in MM, more recent research has shown that these effects can be enhanced or inhibited by complex regulatory networks, depending on the tumor microenvironment (TME) and epigenetic variables.
Although more direct comparison clinical data would support this assertion, IFN-γ’s ability to decrease IL-6 signaling and modify transcription factors supports its therapeutic equivalency to dexamethasone, as suggested in any works.
Studies have reported an increased frequency of CD56+CD3– NK cells in both the bone marrow and peripheral blood of patients with monoclonal gammopathy of undetermined significance (MGUS) and MM. Paradoxically, higher NK cell counts at diagnosis have been associated with poorer prognosis in MM patients [85]. This observation suggests that the immune system’s inability to effectively control MM cell proliferation may be linked to the increased presence of dysfunctional NK cells [85, 102].
Elevated serum immunoglobulin levels in MM can impair NK cell function, diminishing their cytotoxic capacity. Morphological and functional abnormalities—such as the presence of intracellular vacuoles, reduced ADCC, and decreased cytolytic granule content—further indicate compromised NK cell activity [103]. Moreover, monomeric IgG, as well as monoclonal IgA and IgG proteins commonly found in MM patients, have been shown to negatively affect NK cell function [97].
Although data on the role of ILC1s in the development and progression of MM remain limited, recent findings underscore their potential significance. In patients with plasmacytosis, an increased proportion of ILC1s has been observed in the bone marrow. Under normal conditions, ILC1s produce IFN-γ in individuals with MGUS; however, this production is markedly reduced in patients with asymptomatic MM [104].
Elevated expression of Ikzf3 (Aiolos)—a transcription factor essential for B-cell development and a known target of immunomodulatory drugs (IMiDs)—has been identified in human ILC1 subsets. IMiDs such as pomalidomide, which are used in MM treatment, have been shown to stimulate IFN-γ production by ILC1s, suggesting a potential therapeutic mechanism involving modulation of ILC1 activity [105].
ILC2s have been identified as a cell subset with potential pro-tumorigenic properties [105]. The ILC2/IL-13/MDSC axis contributes to the establishment of an immunosuppressive microenvironment that may facilitate tumor progression [106, 107]. Elucidating the role of this axis in MM could provide valuable insights into disease pathogenesis. However, current data on the involvement of ILC2s in MM remain limited.
In patients with plasmacytosis, a reduction in bone marrow-resident ILC2s and a concomitant increase in circulating ILC2 subsets have been observed. While ILC2s from MGUS patients retain the ability to secrete IL-13, this function appears to be lost in asymptomatic MM patients [105].
Studies suggest that ILC2s in MM are activated by IL-33, which induces phenotypic changes and upregulation of maturation markers. Nevertheless, these cells exhibit a diminished capacity to produce cytokines in response to IL-2 and IL-33 stimulation. These findings do not strongly support a direct role for ILC2s in promoting MM cell proliferation. Rather, IL-33 appears to suppress type 1 immune responses against MM and promote the expansion of circulating inflammatory populations, such as KLRG1 high ILC2s [107].
The observed depletion of ILC2s in MM may be mediated by PD-1/PD-L1 interactions, as ILC2s express the PD-1 immune checkpoint receptor, and PD-L1 is abundantly expressed within the MM microenvironment [108, 109].
Despite the fact that ILC2s have been linked to the development of an immunosuppressive milieu in MM, the available data are still few and inconsistent. Although PD-1-mediated malfunction and IL-33-driven expansion point to a regulatory role, the stage-dependent changes in ILC2 function and inconsistent cytokine responsiveness rule out a direct pro-tumorigenic effect. To elucidate their role in MM pathophysiology and therapeutic targeting, more research is required.
ILC3s have demonstrated pro-tumorigenic activity in various cancers through the release of proinflammatory cytokines such as IL-22, IL-17, and IL-23 [110–115]. Although data on the role of ILC3s in MM are currently limited, these cells may play a critical role in disease progression, particularly given the involvement of several regulatory cytokines known to influence ILC3 activation.
The participation of ILCs in MM—encompassing both their dysfunctions and their potential roles in promoting or restraining tumor development—underscores the complexity of the immune microenvironment in this disease. A deeper understanding of these interactions could pave the way for novel therapeutic strategies aimed at targeting specific ILC subsets to enhance anti-tumor immunity or mitigate tumor-promoting effects (see Table 2).
Role of innate lymphoid cell populations in MM.
| Population | MM |
|---|---|
| ILC | Anti-tumor or tumor-promoting activities have been reported [84]. |
| NK cells | Anti-tumor activity was described [86, 87]. |
| ILC1 | This cell population is able to prevent cancer by producing IFN-γ [89–91, 93, 95–97].It causes induction of programmed cell death [92]. |
MM: multiple myeloma; ILC: innate lymphoid cell; NK: natural killer.
The role of ILCs in lymphomas remains incompletely understood. Nevertheless, several studies have begun to elucidate how ILCs contribute to these malignancies, offering insights that may inform the development of novel therapeutic strategies [116–117].
In patients with NHL, the cytotoxic activity of NK cells is significantly reduced compared to HDs. Multiple studies have identified three NK cell subtypes in the peripheral blood of NHL patients: CD56 bright NK cells, CD16+ NK cells, and unconventional CD56 dim (uCD56 dim) NK cells [116–117]. Among these, CD16+ NK cells—known for their potent cytotoxic capabilities—are notably less prevalent in NHL patients than in HDs, suggesting a downregulation of this critical subset. Furthermore, CD16+ NK cells in NHL patients exhibit increased expression of CD73 and CD39, ectoenzymes involved in ATP hydrolysis and commonly upregulated in inflammatory conditions. Conversely, expression of activation markers such as CD69 and KLRG1 is reduced [116]. In CD56 dim NK cells, only CD69 expression is significantly lower in patients compared to HDs. These findings suggest that the TME in NHL may impair NK cell-mediated antitumor responses.
Within neoplastic lymph nodes of NHL patients, NK cells—particularly CD56 bright and CD16+ subsets—show elevated expression of immunosuppressive markers CD73 and CD39 compared to their counterparts in peripheral blood, further supporting the notion that the TME contributes to NK cell dysfunction [116].
In the context of B-cell lymphomas, both type I and type II natural killer T (NKT) cells have been investigated for their roles in lymphoma pathogenesis. Type I NKT cells appear to exert protective antitumor effects, whereas type II NKT cells may suppress immune responses. Although research on NKT cell involvement in hematologic tumor immune evasion is still in its early stages, findings from a murine T-cell lymphoma model indicate that type I NKT cells can inhibit tumor growth. CD1d, a molecule expressed on various human hematopoietic cells, plays a role in antitumor immunity, although its precise function remains to be fully defined. Some hematologic malignancies secrete glycolipids that interfere with CD1d-mediated antigen presentation to NKT cells, thereby facilitating immune escape. Notably, recent studies have shown that type I NKT cells can eliminate EL-4 T-cell lymphoblastic lymphoma cells both in vitro and in vivo in a CD1d-dependent manner [117].
The data show that NHL patients have a significant deficit in NK and NKT cell-mediated immune surveillance. A tumor-driven reprogramming of innate immunity is suggested by the decreased frequency and changed phenotype of cytotoxic CD16+ NK cells, which are characterized by decreased activation markers (CD69, KLRG1) and increased expression of inhibitory ectonucleotidases (CD73, CD39). In the TME, where NK cells display an even more marked immunosuppressive character, this dysfunction is further aggravated. A complicated regulatory axis is further highlighted by the distinct roles that type I and type II NKT cells play in regulating antitumor responses, with type I NKT cells showing promise as antineoplastic immune effectors. All of these results suggest that the TME plays a crucial role in regulating the activity of innate lymphocytes.
The relationship between GI lymphomas and ILCs is an emerging area of research, with intriguing yet inconclusive findings. The GI mucosa represents a highly dynamic environment, constantly exposed to a wide array of microorganisms, and relies heavily on the rapid and precise immune responses mediated by ILCs to maintain homeostasis. ILCs serve as a first line of defense against pathogens such as Helicobacter pylori (H. pylori), a well-established etiological agent of gastric lymphoma [118].
NK cells are abundant in GI tissues, particularly within the lamina propria and intraepithelial compartments [119, 120]. Their activation thresholds are modulated by environmental cues and interactions with commensal microbiota through the regulation of activating and inhibitory receptor expression [121, 122]. The functional maturation of mucosal NK cells is dependent on microbial priming via dendritic cell interactions [123, 124], and in germ-free mice, NK cell function is significantly impaired due to the absence of microbial stimulation [125]. In the context of gastric mucosal inflammation, appropriately activated NK cells can exert direct cytotoxic effects against pathogens and tumor cells, while also amplifying inflammation through cytokine and chemokine production—processes that may influence the development of lymphoproliferative disorders.
Among the helper-like ILC subsets, ILC2s have garnered particular attention in gastric mucosal and tumor immunology. ILC2s are essential for maintaining mucosal integrity and promoting tissue remodeling [126]. They are predominantly localized in the gastric mucosa and rely on signals from the stomach microbiota for their development, particularly via IL-7 receptor (IL-7R) signaling [127]. Unlike ILC2s in the intestines or lungs, which are relatively unaffected by the absence of commensal bacteria, gastric ILC2s are significantly reduced in germ-free mice [128, 129]. These cells exhibit high IL-7R expression, underscoring their dependence on IL-7 signals derived from commensal bacteria [130]. Moreover, commensal microbes stimulate the secretion of IL-7 and IL-33 in the gastric mucosa, which in turn activate ILC2s to mount immune responses against pathogens such as H. pylori [131].
Targeting ILC2s may offer novel therapeutic strategies for managing gastric inflammation and preventing tumor development. However, ILC2s may also contribute to gastric oncogenesis by promoting chronic inflammation, supporting tumor growth, polarizing macrophages toward an M2 phenotype, and interacting with other immunosuppressive cells such as MDSCs [126, 131]. Notably, the absence of ILC2s has been associated with a reduced risk of gastric cancer.
ILC3s, due to their abundance and close relationship with the gut microbiota, are key players in intestinal mucosal immunity, although their role in the gastric mucosa is less well defined. ILC3s produce IL-22 and IL-33 in response to commensal bacteria, promoting the secretion of antimicrobial peptides and indirectly activating ILC2s via IL-33 [131, 132]. Additionally, ILC3s are capable of presenting antigens to CD4+ T cells, potentially enhancing adaptive anti-tumor immune responses [133–135]. However, under the influence of TGF-β, ILC3s can transdifferentiate into regulatory ILCs or ILC1s, thereby reducing their pro-inflammatory and anti-tumor functions [135].
Collectively, accumulating evidence suggests that ILCs contribute to pro-inflammatory immune responses, direct cytotoxicity, and the initiation of adaptive immunity following H. pylori infection—functions that are critical in reducing the risk of gastric lymphoma [136].
A recent study has demonstrated direct interactions between ILCs and malignant cells in HL [137]. The research showed that a dual cytokine fusion protein, IL-12–IL-2, activates T cells and NK cells more effectively than single-cytokine formulations. CD3+ T cells and CD16+ NK cells isolated from peripheral blood exhibited enhanced proliferation in response to the IL-12–IL-2 fusion protein compared to IL-12 alone, underscoring the synergistic effect of these cytokines in amplifying immune responses.
Further analysis revealed that the fusion protein could stimulate T and NK cells upon binding to the membrane of CD30+ target cells. Notably, the IL-12–IL-2 fusion protein did not induce IFN-γ release from T cells in the absence of target cells, indicating that its immunostimulatory effects are context-dependent. When targeted to CD30+ cells via a combined antibody, the fusion protein retained its biological activity and effectively stimulated T cells, reinforcing its therapeutic potential [137].
The effects of NK cells in both NHL and HL are summarized in Table 3.
Anomalies of ILCs in NHL and HL.
| Population | NHL | HL |
|---|---|---|
| NK cells | CD56 bright NK cells showed an increased downregulation of the maturation molecule KLRG1, the activation markers CD38, CD62L, and CD94, and an upregulation of CD73 [120].CD73 and CD39 expression increased in CD16+ NKs [115].Type II NKT cells had a suppressive role in the immune response against cancer, while type I NKT cells had a protective role [93]. | When joined in an IL-12–IL-2 fusion protein, the IL-2 and IL-12 cytokine domains displayed reciprocal activity to activate T-cells; they also maintained their activity when connected to CD30+ target cells via a fused antibody.The growth of cancer in mice given saline solution as a control was prevented by a dual cytokine fusion protein. Since the HRS3-IL12-Fc-IL2 fusion protein had no effect on the growth of the C10 hybridoma, the inhibition of cancer growth was target-antigen-specific [136]. |
ILCs: innate lymphoid cells; NHLs: non-Hodgkin lymphomas; HLs: Hodgkin lymphomas; NK: natural killer; KLRG1: killer cell lectin-like receptor G1. These findings highlight the complex roles of ILCs in lymphomas and suggest promising therapeutic avenues that involve modulating ILC activity to enhance anti-tumor immunity or mitigate tumor-promoting effects.
While chimeric antigen receptor (CAR) T-cell therapy has shown considerable success in treating B-cell malignancies expressing CD19, its application in AML has been limited. This is due to fundamental biological differences between these diseases, a narrow therapeutic window, the risk of severe adverse effects, and the challenge of identifying universal surface antigens suitable for targeted therapy [138–145].
Allogeneic NK cell therapy represents a distinct and promising form of adoptive cell therapy, particularly when combined with allo-HSCT in patients with relapsed or refractory AML. Notably, one study demonstrated the successful use of IL-2-stimulated allogeneic NK cells [144]. In another approach, NK cells were induced and expanded ex vivo from CD34+ HSPCs derived from HLA-matched umbilical cord blood (UCB) [145]. In this study, six out of ten AML patients relapsed at a median of 364 days post-infusion, while four patients remained alive. Importantly, the infused NK cells continued to mature in vivo, acquiring KIRs and CD16 expression.
These findings underscore both the safety and therapeutic potential of NK cell-based therapies. Adverse events of grade 2 or higher were rare [146]. Unlike CAR T-cell therapy, clinical trials involving CAR-engineered NK cells have not reported dose-limiting toxicities, even at doses as high as five billion cells per patient [147]. However, a limitation of NK-92 cells—a continuously growing NK cell line used in some therapies—is their requirement for irradiation prior to infusion to prevent uncontrolled proliferation, necessitating repeated dosing for sustained efficacy. Future strategies may involve genetically engineering NK-92 cells with a “kill switch” to eliminate the need for irradiation.
In a phase I clinical trial, NK cells expanded ex vivo using K562 feeder cells expressing membrane-bound IL-21 (mbIL-21) were well tolerated, with only minor injection-related reactions and limited GVHD symptoms reported [148]. At a median follow-up of 14.7 months, all 13 patients remained in remission, with only one experiencing relapse. The use of mbIL-21 significantly enhanced NK cell proliferation and in vivo persistence. These encouraging results have led to a phase II trial evaluating CSTD002, a haploidentical donor-derived NK cell product generated ex vivo using PM21 nanoparticles in combination with mThbIL-21 and 4-1BB ligand (4-1BBL) [149].
Additionally, research into induced pluripotent stem cell (iPSC)-derived anti-CD19 CAR-NK cells has shown promising results in preclinical models of CD19-expressing lymphoid malignancies [150].
In conclusion, given their intrinsic and particular anti-tumor activity, accessibility as an “off the shelf” cellular therapy, lower costs, and enhanced safety, CAR-NK cells may have advantages over CAR-T cells [151].
Future efforts will focus on customizing CAR-NK cells to specifically target AML cells (Table 4).
Some of the ongoing studies on the treatment of acute myeloid leukemia with NK cells.
| Study title | NCT number | Interventions | Status | Study type |
|---|---|---|---|---|
| Haploidentical NK-cell Infusion in Acute Myeloid Leukemia | NCT01947322 | Drug: allogenic NK cells infusion | Copleted | Interventional |
| Cytokine-Induced Memory-Like Natural Killer Cells (CIML-NK) for Relapsed & Refractory Acute Myeloid Leukemia (AML) | NCT05580601 | Drug: CIML-NK cells | Recruiting | Interventional |
| CD123-CD16-NK Cells Immunotherapy for AML | NCT06835140 | Drug: donor-derived CD123-CD16 bispecific antibody-modified NK cells | Recruiting | Interventional |
| Interleukin-21 (IL-21)- Expanded Natural Killer Cells for Induction of Acute Myeloid Leukemia | NCT02809092 | Biological: NK cells + chemotherapy starting | Unknown status | Interventional |
| NK Cells as Consolidation Therapy of Acute Myeloid Leukemia in Children/Adolescents | NCT02763475 | Drug: cyclophosphamideDrug: fludarabineProcedure: NK cell infusion | Completed | Interventional |
| NK Cell Infusion for Remission Consolidation in AML: A Phase II Trial | NCT06783478 | Biological: NK cell infusionOther: placebo | Not yet recruiting | Interventional |
| Safety and Efficacy of Allogenic NK Cells in Combination With Chemotherapy in the Treatment of r/r AML After Allo-HSCT | NCT05744440 | Drug: allogenic NK cells | Unknown status | Interventional |
| NK Cell Infusion for Patients With Acute Myeloid Leukemia | NCT04221971 | Drug: chemotherapy combined with NK cell infusion | Unknown status | Interventional |
| Natural Killer (NK) Cell Transplantation for AML | NCT00187096 | Drug: cyclophosphamide, fludarabine, clofarabine, etoposide, interleukin-2Procedure: NK cell infusionDevice: CliniMACS system | Completed | Interventional |
| Study of Anti-CD33/CLL1 CAR-NK in Acute Myeloid Leukemia | NCT05215015 | Biological: anti-CD33/CLL1 CAR-NK cells | Unknown status | Interventional |
| Expanded Haploidentical Natural Killer Cells as Consolidation Strategy for Children/Young Adults With AML | NCT05334693 | Biological: expanded haploidentical NK cells | Recruiting | Interventional |
| Allogenic CD123-CAR-NK Cells in the Treatment of Refractory/Relapsed Acute Myeloid Leukemia | NCT05574608 | Biological: CD123-CAR-NK cells | Unknown status | Interventional |
| CIML NK Cells With Venetoclax for AML | NCT06152809 | Biological: cytokine-induced memory-like natural killer cellsBiological: interleukin-2Drug: venetoclax | Recruiting | Interventional |
Reference from: www.clinicaltrials.gov (accessed August 30, 2025). NK: natural killer.
CARs are crucial in CAR-T cell therapy, enabling T cells to detect cancer antigens independently of HLA and recognize a broader range of antigens compared to natural T-cell surface receptors (TCRs) [152–167].
CAR-NK cell therapy offers several advantages over CAR-T cell therapy. CAR-NK cells can be derived from bone marrow or peripheral blood mononuclear cells and are less likely to induce GVHD due to their HLA-restricted nature. Moreover, they exhibit a distinct cytokine profile that reduces the risk of cytokine release syndrome (CRS) and neurotoxicity. CAR-NK cells can also be generated from iPSCs and immortalized cell lines such as NK-92. Notably, CAR-NK cells can bypass tumor immune evasion mechanisms by engaging alternative cytotoxic pathways, including those mediated by CD16 and NKG2D [168].
Several promising CAR targets have been identified for MM [169, 170]. For instance, CD138-targeted CARs expressed in NK-92 cells have demonstrated superior efficacy against MM compared to unmodified NK-92 cells [171]. Additionally, elotuzumab, which targets CS1, has shown potential as a CAR-NK target in preclinical models [172–174].
Importantly, CAR-NK cell therapy appears to have a more favorable safety profile than CAR-T cell therapy [175–182].
In the immunological landscape of hematologic malignancies, ILCs have become important players. They play a crucial role in the pathophysiology of certain blood malignancies due to their quick reaction to environmental stimuli and capacity to influence immunological responses. Disease development, progression, and immune evasion strategies have all been linked to changes in the distribution, phenotype, and function of ILC subsets. Furthermore, certain ILC signatures—like surface marker expression and cytokine profiles—are becoming more widely acknowledged as possible biomarkers for risk assessment and illness classification. In diseases such as AML and lymphomas, where their presence is correlated with inflammatory status, immunological suppression, and clinical outcomes, recent research has demonstrated the prognostic significance of ILC dysregulation [183].
Thus, a better comprehension of ILC biology in the hematopoietic setting may improve the accuracy of diagnosis and aid in the creation of more accurate prognostic models. To completely understand their role in the dynamics of hematologic diseases, more research into their ontogeny, plasticity, and interactions with the TME is necessary.
ILCs are increasingly recognized as a critical component of CAR-based cellular therapies and are considered pivotal to the future of onco-hematology [184, 185]. CAR-NK cells, in particular, show promise as a bridging therapy to allogeneic hematopoietic cell transplantation (allo-HCT) or in combination with other agents for patients with AML, including those with relapsed or refractory disease.
Ongoing research and clinical trials are exploring various generations of CAR constructs and their therapeutic efficacy. However, overcoming adverse effects such as CRS and improving target selectivity remain essential for optimizing CAR-cell therapy. Future strategies should aim to enhance therapeutic efficacy while minimizing toxicity. In this context, investigating the interplay between CAR therapy and oxidative stress may prove crucial. Elevated levels of reactive oxygen species (ROS), produced by both cancer cells and tumor-infiltrating immune cells, contribute to a hostile TME that impairs immune function [186–191].
Reducing ROS levels may help counteract this immunosuppressive effect. For example, studies have shown that agents such as histamine or ceplene can reduce ROS production by monocytes, thereby preserving the cytotoxic function of NK and T cells [192–197]. Additionally, enhancing the production of molecules that promote anti-inflammatory cytokines while suppressing pro-inflammatory cytokines could further improve clinical outcomes.
Overall, CAR-NK cell therapy holds significant potential to advance clinical practice and improve prognosis in hematologic malignancies.
ADCC: antibody-dependent cellular cytotoxicity
allo-HSCT: allogeneic hematopoietic stem cell transplantation
AML: acute myeloid leukemia
BMME: bone marrow microenvironment
CAR: chimeric antigen receptor
CLL: chronic lymphocytic leukemia
CRS: cytokine release syndrome
CRTH2: chemoattractant receptor-homologous molecule expressed on Th2 cells
ECM: extracellular matrix
GI: gastrointestinal
GVHD: graft-versus-host disease
HDs: healthy donors
HLA: human leukocyte antigen
HLs: Hodgkin lymphomas
HSCT: hematopoietic stem cell transplantation
HSPCs: hematopoietic stem and progenitor cells
IFN-γ: interferon-gamma
ILCs: innate lymphoid cells
IMiDs: immunomodulatory drugs
iPSC: induced pluripotent stem cell
KIRs: killer cell immunoglobulin-like receptors
LTi: lymphoid tissue inducer
mbIL-21: membrane-bound IL-21
MDS: myelodysplastic syndrome
MDSCs: myeloid-derived suppressor cells
MGUS: monoclonal gammopathy of undetermined significance
MHC: major histocompatibility complex
MM: multiple myeloma
MSCs: mesenchymal stem cells
NCR+: natural cytotoxic receptor-positive
NHLs: non-Hodgkin lymphomas
NK: natural killer
NKT: natural killer T
PGD2: prostaglandin D2
Rag: recombination-activating genes
RORγt: RAR-related orphan receptor gamma T
ROS: reactive oxygen species
STAT4: signal transducer and activator of transcription 4
T-bet: T-box transcription factor TBX21
TGF-β: transforming growth factor-beta
TME: tumor microenvironment
TNF: tumor necrosis factor
Tregs: regulatory T cells
MDG: Conceptualization, Methodology, Formal analysis, Writing—review & editing, Supervision. AP: Conceptualization, Formal analysis, Writing—original draft, Writing—review & editing. GM: Methodology, Writing—review & editing. LdV: Methodology, Formal analysis. FS: Methodology, Formal analysis, Writing—review & editing, Supervision. SG: Conceptualization, Writing—review & editing, Supervision. AA: Conceptualization, Writing—original draft, Writing—review & editing, Supervision. All authors have read and agreed to the published version of the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 2783
Download: 35
Times Cited: 0
