 Review
Review

Affiliation:
Laboratory for Neurophysiology, Department of Cell and Chemical Biology, Leiden University Medical Center, 2300 RC Leiden, The Netherlands
ORCID: https://orcid.org/0009-0000-3570-4153
Affiliation:
Laboratory for Neurophysiology, Department of Cell and Chemical Biology, Leiden University Medical Center, 2300 RC Leiden, The Netherlands
ORCID: https://orcid.org/0000-0001-5506-5037
Affiliation:
Laboratory for Neurophysiology, Department of Cell and Chemical Biology, Leiden University Medical Center, 2300 RC Leiden, The Netherlands
Email: t.de_boer@lumc.nl
ORCID: https://orcid.org/0000-0002-6402-6248
Explor Endocr Metab Dis. 2025;2:101437 DOI: https://doi.org/10.37349/eemd.2025.101437
Received: March 18, 2025 Accepted: May 28, 2025 Published: July 15, 2025
Academic Editor: Charlotte Steenblock, University Clinic Carl Gustav Carus, Germany
The article belongs to the special issue The HPA Axis in Health and Disease
Circadian rhythms are present in almost every cell of the body and play important roles in various physiological processes. The central circadian clock in the suprachiasmatic nucleus (SCN) is synchronized to the environmental light-dark cycle and ensures a temporal order for the peripheral clocks, which in turn modulate tissue and organ function. This temporal organization is crucial for the precise timing of bodily processes, including sleep, glucocorticoid release, and the function of the glymphatic system. Sleep and the glymphatic system are significantly impacted by the rhythmic secretion of glucocorticoids. One important function of the glymphatic system is the clearance of waste metabolites, which most likely happens during sleep. Disruptions within the SCN, glucocorticoid rhythms, sleep, or glymphatic clearance have been implicated in compromised brain health. This review explores the current knowledge on the interdependence of the SCN, glucocorticoids, sleep, and the glymphatic system, and emphasizes their importance in homeostasis and pathology; in particular, Alzheimer’s disease.
Life has evolved under cyclic environmental conditions, driving organisms to adapt their active period to align with the 24-hour light-dark cycle. As a result, most physiological and biochemical processes, like body temperature, blood flow, and hormone release, are highly regulated and optimized throughout the day [1]. Importantly, these rhythms persist in the absence of external time cues and, since they have a period of approximately but not exactly 24 hours, are termed circadian rhythms. These circadian rhythms are present in nearly every tissue and organ system and are controlled by molecular circadian clocks. To align or entrain all these internal biological clocks to the external light-dark cycle and to maintain the right timing between physiological processes, mammals rely on a central circadian clock in the suprachiasmatic nucleus (SCN), which is located in the hypothalamus [2]. The SCN receives photic input from specialized ganglion cells in the retina to entrain its own circadian rhythm to the light-dark cycle, and it makes use of several neuronal and hormonal pathways to convey this timing information to peripheral tissues (Figure 1). This entrainment ensures that physiological processes occur at optimal times for performance and health.
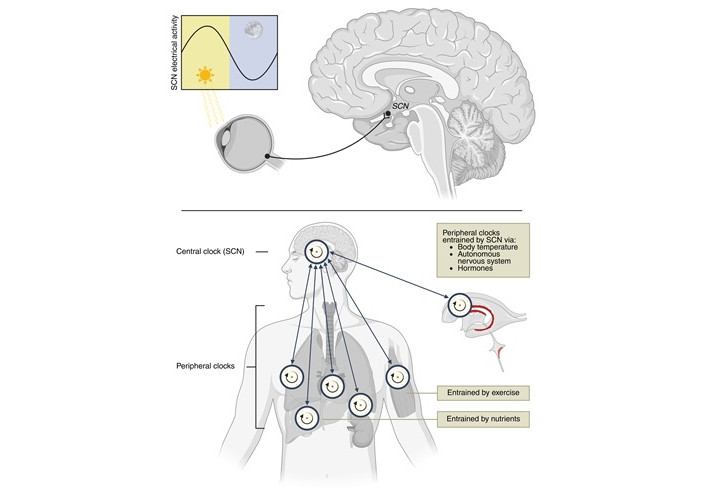
Overview of the circadian system. Upper: the suprachiasmatic nucleus (SCN), located in the hypothalamus, receives light information from specialized retinal ganglion cells to entrain its circadian rhythm to the external light-dark cycle. The electrical activity of the SCN is highest when there is light input. Lower: the SCN entrains peripheral clocks in structures like the choroid plexus (ChP) (red), lungs, heart, liver, adrenal gland, and muscles through body temperature rhythms, autonomic nervous system signals, and hormones. Peripheral clocks can also be entrained by external zeitgebers, such as exercise (muscle) and nutrient intake (liver). Reciprocally, peripheral clocks interact with the SCN. Created in BioRender. van der Zwet, L. (2025) https://BioRender.com/2t7oo3q
One of the systemic signals that is regulated by the SCN and helps to keep temporal order in physiological processes is the rhythmic release of glucocorticoids (GCs) from the adrenal gland cortex. In humans, GC levels peak at the beginning of the day, likely to anticipate and prepare the body for a period of wakefulness. In nocturnal mice, the GC level also peaks shortly before the active period, and it has been shown that this circadian rhythm in GCs is dependent on a functional SCN [3]. GCs will also cross the blood-brain barrier in a controlled way [4] and modulate a multitude of brain functions.
One brain area under the control of circadian rhythmicity and influenced by GC is the choroid plexus (ChP) [5, 6]. This epithelial tissue lines the walls of the ventricles and is the main source of cerebrospinal fluid (CSF) in the brain, an important component of fluid homeostasis in the brain [7]. The produced CSF eventually moves through the glymphatic system, a network of perivascular spaces that facilitate directional motion of fluid in the brain. This CSF movement is thought to be primarily important for clearing waste metabolites and proteins [8, 9]. In addition, the glymphatic system seems to distribute compounds important for signalling and modulating brain function [8]. The cyclic signals released by the ChP may therefore be used to synchronize circadian rhythms of cellular processes in different brain areas [5, 10, 11]. Recent research has highlighted the connections between the SCN, GCs, and the glymphatic system. For instance, Drapšin et al. [5] demonstrated that disruption of the SCN affects circadian GC regulation and that the ChP’s internal circadian clock is highly sensitive to GCs [6].
Glymphatic influx produced by the ChP and waste clearance are thought to reach peak efficacy during sleep [12], which in turn is also modulated by the central circadian clock. GCs, among others, can influence sleep patterns, and sleep disturbances are commonly reported as side effects by patients treated with exogenous GCs, as well as by patients with Cushing’s syndrome, which is characterized by excessive cortisol production [13]. As the efficacy of the glymphatic system has been associated with sleep in humans [14], sleep-wake cycles may be an important link in the interplay between the SCN, GC release, and the glymphatic system. Disruptions in the crosstalk between these systems may contribute to the etiopathology and pathogenesis of neurodegenerative disease, where less efficient clearing by the glymphatic system may lead to increased deposition of tau, α-synuclein, and amyloid-β, leading to neurological diseases like Parkinson’s and Alzheimer’s disease (AD), highlighting the importance of further investigation. However, a comprehensive review illustrating and critically analysing these interactions has not yet been presented.
So, while the elements of brain homeostasis in health and disease are being thoroughly investigated, the interactions between them, and particularly the influence of the circadian system as a potential regulator and at the same time therapeutic target, receive less attention. Therefore, this review aims to explore the interactions between the SCN and circadian rhythms, GCs, sleep, and the glymphatic system. The first section examines the SCN and its influence on sleep. The second section addresses circadian GC release and explores mechanisms by which GCs affect peripheral clocks. In the third section, the focus will shift to the ChP and the glymphatic system, linking prior sections to discuss how circadian and sleep-driven influences optimize glymphatic clearance. The final section synthesizes the connection between the SCN, GCs, sleep, and the glymphatic system and examines how dysregulation of these systems may contribute to neurodegenerative processes.
Circadian rhythms in mammals are coordinated by the SCN, a hypothalamic bilateral nucleus located just above the optic chiasm [15]. The SCN receives and processes photic information, which it uses to convey external time information to peripheral clocks, thereby synchronizing them to the environment [16]. The next section will examine the SCN and its interactions with sleep.
In the SCN, as well as in most peripheral cells, a transcriptional-translational feedback loop (TTFL) occurs in the expression of a handful of clock genes, which lasts ~24 hours [17]. The TTFL consists of a positive and negative feedback loop (Figure 2). In the negative feedback loop, a heterodimer composed of BMAL1 and CLOCK or NPAS2 [18] binds to the E-box of three different period (Per) genes and two different cryptochrome (Cry) genes. After translation and post-translational modification, PER and CRY proteins translocate back into the nucleus, where CRY interacts with the CLOCK-BMAL1 heterodimer to inhibit its own transcription. As a result of this negative feedback loop, Bmal1 mRNA reaches peak levels twelve hours in antiphase with Per and Cry mRNA levels [2].
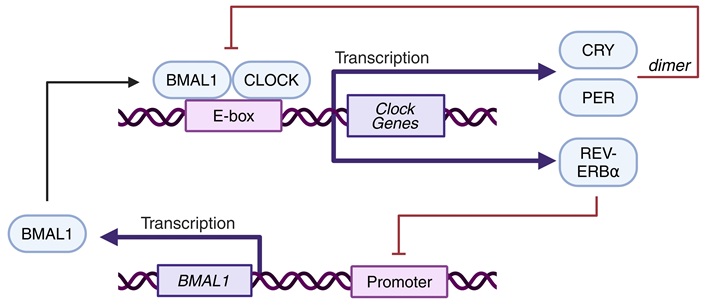
Simplified schematic representation of the transcriptional-translational feedback loop. The CLOCK-BMAL1 heterodimer activates transcription of several clock genes (Cry, Per, Rev-Erbα) by docking onto an E-box, a dimer of CRY and PER, in turn, inhibits CLOCK-BMAL1 heterodimer formation. Created in BioRender. van der Zwet, L. (2025) https://BioRender.com/klj0tg1
Besides Per and Cry, the CLOCK-BMAL1 heterodimers activate Rev-Erbα expression, starting the positive feedback loop. REV-ERBα represses the transcription of Bmal1. Therefore, when CRY inhibits the CLOCK-BMAL1 heterodimer, it also inhibits the expression of Rev-Erbα. This inhibition leads to the de-repression—thus activation—of Clock and Bmal1 transcription and resets the TTFL [2].
The TTFL does not complete in exactly 24 hours. Therefore, the SCN receives afferent information from external zeitgebers to synchronize its clock gene transcription to the time of day [19]. This entrainment allows the SCN to convey accurate temporal cues, in synchrony with the environment, to peripheral clocks.
Light is one of the most potent zeitgebers for the SCN [20]. A subset of intrinsically photosensitive retinal ganglion cells (ipRGCs) expresses melanopsin, a photosensitive pigment. When these ipRGCs detect light or receive photic information from photoreceptor cells in the retina, they process and relay this information to the SCN through the retinohypothalamic tract [21–23]. Glutamate, which is the main neurotransmitter of this tract, stimulates NMDA receptors and consequently L-type voltage-dependent Ca2+ channels on the SCN neurons, leading to an increase in intracellular calcium concentration ([Ca2+]i) [2, 24–26]. This increased [Ca2+]i can adjust transcription of the clock genes in the TTFL by activating the cAMP response element-binding protein (CREB)/CRE transcriptional pathway [27]. Besides photic input, non-photic input like behavioural activity is also known to be able to modulate the SCN phase [28, 29].
The SCN has historically been considered as the ‘master oscillator’ that forces circadian timing on ‘slave’ peripheral clocks [2]. However, recent perspectives, like that of Starnes and Jones [30], are more nuanced, proposing that interactions between the SCN and peripheral clocks are often bidirectional. Moreover, while some peripheral clocks need constant SCN input, others only require periodic cues to align their rhythm to the environment.
The two-process model of sleep regulation [31, 32] is often used to explain the timing of sleep within a circadian framework. It describes the interaction between two regulatory processes of sleep: a circadian mechanism (process C) and a sleep-dependent homeostatic mechanism (process S). Process S, which represents the sleep pressure, accumulates during wakefulness and decreases during sleep. Process S is thought to be reflected in the activity of the slow waves in the non-rapid eye movement (NREM) sleep electroencephalogram (EEG). Process C is responsible for sleep/wake initiation and is controlled by the SCN [31]. Sleep onset occurs when the rising process S passes the upper sinusoidal sleep-initiating threshold of process C. During sleep, process S decreases until it intersects with the lower wake-inducing threshold of process C, and wakefulness is initiated. While processes S and C work independently, sleep is optimal when the homeostatic drive for sleep is aligned with the circadian timing of sleep [33, 34].
This entanglement of sleep homeostatic and sleep-regulating circadian processes makes that sleep can be changed by factors that influence the clock. Exposure to light during the night can delay or advance the phase of the circadian clock depending on the timing of light exposure. Light at the beginning of the subjective night will cause a phase delay, while light at the beginning of the subjective day causes a phase advance [20]. Consequently, looking at the model, the sleep homeostatic process S might intersect sooner or later during the day with the thresholds of process C, thus resulting in advanced or delayed sleep onset, respectively.
The eventual transition from wakefulness to sleep and vice versa depends on a bistable mutually inhibitory system. During wakefulness, arousal-promoting regions in the reticular formation and posterior hypothalamus, like the Raphe nucleus, Locus Coeruleus, and the lateral hypothalamus (LH), are active [35–38]. These centres, collectively known as the ascending reticular activation system, inhibit the sleep-promoting neurons in the ventrolateral preoptic nucleus (VLPO) of the hypothalamus [39]. However, when the VLPO is more active, it inhibits the arousal-promoting regions using GABA and rapidly induces sleep [40]. This ‘flip-flop switch’, as it was named by Saper et al. [41], efficiently promotes fast switching between sleep and wakefulness and prevents intermediary states.
The flip-flop switch is thought to be stabilized by orexin, a neuropeptide produced in the LH. Orexin stimulates the arousal-promoting regions, thus reinforcing wakefulness and biasing the switch against sleep. This wake-promoting action is counteracted by the VLPO, which, when active, inhibits the orexin neurons in the LH [42].
The SCN plays an important role in sleep-wake transitions, but has only a few direct neuronal projections to the VLPO or orexin neurons [43–45]. Instead, the SCN influences these areas through an indirect, multi-synaptic pathway. The SCN projects to the subparaventricular zone [45, 46], which subsequently relays to the dorsomedial nucleus of the hypothalamus (DMH) [47]. The DMH has extensive connections to the VLPO and orexin neurons [43, 44] and is essential in relaying SCN information to the sleep-wake regulating network [48].
During the day, when SCN neuronal activity is high, the human SCN is thought to indirectly inhibit the VLPO and stimulate orexin production [49, 50] (the opposite then is thought to occur in nocturnal rodents). Both mechanisms drive the switch toward the waking state. By the end of the day, when SCN activity decreases and sleep pressure is highest, the VLPO is activated and sleep is initiated.
Sleep consists of two distinct states: rapid eye movement (REM) sleep and NREM sleep. 75% of sleep is spent in NREM sleep, during which the parasympathetic system is most active, leading to decreased heart and respiration rates [51, 52]. During REM sleep, the activity of the sympathetic nervous system dominates, and respiration and heart rate are enhanced [51, 52]. The duration of this ‘dreaming’ state increases through the night and occupies approximately 25% of total sleep [53]. Sleep onset typically begins with a period of NREM sleep, which ends in wakefulness or REM sleep. Humans usually experience 4 to 5 cycles of alternating NREM and REM states during the night [53, 54].
Previously, we have demonstrated that REM sleep significantly increases SCN neuronal activity, and that this enhancement decreases at the end of REM sleep. This effect was observed independent of the circadian phase, and SCN neuronal activity was shown to correlate with EEG slow wave activity [55]. Therefore, the interactions between the SCN and sleep homeostatic mechanisms appear to be bidirectional.
Besides timing sleep/wake cycles, the SCN regulates the hypothalamic-pituitary-adrenal axis (HPA-axis), which controls GC production. Although GCs like cortisol are commonly known as stress hormones, they are also of vital importance in non-stress-related physiology. For example, GCs maintain normal metabolism and have potent anti-inflammatory and immunosuppressive properties [56–59]. Furthermore, their rhythmic release can synchronize circadian rhythms of peripheral clocks [60, 61]. In the following part of the review, the mechanisms and patterns of GC release will be explored. Then, the influence of the SCN on the HPA-axis will be discussed, followed by a description of GC effects on peripheral clocks.
GCs are produced from cholesterol in the zona fasciculata of the adrenal gland cortex through a series of enzymatic conversions [62]. The regulation of GC synthesis and secretion starts in the paraventricular nucleus (PVN), which releases corticotropin-releasing hormone (CRH) into the median eminence either in response to stress or the circadian clock. In the anterior pituitary, CRH binds to its G-protein coupled receptor (GPCR) on corticotropic cells, activating Gαs and raising cAMP concentration, which ultimately causes exocytosis of adrenocorticotropic hormone (ACTH) into the bloodstream [63, 64]. When ACTH binds to its GPCR in the zona fasciculata of the adrenal cortex, intracellular movement of steroid precursors between cytosolic stores and mitochondria is accelerated, and the production of GCs is increased [62]. In humans, the most important GC hormone is cortisol, while corticosterone is the main GC in rodents [65].
To prevent excessive GC production and optimize secretion in unstressed situations, cortisol/corticosterone exerts negative feedback on the PVN and the anterior pituitary [66]. Research in rodents has revealed two types of corticosterone-related negative feedback: a fast nongenomic mechanism and a slower genomic mechanism. The nongenomic mechanism involves GC-mediated suppression of CRH and ACTH release by the PVN and anterior pituitary, respectively [66]. Studies have demonstrated that bilateral adrenalectomy in rats rapidly increases circulating ACTH levels and CRH immunoreactivity in the PVN [67, 68]. Levin et al. [69] add to this observation by demonstrating that exogenously administered corticosterone acts at the PVN to decrease ACTH levels in adrenalectomized rats. Lesioning the PVN before administering corticosterone, in accordance with the proposed mechanism, increases plasma ACTH levels [70].
The genomic mechanism suppresses CRH gene expression and pro-opiomelanocortin—the precursor to ACTH—transcription in pituitary corticotroph cells [66]. The exact mechanisms behind genomic suppression are largely unknown [66], but several rodent studies have shown that GCs inhibit pro-opiomelanocortin gene expression [71–73].
CRH is secreted in an ultradian rhythm. The amplitude and frequency of these ultradian peaks are determined by the phase of the circadian rhythm in GC secretion [74]. GC levels are highest just before an animal’s active period, which is early morning for diurnal species and early evening for nocturnal species [75–78]. This latter GC peak may permit the body to respond and prepare effectively for physical stressors during the active period [79]. On top of the ultradian rhythm, stressors like hypovolemia or fear can cause acute (~1 hour) peaks in GC release by enhancing the PVN’s CRH secretion [80].
The SCN is well-established as the regulator of the circadian rhythm in GC release, as studies have consistently demonstrated that lesions to the SCN and exposure to constant light result in the loss of circadian GC rhythmicity [81–84]. Despite this knowledge, the precise mechanisms underlying the SCN’s involvement in GC production remain unclear [85].
Several studies with different neuronal tracers have established connectivity between the SCN and the nuclei of the HPA-axis. All showed that the SCN has projections to the PVN/DMH, although the density of these projections remains a topic of debate [45, 46, 86, 87]. Moreover, these PVNCRH neurons show circadian rhythms in clock gene expression and neuronal activity, coordinating peak CRH release around waking [88]. Deletion of BMAL1 in PVNCRH neurons results in irregular GC secretion. Besides clock gene expression, circadian GC release also depends on SCN input to the PVNCRH neurons with inhibitory vasoactive intestinal peptide. GC release rhythms are lost without SCN input [88], thus affirming the tight causal link between the SCN and PVN in regulating circadian GC rhythms.
As some projections from the SCN use arginine vasopressin (AVP) as their neurotransmitter [46, 89], Kalsbeek et al. [90] set out to investigate the effects of AVP on corticosterone release in rats. Following bilateral lesioning of the SCN, leaving the animal arrhythmic, they implanted cannulas aimed at the PVN/DMH area. After perfusing different AVP agonists and antagonists into the PVN/DMH area, blood samples were taken at different time points. The results indicate that AVP inhibits corticosterone release in nocturnal rodents; administration of AVP suppressed corticosterone levels, while AVP antagonists stimulated corticosterone release [90]. The experiment was repeated in the diurnal rodent Arvicanthis ansorgei, which, in contrast to the nocturnal rat, responded to AVP with an elevation in corticosterone levels [91]. These results suggest that the SCN modulates the PVN/DMH induced GC release with AVP differentially in diurnal and nocturnal animals, possibly relating to the difference in temporal patterns in rhythms of GC levels.
Control over rhythmic GC secretion by the SCN seems to involve additional pathways independent of the hypothalamus and ACTH. Studies of ACTH and GC levels have revealed that while GC levels rise drastically before the active period, this is accompanied by only a minor and often insignificant rise in ACTH levels [92–94]. The existence of an ACTH-independent pathway is supported by findings that hypophysectomised rats, implanted with constantly releasing ACTH- and thyroxine pellets to maintain basal adrenal function, still display rhythmic corticosterone release [95].
An alternative pathway suggested for HPA-axis-independent control of GC secretion by the SCN is through the splanchnic nerves [85]. As part of the sympathetic autonomic nervous system, the splanchnic nerves facilitate communication between the central nervous system and visceral organs, like the adrenal gland [96]. The evidence for SCN-controlled GC release through the splanchnic nerve involves two key mechanisms: (1) photic input can modulate GC release through the sympathetic innervation of the adrenal gland, and (2) this sympathetic innervation can modulate adrenal sensitivity to ACTH [85, 97, 98] (Figure 3).
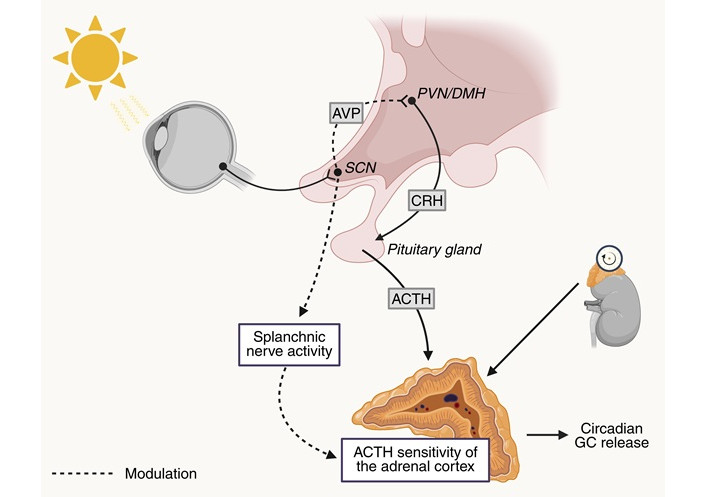
Interactions between the SCN and the HPA-axis. Photic input can modulate the sensitivity of the adrenal cortex to adrenocorticotropic hormone (ACTH) through the splanchnic nerve. Using arginine vasopressin (AVP) as a neurotransmitter, the suprachiasmatic nucleus (SCN) modulates paraventricular nucleus (PVN)/dorsomedial nucleus of the hypothalamus (DMH)-induced glucocorticoid (GC) release. Circadian timing of GC release also depends on the internal molecular clock of the adrenal cortex. Created in BioRender. van der Zwet, L. (2025) https://BioRender.com/ja7kadf
Niijima et al. [97] demonstrated that photic stimuli can alter the activity of the adrenal branch of the splanchnic nerve in rats during the active period. Following light exposure, they observed an increased discharge rate in the adrenal nerve, which was not present in rats with bilateral SCN lesions. Although their very limited sample size of SCN-lesioned animals calls for cautious interpretation, the results suggest that the modulation of the splanchnic nerve activity by photic information is mediated by the SCN [97]. Building on these results, Ishida et al. [98] later showed that photic stimulation in mice during the active period not only elevates the discharge rate in the splanchnic nerve but also rapidly increases corticosterone levels. The absence of a corresponding rise in ACTH reinforces the notion that the hypothalamus and pituitary are not involved in this pathway. Moreover, as their SCN-lesioned mice and mice without splanchnic nerves did not display increased corticosterone levels in response to photic stimuli, Ishida et al. [98] confirm the role of the SCN and splanchnic nerve in light-evoked GC rises.
The GC rise following splanchnic nerve stimulation may be attributed to the role of the splanchnic nerve in modulating adrenal sensitivity to ACTH, a mechanism that has been demonstrated across various animal models [99–101]. The sensitivity of the adrenal gland to ACTH is also modulated by a circadian clock. Adrenal sensitivity in rats peaks during their active (dark) phase, while it is low during their inactive (light) phase [94, 102]. Lesioning the SCN eliminates these day/night differences in ACTH sensitivity [103]. Notably, transection of the splanchnic nerve attenuates corticosterone levels in rodents without affecting ACTH levels [101], suggesting that the splanchnic nerve indeed sensitizes the adrenal gland to ACTH.
Altering the activity of the splanchnic nerve, along with direct control of the HPA-axis through the PVN/DMH, allows the SCN to utilize both neural and hormonal signals to fine-tune adrenal responses depending on time of day. However, some doubts remain about the control of the adrenal gland by the SCN. For instance, rats with bilateral SCN lesions still show rhythmic corticosterone secretion [104], and time-restricted feeding produces two GC release peaks, of which only one is SCN-dependent [105]. These results indicate that there are extra-SCN processes that can mediate GC secretion.
It is important to recognize that most cells possess intrinsic molecular clocks, including those in the adrenal gland [106], and can maintain their own autonomous rhythm. Therefore, the main task of the SCN is to synchronize the peripheral rhythms with external stimuli such as light. For example, in the adrenal gland, the CLOCK:BMAL1 heterodimer keeps Steroidogenic Acute Regulatory protein—the rate-limiting enzyme in GC synthesis—under transcriptional control [106]. The expression of this enzyme is unaffected by splanchnic nerve transection [101], demonstrating that the adrenal gland can independently regulate circadian GC production through its molecular clock as well.
The earlier presented evidence shows that the SCN and the adrenal molecular clock can regulate GC secretion. GCs themselves appear to act as a mediator to align the rhythms of peripheral clocks to that of the SCN. For instance, when rats are given dexamethasone—a potent GC receptor (GR) agonist—in constant darkness, this injection acts as a zeitgeber for the regulation of intraperitoneal temperature [107]. Furthermore, GC rhythms play a role in coordinating day-night cycles of bladder capacity [108], heart function [109], and metabolism [110].
GCs exert their effects through binding to the GR, a nuclear receptor known as NR3C1. When bound, the complex migrates from the cytoplasm to the nucleus, where it functions as a stimulatory transcription factor by binding to GC response elements (GREs) in the promotor of certain genes. Clock genes Per1 and Per2 were found to contain GREs [111], suggesting a direct impact on the phase of the peripheral molecular clock by GCs. Therefore, it is not surprising that the genes integral to circadian rhythm regulation are particularly sensitive to the loss of GR function, as found in zebrafish models [112].
Interestingly, studies have shown that clock proteins can interact with the GR to alter its function. For example, REV-ERBα affects the subcellular localization of the GR [113], and CLOCK-BMAL1 can acetylate the GR, thereby inhibiting its activity [114]. CRY1 and 2 also seem to oppose the actions of the GR [115]. These mechanisms might help with adjusting the phase of peripheral clocks to that of the external environment. For instance, when GC levels are acutely elevated due to stress, they can disrupt the normal synchronization of peripheral clocks to the SCN [116]. By inhibiting the GR through e.g., CLOCK-BMAL1, unwanted desynchronisation can be mitigated [114].
The GR is expressed in many peripheral cells and in the brain, but has not been found in adult SCN neurons [61, 117]. This finding suggests that GC levels do not influence the rhythmic activity of SCN neurons. However, GRs have been identified in the foetal/neonatal SCN, gradually disappearing as gestation progresses [118]. As maternal GCs can pass the placenta [119], they can entrain the foetal SCN to the maternal circadian rhythm by acting at the GRs [120].
In contrast to adult SCN neurons, GRs were found in the glial cells associated with the SCN and PVN [121, 122]. These glial cells actively control SCN activity by regulating extracellular GABA and glutamate concentrations [123]. GCs have been shown to stimulate the expression of glial fibrillary acidic protein (GFAP), an integral component of glial filaments, in SCN and PVN-associated glial cells [122]. This upregulation suggests a role for GCs in modulating glial plasticity, possibly impacting the SCN network.
Stress has profound effects on sleep/wake behaviour, often altering sleep architecture and increasing arousal. In animal models, acute stress and hydrocortisone administration have been shown to suppress sleep [124]. Moreover, conditions characterized by excessive cortisol production, like Cushing’s syndrome, are often associated with sleep disturbances [13]. Chronic corticosterone administration in rodents has been shown to disrupt sleep by influencing the brain regions involved in sleep initiation and/or wake maintenance; corticosterone activates noradrenergic neurons in the locus coeruleus, one of the arousal-promoting areas, and inhibits the GABAergic neurons of the VLPO, thereby promoting wakefulness. These GC-induced sleep disruptions are mediated through the GR, as a GR antagonist can reverse the sleep disturbances [125].
The distribution of temporal information from the central clock has been described above using mainly neuronal pathways. However, the SCN also produces and releases a number of neuropeptides, which help to regulate circadian rhythms in physiology and behaviour [126]. The released signals from the SCN can reach peripheral clocks through a specialized vascular circulation pathway to the lamina terminalis, which has recently been described to function as a portal vein [127]. Additionally, the CSF may take up and distribute signalling molecules from the SCN through a system of perivascular channels known as the glymphatic system (Figure 4) [128]. The main function of the glymphatic system was suggested to be the clearance of waste from the brain, the failure of which supposedly contributes to neurological diseases like AD [129]. The SCN, sleep, and GC signalling all seem to influence the glymphatic system, which immediately raises the question about the role of temporal alignment for the proper functioning of the glymphatic system. This part will focus on the function of the glymphatic system and the circadian rhythmicity of an important component, the ChP. Subsequently, the influence of GCs, circadian rhythms and sleep on the glymphatic system will be discussed.
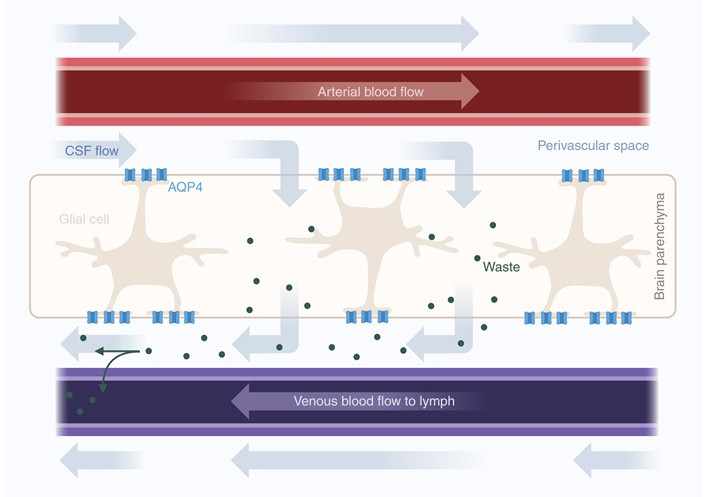
Glymphatic clearance in the brain. Cerebrospinal fluid (CSF) in perivascular spaces enters the brain parenchyma through aquaporin-4 (AQP4) channels located on the endfeet of glial cells. Convective flow through the brain parenchyma facilitates the clearance of waste, which is transported to the perivenous space and into the vein toward the lymphatic system. Arrows indicate the movement of fluids and solutes. Created in BioRender. van der Zwet, L. (2025) https://BioRender.com/bhqajuf
To support the weight of the brain and to nourish it with nutrients, growth factors, ions, and peptides, the brain is bathed in CSF [130]. CSF is produced by the ChPs, which are tufts of highly vascularized, villous structures covered by epithelia that line the brain’s ventricles [131, 132]. The epithelial cells in the ChP are connected by tight junctions, thereby creating a blood-CSF barrier and restricting unwanted movement of water-soluble molecules from the systemic blood circulation into the CSF [133]. Due to the tight junctions and adherence junctions, ChP epithelial cells are polarized and can create osmotic gradients [134]. The extensive system of fenestrated capillary loops in the ChP and the osmotic gradient govern the passive ultrafiltration of plasma into the interstitial fluid of the ChP [135, 136]. To secrete the produced fluid into the ventricles, the ChP epithelium contains aquaporins (AQPs), which are water channels [137]. Ions are actively transported into the CSF by ion transporters [138, 139].
However, increasing buoyancy and nourishing the brain are not the only functions of CSF. A study from 2012 by Iliff et al. [128] demonstrated that CSF from the subarachnoid space can enter the brain parenchyma, mix with interstitial fluid, and drain into the peripheral lymphatic system through perivascular spaces and meningeal lymphatic vessels [140]. This CSF efflux depends on astrocytic AQP4 and is likely modulated by circadian rhythms and sleep/wake cycles [12, 141]. As the system seems to have similar functions as the lymphatic system—clearing waste—and depends on glial cells, it was termed the glymphatic system [128]. The glymphatic system has been shown to clear tau, α-synuclein, and amyloid-β, which are proteins associated with neurodegenerative diseases [128, 142, 143].
A waste-clearing system is most efficient if it has directionality. In the glymphatic system, this direction is from the brain parenchyma toward the peripheral lymphatic system. Several different factors contribute to the directionality of CSF movement. For instance, the rhythmic expansion and contraction of the cerebral arteries propels the fluid through the perivascular space [144, 145], while spontaneous changes in the muscle tone of vessels (vasomotion) support steady CSF movement [146, 147]. At the same time, respiratory cycles create pressure changes that further facilitate CSF flow [148].
The glymphatic system can be compared to a closed hydraulic system, as it relies on fluid pressure and directed flow to clear waste efficiently. Just as poking a hole in a hydraulic system would disrupt its function and pressure, any disruptions in the glymphatic system can compromise its ability to clear waste. Uncareful introduction of tracers or cannulas into the glymphatic system can create leaks that disrupt the closed nature of the system and thereby inactivate it [141, 149]. Likewise, perivascular structures collapse and fill with other fluids postmortem, rendering most histological samples of this system useless [145]. These difficulties in maintaining the glymphatic system’s integrity have caused significant discussion about its existence [150]. The most recent research on the glymphatic system therefore increasingly relies on in vivo imaging techniques like diffusion tensor imaging and positron emission tomography, as this can maintain the system’s integrity and function [151, 152].
Besides the glymphatic system, other pathways have been proposed for clearing brain waste. The intramural periarterial drainage theory suggests that perivascular spaces do not exist. Instead, the CSF enters the brain via arterial pial-glial basement membranes, mixes with interstitial fluid, and leaves along the basement membrane of the smooth muscle cells in the artery [153]. Another theory proposes that convective mixing mechanisms drive the efflux of solutes from the brain parenchyma [154]. As the glymphatic pathway is the most widely accepted waste-clearing theory, this review will exclusively focus on this pathway.
The ChP exhibits many functional daily rhythms. CSF production, for instance, was observed in a diffusion MRI study to peak during the night in humans [155]. Moreover, the composition of CSF varies across the day, with Na+ concentrations in human subjects reaching peak values throughout the day and reaching lowest levels during the night [156]. These daily fluctuations can be linked to rhythmic gene expression within the ChP. For example, SNAT3 and NCX4, both sodium transporters, show upregulation during the light phase [157], aligning with the diurnal changes in CSF composition. Additional genes showing high circadian expression are related to the endoplasmic reticulum stress response. Transcriptomic data shows that ER stress response markers peak during the late subjective night in the ChP, optimizing the preparation for stress due to the rapid processing and accumulation of secreted proteins, such as transthyretin [11]. This anticipatory upregulation may prevent ER-stress-related cytotoxicity [158] and fine-tune circadian secretion.
Like most cells in the body, the cells in the ChP contain a molecular clock that can coordinate rhythmic gene expression [159]. However, unlike most peripheral clocks, the cells in the ChP can maintain rhythmic expression of clock genes in vitro without input from the SCN. This autonomous rhythmicity has been demonstrated in organotypic explants of the ChP from PER2::LUC mouse models, which showed consistent rhythmic expression over several days [10, 160]. The clock gene expression in the ChP is not only persistent, but can even show stronger oscillations than the SCN itself [10]. This observation is likely explained by the presence of gap junctions between ChP cells, which allow for strong intercellular coupling [161]. This notion is supported by the finding that inhibiting the ChP gap junctions prolongs the circadian period and reduces the PER2::LUC amplitude [10].
Although the SCN appears to be redundant for the rhythmic expression of ChP clock genes in organotypic slices, in vivo studies indicate that the SCN does play a role in sustaining ChP oscillations [11]. Sládek et al. [11] examined transcriptome profiles in control and SCN-lesioned mice, revealing significantly decreased amplitudes in Clock and other rhythmically expressed ChP genes in the SCN-lesioned group. Besides the SCN-to-ChP communication, a coculturing study has found that the ChP also influences the SCN, likely through signalling molecules in the CSF [10]. This reciprocal influence might enhance the synchronization between the SCN and peripheral clocks.
While the precise mechanism behind the SCN-to-ChP communication remains unknown, evidence suggests a role for GCs as a mediator. As described before, GC synchronizes circadian clocks through the GR. When GCs are bound to the GR, it will function as a transcription factor for genes with a GRE, like Per1 and Per2 [111]. Cells of the ChP contain GRs and are therefore susceptible to clock modulation by GCs [6, 162]. This modulation has been meticulously investigated by Liška et al. [6] in a series of in vivo and in vitro experiments. Indeed, after adrenalectomy, the rhythmic expression of Per1 and Per2 in the ChP of mice is abolished and significantly dampened, respectively. However, when these mice were given dexamethasone injections, the rhythmicity in clock genes was restored [6]. Dexamethasone was also able to restore rhythmic clock gene expression in SCN-lesioned mice [11]. In vitro, dexamethasone either delayed or advanced the Per2 rhythm in the ChP depending on the timing of administration [6].
Moreover, the ChP expresses 11β-HSD1, which locally catalyses the conversion from inactive GCs to cortisol/corticosterone [163]. 11β-HSD1 has been found to show circadian expression in the rat’s hippocampus with peak levels occurring around activity onset [164]. Through 11β-HSD1, the ChP might adjust the signals from the HPA-axis to maintain circadian CSF homeostasis. However, as there has been no detailed investigation on diurnal oscillations of 11β-HSD1 in the ChP, the functional role of this protein in ChP physiology remains speculative.
While the glymphatic system is increasingly being accepted as an integral part of central nervous system physiology, the diurnal rhythm of its clearance function is still under debate. To date, the majority of evidence suggests a higher flow of CSF and clearance during sleep [12, 155, 165, 166]. Since sleep regulation and the central circadian clock are closely intertwined, as described above, it remains unclear whether this increased clearance is sleep-driven or circadian-driven, and studies are needed to distinguish the effect of sleep states from the potentially direct effects of the circadian clock on glymphatic flow.
Before exploring the efficacy of the glymphatic system in different vigilance states, it is important to discuss the use of anaesthetics. Most animal experiments investigating the interaction between sleep and the glymphatic system rely on anaesthetic drugs to mimic sleep. However, this approach has limitations. First, although some anaesthetics induce EEG patterns that resemble those seen in NREM sleep [167], sleep and anaesthesia remain two distinct states. For instance, anaesthesia fully inhibits locomotion and autonomic reflexes and produces a more extensive disruption of brain connectivity [168]. Second, different anaesthetic drugs were demonstrated to alter the glymphatic system in different ways, potentially introducing confounding factors to the experiments. Dexmedetomidine, propofol, and pentobarbital were shown to increase glymphatic function in rodents [169, 170], while inhaled isoflurane and ketamine inhibit glymphatic function [171]. Both increases and decreases in glymphatic influx have been observed when ketamine is combined with different doses of xylazine [165, 171]. Thus, the effects of anaesthetic drugs on glymphatic function are both drug- and dose-dependent and cannot be directly compared to a natural sleeping state. Anaesthetics can, however, be used to investigate circadian effects by applying them at different circadian phases.
In 2013, Xie et al. [165] elegantly investigated CSF influx in sleeping and awake mice using in vivo two-photon imaging. While recording electrocorticography (ECoG) and electromyography to determine the mice’s vigilance state, they injected fluorescent tracers into the subarachnoid CSF. Compared to sleeping mice, parenchymal and periarterial tracer influx was sharply reduced in awake mice. This suppressed influx in awake mice appeared to be linked to decreases in interstitial space volume (ISV), as sleeping mice were observed to have an expanded ISV compared to awake mice [165]. The observed changes in ISV might explain the change in CSF influx and the higher efficiency of the glymphatic system during sleep, as increased ISV may decrease resistance to CSF movement, thereby increasing CSF influx [165].
The ECoG of sleeping mice exhibits significantly more slow waves than the ECoG of awake mice. A subsequent study revealed a positive correlation between the ECoG power density of these slow waves and CSF tracer influx in anaesthetized rodents [170]. Notably, the authors used six different anaesthetics, which have varying actions in increasing and decreasing glymphatic influx and likewise in modulation ECoG slow-waves, thereby increasing the reliability of their findings. A study by Fultz et al. [14] has also found coupling between neuronal activity, hemodynamic oscillations, and CSF flow. Additionally, a decreased heart rate, which is a NREM sleep characteristic, was found to positively correlate with tracer influx [170].
Recently, a study from Miao et al. [172] questioned the general consensus on the effects of sleep on brain clearance by stating that brain clearance is reduced during sleep and anaesthesia. Here, the authors measured the diffusion coefficient of their neuronal tracer by tracking its movement from the caudate putamen to the frontal cortex and validated the result in agarose gels. The authors found no difference in diffusion coefficient between waking, sleep, and anaesthesia. Yet, when measuring the clearance of the tracer, a significant clearance reduction was observed in sleeping mice compared to awake mice [172].
However, with their experiments, Miao et al. [172] deviate from commonly used methods to assess glymphatic function. Tracer studies are often performed by injecting the tracer into a ventricle and following its path through the glymphatic system [12, 165, 170]. In contrast, Miao et al. [172] injected their tracer directly into the caudate putamen, which they argue better mimics the location of waste molecules in physiological conditions. Moreover, they used a smaller tracer (4 kDa), compared to previously used tracers (66 kDa) [12, 170], and proteins like the amyloid-β precursor (110–135 kDa) [173]. These methodological discrepancies hamper the comparison to earlier work. Therefore, future research should focus on using similar methods and similar-sized tracers to elucidate whether there are genuine discrepancies in the results obtained or only in the interpretations of the results between the different laboratories.
The evidence that links the efficacy of the glymphatic system to circadian rhythms is mainly focussed on AQP4. AQP4 is a bidirectional water channel in the brain that is most abundant in astrocytes [174]. The importance of AQP4 was established by Iliff et al. [128], who demonstrated through AQP4 KO mice that AQP4 is crucial for coupling CSF influx from para-arterial spaces to clearance of the interstitial fluid. To establish efficient coupling, AQP4 needs to be polarized to the endfeet of the astrocytes [12]. Other studies confirm that changes in AQP4 expression or polarization disturb the glymphatic system [175, 176]. Indeed, both Aqp4 gene expression and polarization show diurnal variation in nocturnal rodents; during their resting phase, Aqp4 expression is upregulated compared to the active phase, and AQP4 is localized at the endfeet of astrocytes [12]. This temporal polarization is likely dependent on the circadian expression of the proteins within the dystrophin-associated complex, a membrane-associated scaffold [12, 177, 178].
Hablitz et al. [12] demonstrated that circadian AQP4 polarization is preserved in constant light conditions. In this experiment, mice were kept in constant light for ten days before being anaesthetized with consistent doses of ketamine/xylazine during the subjective mid-resting or mid-active period. AQP4 polarization and glymphatic influx were found to be highest during the mid-resting phase [12], indicating that AQP4-mediated glymphatic clearance is circadian-dependent.
Moreover, various other circadian-regulated processes may promote glymphatic efficacy during the resting period. For example, CSF production by the ChP is significantly higher during the resting phase compared to the active phase [155]. One problem is that the number of studies investigating circadian functioning of the glymphatic system is limited and none of them is able to separate circadian changes from sleep regulatory influences. Therefore, these studies should be interpreted with caution.
However, although certain aspects that influence glymphatic efficacy can be attributed to circadian or sleep-related processes, it is essential to emphasize that these processes are intimately related. As discussed, the circadian rhythm of the SCN mainly influences sleep timing. Sleep deprivation decreases SCN neuronal activity [179], alters the circadian expression of clock genes, and decreases their amplitude [180–182]. Given this interdependence between sleep homeostatic mechanisms and circadian sleep timing, it is necessary to apply sleep-wake protocols where sleep timing is separated from the circadian cycle [183–185] to investigate this further.
In previous parts of this review, the individual interactions between the SCN, circadian rhythms, GCs, sleep, and the glymphatic system were explored. The following paragraphs will review how these systems work in concert to maintain brain homeostasis in humans, and how disruptions may contribute to neurodegenerative diseases like AD (Figure 5).
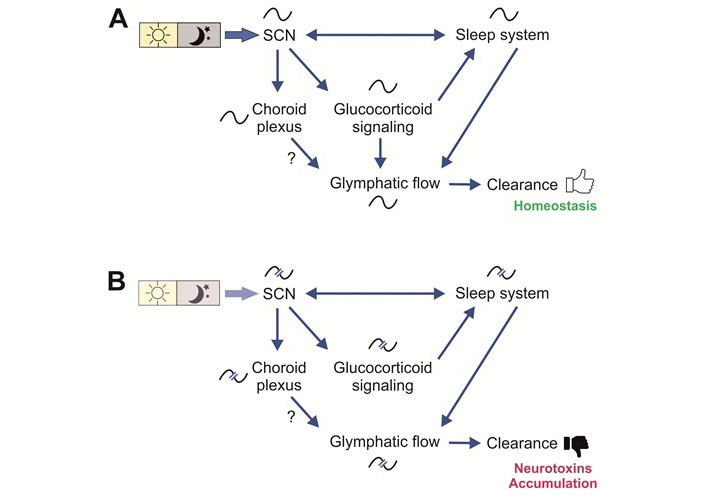
Interactions in health and disease. (A) Interactions in health. The circadian rhythm in the suprachiasmatic nucleus (SCN) receives light input and is thereby synchronized (entrained) to the environmental cycle of day and night. The SCN coordinates circadian rhythms in peripheral clocks by means of neuronal or hormonal signalling pathways to create a temporal order. The sleep system is using the time information issued by the SCN to adjust sleep/wake patterns and it also reports its status back to the SCN. The rhythm in glucocorticoid (GC) level is entrained by SCN signals which are mediated through the HPA-axis. GC signalling influence the sleep system as well as the rhythmic production of CSF in the choroid plexus (ChP). The rhythms in the ChP also dependent on SCN function. Rhythmic GC signalling as well as sleep stages (NREM) contributes to a day–night difference in glymphatic flow, which was suggested to lead to a nightly clearance of neurotoxic substances and foster brain homeostasis. The rhythmic production of CSF in the ChP may also contribute to the modulation of flow. (B) Interactions in disease. In diseases like Alzheimer’s, early symptoms include disturbance in the sleep/wake pattern, which lead to a disruption of the rhythms in GCs and glymphatic flow. Circadian disorders caused by irregular activity schedules and light regimes as well as by weakened circadian system as found in the elderly, will contribute to this downward spiral, leading to a less efficient clearance and accumulation of neurotoxins
One of the primary signalling pathways the SCN uses to synchronize peripheral clocks to the external environment is rhythmic GC release [81–83]. In the early morning, the SCN stimulates GC production by the HPA-axis through the PVN/DMH, and via sympathetic activation of the splanchnic nerve [88, 97, 98]. Notably, the adrenal gland possesses an intrinsic clock, which is aligned by the SCN, but can also independently release GCs [106]. This morning increase in GCs activates the arousal-promoting areas and inhibits the sleep-promoting areas (VLPO) [124, 125]. Simultaneously, the homeostatic sleep pressure reaches its trough and the circadian process promotes wakefulness [31, 32].
During the day, when its activity is highest, the SCN inhibits the VLPO and stimulates orexin production, stabilizing wakefulness throughout the day [49, 50]. As the evening approaches, the activity of the SCN gradually declines, and the inhibition of the VLPO wanes. In addition, the pineal gland, whose circadian rhythm is entrained by the SCN, starts producing melatonin [186, 187]. Melatonin can further attenuate SCN firing [188–190], amplifying the decreased inhibition of the VLPO. In addition, sleep pressure, which has been accumulating throughout the day, is thought to reach the sleeping threshold of process C. This series of events facilitates the transition from wakefulness to sleep.
During sleep, the ChP, entrained by GCs, increases CSF production [155]. The prevailing hypothesis indicates that the glymphatic system reaches peak efficacy during sleep, clearing the brain of amyloid-β, tau, and α-synuclein [128, 165]. As the night progresses and ends, the sleep pressure gradually declines, and GC release and SCN activity rise, allowing the cycle to restart.
In AD, those processes are disrupted. AD is a progressive neurodegenerative disorder characterized by dementia. The severity of dementia correlates with the amount and distribution of neurofibrillary tangles, which likely cause the clinical symptoms of the disease. The tangles are mainly composed of cytoskeleton-associated tau proteins [191]. The cascade of events that leads to neurofibrillary tangle formation is thought to be initiated by atypical amyloid-β secretion [192].
In addition to cognitive decline, AD is also associated with sleep and circadian dysfunction [193, 194]. Rather than solely presenting as symptoms, accumulating evidence suggests that these disruptions contribute to AD pathogenesis [195]. Sleep deprivation increases amyloid-β deposition in both healthy humans and AD mouse models [196, 197]. In a mouse model for AD, depositions could be reduced by injecting the mice with orexin antagonists, which stimulate sleep [197]. Moreover, in humans, high orexin concentrations in CSF have been associated with elevated levels of phosphorylated tau, present in the neurofibrillary tangles [198].
As the glymphatic system seems to clear amyloid-β and tau from the interstitial space most efficiently during sleep, disruptions of sleep and circadian rhythms may contribute to AD pathogenesis through dysfunction of the glymphatic system. Indeed, in AD patients, CSF production is lower than in healthy humans [199], the ChP shows altered secretion of neuroprotective peptides related to amyloid-β [200], and AQP4 polarization to the perivascular endfeet is lost [201].
Furthermore, a post-mortem study has demonstrated that the volume of the SCN and the number of SCN neurons in AD patients are decreased compared to non-AD patients [202]. In humans, the loss of SCN neurons is associated with amplitude-dampened and fragmented locomotor activity [203]. In addition, different brain regions of AD patients lose cohesion in circadian clock gene expression [204], indicating reduced functionality of the SCN. Molecular findings implicate a role of amyloid-β in the desynchronization of brain regions, as it facilitates BMAL1 degradation in neurons [205]. BMAL1 has been shown to regulate the expression of redox defence genes, so disruption of this clock gene can cause neurodegeneration due to oxidative stress [206].
Perhaps due to SCN deterioration, AD patients show reduced melatonin levels and rhythmicity [202, 207]. In addition, the SCN of late-stage AD patients expresses fewer melatonin type 1 receptors [207], the receptor that suppresses SCN firing activity and thereby allows VLPO activation. The reduced melatonin levels, rhythmicity, and ability to give feedback to SCN likely contribute to the sleep disruption observed in AD patients. Importantly, melatonin has been shown to reduce the production of amyloid-β [208]. Loss of proper melatonin levels and rhythmicity may therefore be detrimental to neuronal health in AD.
Dysregulation of the HPA-axis also plays an important role in the development of AD. AD patients show high basal cortisol levels [209, 210] and higher awakening cortisol levels, which are associated with worse memory performance [211]. In a 6-year follow-up study, AD patients with high cortisol levels declined faster in global cognition, episodic memory, and executive scores compared to AD patients with low cortisol levels [212]. Green et al. [213] have shown that GCs increase amyloid-β formation and neurofibrillary tangle formation in AD mouse models. As GCs also alter sleep architecture and increase arousal, the resulting loss of sleep might be harmful for the maintenance of amyloid-β and tau clearance by the glymphatic system.
Moreover, chronically high GC levels result in a loss of hippocampal volume [214], which is an early biomarker of AD progression [215]. In health, the hippocampus inhibits CRH and thus GC release [216, 217]. Therefore, the withering of the hippocampus results in a vicious cycle of GC release, exacerbating HPA-axis dysfunction. The presence of elevated HPA-axis activity in AD patients is corroborated by the finding that AD patients show increased adrenal sensitivity to exogenous ACTH [218].
In addition, GCs stimulate the expression of GFAP, a marker for abnormal astrocyte proliferation (reactive astrogliosis) [219]. Indeed, abnormally high GFAP levels have been observed in humans with high brain amyloid-β load [219, 220]. GFAP expression correlates with amyloid-β plaque density in AD brain tissue. As astrocytes can degrade amyloid-β [221], the increased GC levels in AD patients may initially serve as a protective mechanism. However, reactive astrogliosis is also closely associated with a decline in glutamate transporters in astrocytes [222], leaving the proximal neurons vulnerable to excitotoxicity.
In conclusion, sleep, circadian rhythms, the glymphatic system and GCs seem to be closely related in the pathogenesis of AD [129]. Further experiments are needed to gain more mechanistic insight on this as the evidence is now mainly correlative. Given the interdependencies of the circadian clock, sleep system, HPA signalling and the glymphatic function, the task is challenging but also holds great therapeutic potential. Treatment of circadian disorders with chronotherapy could normalize sleep patterns and GC signalling and have a positive effect on amyloid clearing by restoring glymphatic flow pattern. This could lead to either delayed onset or slower progression of not only AD, but also other neurodegenerative diseases like Parkinson’s and Huntington’s [223, 224].
Maintaining correct temporal organization among the discussed circadian systems is probably essential for brain homeostasis, with disruptions increasing the risk for neurodegenerative diseases. However, the pathways and mechanisms used by the circadian system to maintain a healthy temporal order are still poorly understood. This review explored the intricate interplay between the circadian system, GC signalling, the glymphatic system, and sleep. While the SCN is traditionally regarded as the controlling factor of circadian rhythms across these systems, emerging evidence suggests that the SCN also receives feedback from the peripheral clocks it entrains. This evolving perspective positions the SCN central in a bidirectionally regulated circadian network, instead of hierarchically above peripheral clocks.
Many open questions remain regarding the interactions between the central clock and peripheral clocks. For instance, the ChP has been shown to exhibit circadian patterns in the transport and degradation of endogenous signalling molecules in CSF [11]. As many brain structures, including the SCN, are exposed to CSF, the ChP may function as a conveyor of circadian signals [10]. For example, there is evidence for rhythmicity of prostaglandin D2 (PGD2) in the CSF, which promotes sleep [11], but the nature of circadian-rhythm-altering molecules in the CSF has not yet been identified. An important question that remains is the relative contribution of sleep and the circadian clock to glymphatic flow, which is important for future treatment possibilities. Should the treatment focus be on enhancing sleep quality or on increasing circadian amplitude? Finally, establishing standardized and reliable methodology will help uncover the exact relationship between sleep, circadian rhythms, and the glymphatic system.
An extensive understanding of the crosstalk between the circadian system, GC signalling, the glymphatic system, and sleep is imperative for preventing and treating circadian morbidity. Currently approved AD medicine focuses on symptom management and directly reducing amyloid-β plaques [225]. However, enhancing the function of the glymphatic system by restoring circadian control and improving sleep may offer an alternative approach by indirectly reducing amyloid-β deposition. Future research should therefore focus on increasing the robustness of the circadian system, and exploring the function of cells that are integral to all discussed systems, like glial cells. These findings may uncover novel therapeutic targets for improving brain health and resilience.
ACTH: adrenocorticotropic hormone
AD: Alzheimer’s disease
AQP4: aquaporin-4
AVP: arginine vasopressin
ChP: choroid plexus
CRH: corticotropin-releasing hormone
Cry: cryptochrome
CSF: cerebrospinal fluid
DMH: dorsomedial nucleus of the hypothalamus
ECoG: electrocorticography
EEG: electroencephalogram
GC: glucocorticoid
GFAP: glial fibrillary acidic protein
GPCR: G-protein coupled receptor
GR: glucocorticoid receptor
GREs: glucocorticoid response elements
HPA-axis: hypothalamic-pituitary-adrenal axis
ipRGCs: intrinsically photosensitive retinal ganglion cells
ISV: interstitial space volume
LH: lateral hypothalamus
NREM: non-rapid eye movement
Per: period
PVN: paraventricular nucleus
REM: rapid eye movement
SCN: suprachiasmatic nucleus
TTFL: transcriptional-translational feedback loop
VLPO: ventrolateral preoptic nucleus
LCAvdZ: Conceptualization, Visualization, Writing—original draft, Writing—review & editing. SM: Conceptualization, Supervision, Visualization, Writing—original draft, Writing—review & editing. TD: Project administration, Supervision, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Stefan R. Bornstein ... Waldemar Kanczkowski
Amanpreet Kaur Kalsi ... Jai Bhagwan Sharma
Eva B. van Dijk ... Lara E. Graves
Waldemar Kanczkowski ... George P. Chrousos
John Milton, Alexander Churilov
