 Original Article
Original Article

Affiliation:
1Department of Zoology, Dyal Singh College, Karnal, Haryana 132001, India
Email: kalsi.amanpreetkaur@gmail.com
ORCID: https://orcid.org/0000-0002-6395-0594
Affiliation:
2Department of Reproductive Biology, All India Institute of Medical Sciences, New Delhi 110029, India
Email: ashutoshhalder@gmail.com
ORCID: https://orcid.org/0000-0002-3116-3502
Affiliation:
2Department of Reproductive Biology, All India Institute of Medical Sciences, New Delhi 110029, India
ORCID: https://orcid.org/0000-0002-1613-0753
Affiliation:
3Department of Obstetrics and Gynecology, All India Institute of Medical Sciences, New Delhi 110029, India
Explor Endocr Metab Dis. 2024;1:39–55 DOI: https://doi.org/10.37349/eemd.2024.00006
Received: June 05, 2023 Accepted: August 22, 2023 Published: May 22, 2024
Academic Editor: Charlotte Steenblock, University Clinic Carl Gustav Carus, Germany
The article belongs to the special issue The HPA Axis in Health and Disease
Aim: Epigenetic alterations have been reported in patients with pituitary tumors and those on antipsychotic drugs, which are also responsible for hyperprolactinemia. This suggests a possible role of epigenetics in the etiopathology of hyperprolactinemia.
Methods: The study recruited 83 hyperprolactinemia cases with prolactin > 100 ng/mL and 65 controls. Global DNA methylation status was studied by MethylFlash Methylated DNA Quantification Kit and genome-wide methylation analysis (GWMA) by Infinium Methylation EPIC BeadChip 850K array.
Results: Hyperprolactinemia cases showed significant global DNA hypermethylation compared to controls. Around 66.67% of hypomethylated and 12.9% of hypermethylated cases were on antipsychotics. Gene enrichment analysis of 5-cytosine-phosphate-guanine-3 (CpG) site-associated genes demonstrated significantly enriched major histocompatibility complex (MHC)-related protein classes and cellular components.
Conclusions: The study suggested the role of epigenetics in the etiopathology of hyperprolactinemia.
An important mechanism by which environmental agents can affect gene expression is by changing epigenetics—DNA methylation, histone modification, and microRNA (miRNA) [1]. DNA methylation refers to adding a methyl group to the cytosine base of DNA. The sequence-specific pattern of DNA methylation is required for proper genome functioning. DNA methylation is often associated with transcription repression, although the effect may vary based on genomic context and locus [2, 3]. Studies have reported significant associations between DNA methylation patterns and hormonal variations [4, 5]. Nugent et al. [5] reported that hormone levels affect DNA methylation status, regulating gene transcription throughout the entire lifetime.
Prolactin hormone is synthesized and secreted mainly by the anterior pituitary lactotroph cells and is regulated by a dopaminergic pathway [6]. Dopamine, released from the tuberoinfundibular neurons of the hypothalamus [7], binds to Gi/o coupled D2-type receptors on lactotroph cells of the anterior pituitary and inhibits prolactin synthesis and secretion [6, 8]. Besides dopaminergic regulation, prolactin is under direct regulation of corticosteroid hormones, which bind to glucocorticoid receptors on lactotroph cells and inhibit prolactin secretion [9]. Both receptor types employ different pathways to ultimately target the proximal promoter region of the prolactin gene, inhibiting prolactin gene transcription [10–15]. There is evidence for epigenetic role in dopamine D2 receptor (D2R)-dependent inhibition of the prolactin gene where histone deacetylation and corepressor complex recruitment occur at the prolactin gene promoter region [8]. A reduction in D2R has also been shown to increase prolactin production via epigenetic mechanisms [16].
The pathological condition where prolactin level rises above the normal range is called hyperprolactinemia, which has serious side effects like galactorrhoea, irregular menstrual cycle, secondary amenorrhea, infertility, bone loss, etc. [17]. The prevalence of hyperprolactinemia varies from 40–70% in patients with antipsychotics based on individual antipsychotic medication [18]. Besides antipsychotics, hyperprolactinemia is also caused by pituitary micro/macro adenomas [19], some anti-acidity drugs, other secondary causes, and some idiopathic cases [20]. Most pituitary tumors arise due to gene-specific changes in the epigenome [17, 21–26]. Studies are reporting epigenetic alterations in patients with pituitary tumors and those on antipsychotic drugs, conditions that are also responsible for hyperprolactinemia [24, 25]. Antipsychotic drugs are known to mediate their action by methylation changes in dopaminergic and non-dopaminergic pathways, which function by altering the transcription pattern of target genes [17, 26]. A single genetic cause of hyperprolactinemia was found in three sisters of the same family, showing germline loss-of-function mutation in the prolactin receptor gene [27].
Lack of familial cases and genetic associations of hyperprolactinemia on one side and evidence of epigenetic involvement in prolactin regulation in patients with pituitary tumors, on antipsychotic drugs, and those treated with dopamine agonist, cabergoline, used in treatment of hyperprolactinemia on the other side [28], makes it reasonable to hypothesize that epigenetic factors might be involved in etiopathology of hyperprolactinemia condition.
This study was conducted in the Department of Reproductive Biology, All India Institute of Medical Sciences (AIIMS), New Delhi. Patients having prolactin levels > 100 ng/mL on two occasions of > 1-month intervals were included in the study. Informed consent was obtained from all individual participants included in the study. Prolactin assays were performed using highly specific Chemiluminescence Microparticle Immunoassay (CMIA, 7K76 G6-5314/R06 B7K760) using ARCHITECT PLUS i2000SR automated immunoassay system (Abbott Laboratories, USA). Eighty-three female patients with prolactin levels > 100 ng/mL were prospectively enrolled in the study after obtaining written consent. Detailed clinical data was recorded as per predetermined proforma. The minimum initial evaluation consisted of a complete medical history, physical examination, and hormone measurements besides computed tomography (CT)/magnetic resonance imaging (MRI) to evaluate the pituitary fossa for prolactinoma and/or other pituitary adenoma. Screening for macroprolactinemia was done in all hyperprolactinemia patients using the polyethylene glycol (PEG) precipitation method [29]. Macroprolactinemia was considered when prolactin recovery was < 25% of the initial prolactin value.
Based on the cause of hyperprolactinemia, the cases were classified into drug-induced (n = 37), idiopathic (n = 28), and pituitary micro/macro adenoma (n = 18). Additionally, 65 healthy fertile females aged 20 to 35 years with regular menstrual cycles were included as controls. Approximately 5 mL EDTA blood was taken for manual DNA extraction by Miller’s method, as described further [30]. The blood was taken in a 50 mL Falcon tube. Then, 1X red blood cell (RBC) lysis buffer (RLB) was added to the 50 mL mark, and the contents were inverted and mixed. The Falcon tube was kept as such for 20 min at room temperature. The tube was centrifuged at 4,000 rpm for 10 min at room temperature. The supernatant was discarded. After this, 15 mL 1X RLB was added to the tube, and the pellet was dissolved by tapping. It was then centrifuged at 3,500 rpm for 10 min. The supernatant was discarded, and the tube was kept inverted on a blotting sheet to decant the supernatant completely. Around 12 mL nuclear lysis buffer (NLB) was added to the tube, and the pellet was dissolved by tapping. Following this, 800 µL 10% sodium dodecyl sulphate (SDS) was added, and contents were mixed thoroughly by tapping. In the next step, 50 µL proteinase K was added, and after the contents were thoroughly mixed, the tube was incubated in a water bath at 56℃ for 2 h. When the incubation ended, 4 mL of 6 mol/L sodium chloride (NaCl) was added, and the contents were mixed by tapping. The tube was then centrifuged at 3,100 rpm for 30 min. The supernatant containing DNA was transferred to a new 50 mL Falcon tube. DNA was precipitated by adding 2X volume of 100% ethanol and inverting the tube several times until strands of precipitated DNA were visible. The precipitated DNA was transferred to 70% ethanol in a 1.5 mL Eppendorf tube with the help of a wide-bored pipette tip. The Eppendorf tube was centrifuged at 13,000 rpm for 10 min at room temperature. The supernatant was removed, and the pellet was allowed to dry completely. To the dried DNA pellet, 1 mL elution buffer was added and was left overnight for the pellet to dissolve in the buffer. The next day, the Eppendorf tube was incubated in a water bath at 56℃ for 2 h to ensure complete DNA mixing in the elution buffer. DNA was finally quantified using Nanodrop (NanoDrop 1000, NanoDrop Technologies via Thermo Fisher Scientific USA Serial No. B571 Model ND-1000 UV/Vis).
The global DNA methylation status of these 83 cases and 65 controls were studied by MethylFlash Methylated DNA Quantification Kit (colorimetric) from Epigentek (base catalog# P-1034). The assay was performed according to the manufacturer’s protocol using 100 ng input DNA per sample, bound to the plate wells specifically treated to have a high affinity for DNA. Methylated DNA fraction was detected by capture and detection antibodies and quantified colorimetrically by reading absorbance at 450 nm in a microplate spectrophotometer. The amount of absolute methylated DNA was calculated by making a standard curve of optical density (OD) values and the amount of positive control (ME4) at each concentration point. ME4 is a methylated polynucleotide containing 50% of 5-methylcytosine (5-mC). The slope (OD/ng) of the standard curve was determined using linear regression, and the most linear part of the standard curve was used for optimal slope calculation. The amount and percentage of Methylated DNA (5-mC) in total DNA was then calculated using the formulas:
Where “ME3” represents negative control, is an unmethylated polynucleotide containing 50% of cytosine, “S” represents the amount of input sample DNA in ng, “2” represents a factor to normalize 5-mC in the positive control to 100%, as the positive control contains only 50% of 5-mC.
The mean of 5-mC (%) of controls was calculated, and cases with 5-mC (%) > 2 fold or < 0.5 fold the control mean was said to have significant differences in global DNA methylation and thus were considered hypermethylated and hypomethylated. Statistical significance between groups was determined by comparing P values.
Based upon the global DNA methylation study findings, six significantly hypomethylated and six hypermethylated cases and four controls were selected for genome-wide methylation analysis (GWMA). The details of the selected 12 cases are described in Table 1.
Details of hyperprolactinemia cases selected for GWMA
| Serial No. | Sample No. | Cause of hyperprolactinemia | True hyperprolactinemia or macroprolactinemia |
|---|---|---|---|
| 1 | DBN 6 | Pituitary adenoma | True hyperprolactinemia |
| 2 | DBN 7 | Pituitary adenoma | True hyperprolactinemia |
| 3 | DBN 8 | Idiopathic | True hyperprolactinemia |
| 4 | DBN 16 | Idiopathic | Macroprolactinemia |
| 5 | DBN 30 | Antipsychotics | True hyperprolactinemia |
| 6 | DBN 39 | Idiopathic | Macroprolactinemia |
| 7 | DBN 51 | Pituitary adenoma | Macroprolactinemia |
| 8 | DBN 54 | Idiopathic | Macroprolactinemia |
| 9 | DBN 68 | Idiopathic | True hyperprolactinemia |
| 10 | DBN 75 | Antipsychotics | True hyperprolactinemia |
| 11 | DBN 92 | Antipsychotics | Macroprolactinemia |
| 12 | DBN 94 | Antipsychotics | True hyperprolactinemia |
DBN: data base number
Whole blood DNA samples of 12 hyperprolactinemia cases and four controls were sent to MedGenome Labs Ltd. for GWMA using Infinium Methylation EPIC BeadChip 850K array with Illumina iScan System. The 12 hyperprolactinemia cases and four controls were assessed for genome-wide methylation differences among various conditions (Table 2). Both site-level and region-level differential methylation analyses were performed.
Grouping of hyperprolactinemia cases and controls for differential methylation analysis
| Comparison NO. | Group control (n) | Group test (n) |
|---|---|---|
| 1 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-all (12)DBN 30, DBN 54, DBN 68, DBN 75, DBN 92, DBN 94, DBN 6, DBN 7, DBN 8, DBN 16, DBN 39, DBN 51 |
| 2 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-hypomethylated (6)DBN 30, DBN 54, DBN 68, DBN 75, DBN 92, DBN 94 |
| 3 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-hypermethylated (6)DBN 6, DBN 7, DBN 8, DBN 16, DBN 39, DBN 51 |
| 4 | HPRL- hypomethylated (6)DBN 30, DBN 54, DBN 68, DBN 75, DBN 92, DBN 94 | HPRL-hypermethylated (6)DBN 6, DBN 7, DBN 8, DBN 16, DBN 39, DBN 51 |
| 5 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-antipsychotics, all hypomethylated (4)DBN 30, DBN 75, DBN 92, DBN 94 |
| 6 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-idiopathic, hypomethylated (2)DBN 54, DBN 68 |
| 7 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-idiopathic, hypermethylated (3)DBN 8, DBN 16, DBN 39 |
| 8 | CTRL (4)CTRL 4, CTRL 12, CTRL 31, CTRL 54 | HPRL-pituitary adenoma, all hypermethylated (3)DBN 6, DBN 7, DBN 51 |
| 9 | HPRL-idiopathic, hypomethylated (2)DBN 54, DBN 68 | HPRL-idiopathic, hypermethylated (3)DBN 8, DBN 16, DBN 39 |
HPRL: hyperprolactinemia patients; CTRL: control (for Table 2 only)
Following by comparison between groups, the methylation level of individual cases was compared to the control mean methylation level to find differentially methylated 5-cytosine-phosphate-guanine-3 (CpG) sites in individual cases. All sites with a methylation change of 50% compared to the control mean methylation level were selected for gene ontology (GO) analysis.
GO analysis was performed with the publicly available tool Protein Analysis Through Evolutionary Relationships (PANTHER, http://www.pantherdb.org/). A pathway or biological process was significantly enriched if the false discovery rate (FDR) P value was < 0.05.
Overall, hyperprolactinemia patients showed significant hypermethylation compared to controls (Table 3 and Figure 1). Of 83 cases, 40 hyperprolactinemia cases (48.19%) showed significant changes in global DNA methylation (Figure 2). Of these 40 cases, 9 showed hypomethylation, of which 6 cases (66.67%) were on antipsychotic drugs, one was on anti-acidity drugs, and 2 were idiopathic hyperprolactinemia cases. The remaining 31 cases out of 40 (77.5%) showed global DNA hypermethylation, out of which 4 cases (12.9%) were on antipsychotic drugs, 3 cases (9.6%) were on anti-acidity drugs, 12 cases (38.71%) had idiopathic hyperprolactinemia and 12 cases (38.71%) had hyperprolactinemia due to pituitary micro/macro adenoma. The percentage of cases with global DNA hypomethylation or hypermethylation in each category of hyperprolactinemia patients is presented in Figure 3. There were 25% hypermethylated and 38% hypomethylated cases among antipsychotics induced hyperprolactinemia patients, while 37% had no significant methylation change. In cases with hyperprolactinemia due to drugs other than antipsychotics, 14% were hypermethylated, 5% were hypomethylated, and 81% had no significant methylation change. In idiopathic hyperprolactinemia cases, 43% were hypermethylated, 7% were hypomethylated, and the remaining 50% showed no significant change in global DNA methylation. In cases with hyperprolactinemia due to pituitary adenoma, 67% of cases had significant hypermethylation, and the remaining 33% had no significant change in global DNA methylation.
Comparison of global 5-mC (%) between different categories of hyperprolactinemia cases and controls
| Group (n) | 5-mC (%), mean ± SD (range) | P value |
|---|---|---|
| Controls (65) | 2.82 ± 1.40 (< 0.2–6.74) | NA |
| Hyperprolactinemia patients (83) | 4.98 ± 2.68 (< 0.2–15.98) | < 0.0001** |
| Drug-induced (37) | 3.59 ± 1.91 (< 0.2–7.38) | 0.0216* |
| Idiopathic (28) | 5.94 ± 2.83 (0.63–15.98) | < 0.0001** |
| Pituitary adenoma (18) | 6.34 ± 2.44 (1.8–11.41) | < 0.0001** |
| Macroprolactinemia (20) | 5.47 ± 3.5 (< 0.2–15.98) | < 0.0001** |
| True hyperprolactinemia (63) | 4.87 ± 2.33 (0.74–8.4) | < 0.0001** |
* P < 0.05; ** P < 0.0001; NA: not applicable
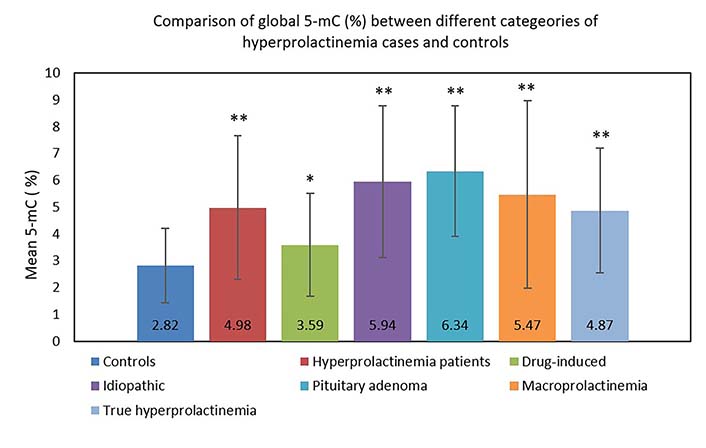
Bar graph showing the comparison of global 5-mC (%) between different categories of hyperprolactinemia cases and controls. * P < 0.05; ** P < 0.0001. 5-mC: 5-methylcytosine
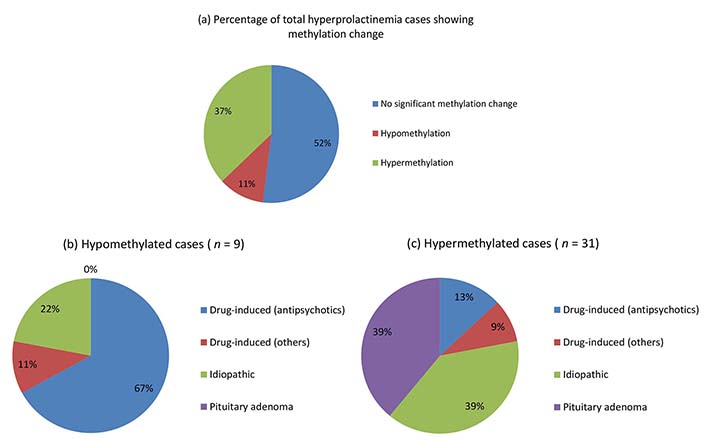
Pie chart showing percentages of hyperprolactinemia cases with hyper/hypomethylation (a), and their categorization into different hyperprolactinemia categories (b and c)
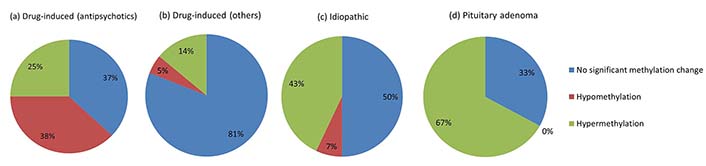
Pie chart showing the percentage of cases with global DNA hypo/hypermethylation in each category of hyperprolactinemia
Site-level GO analysis did not yield statistically significant results for all pair-wise comparisons. Differential methylation for the different regions (Tiling, genes, promoters, and CpG islands) was performed. No significant gene enrichment analysis was found for all comparisons for differentially methylated sites in the CpG island or promoter region.
The CpG methylation level of each of the 12 hyperprolactinemia patients was individually compared with the mean methylation level of controls. The number of differentially methylated CpG sites in each case (more than 50% change in methylation level compared to control mean) are presented in Table 4.
The number of hypomethylated (< 50%) and hypermethylated (> 50%) CpG sites and the number of gene coding CpG sites showing a 50% change in methylation in each of the 12 hyperprolactinemia patients
| Case | CpG sites differentially methylated | CpG sites hypomethylated (< 50%) | CpG sites hypermethylated (> 50%) |
|---|---|---|---|
| DBN 6 | 166 | 122 | 44 |
| DBN 7 | 156 | 116 | 40 |
| DBN 8 | 167 | 113 | 54 |
| DBN 16 | 194 | 155 | 39 |
| DBN 30 | 171 | 139 | 32 |
| DBN 39 | 161 | 119 | 42 |
| DBN 51 | 164 | 118 | 44 |
| DBN 54 | 181 | 153 | 28 |
| DBN 68 | 164 | 118 | 46 |
| DBN 75 | 182 | 149 | 33 |
| DBN 92 | 169 | 109 | 60 |
| DBN 94 | 152 | 105 | 47 |
DBN: data base number; CpG: 5-cytosine-phosphate-guanine-3
In order to have further insight into the cellular components, molecular processes, biological processes, protein classes, and pathways associated with the aberrant methylated genes in individual cases, gene set enrichment analysis was performed for these genes using the PANTHER classification system. The gene set enrichment analyses for the aberrantly methylated genes in individual hyperprolactinemia patients compared to control mean methylation are shown in Figures 4 and 5.
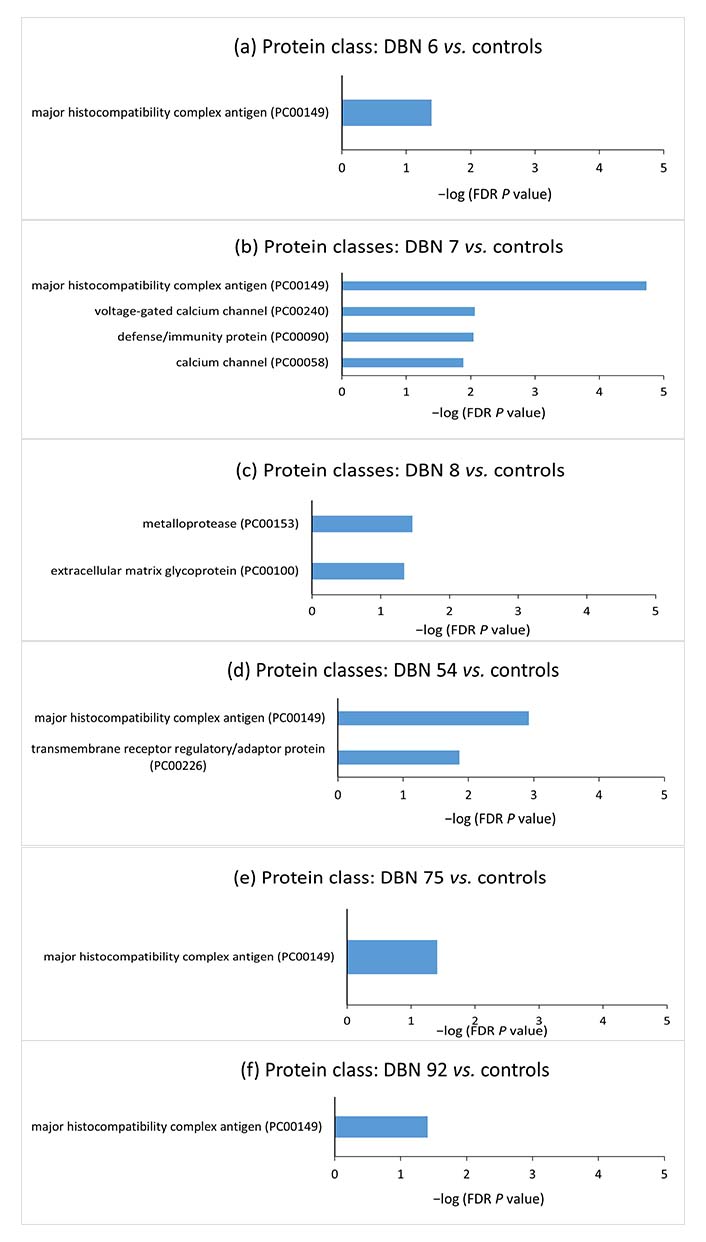
Gene set enrichment analyses for the aberrantly methylated genes belonging to protein classes between individual hyperprolactinemia cases and the controls. The terms were sorted according to the −log (FDR P value). DBN: data base number; FDR: false discovery rate
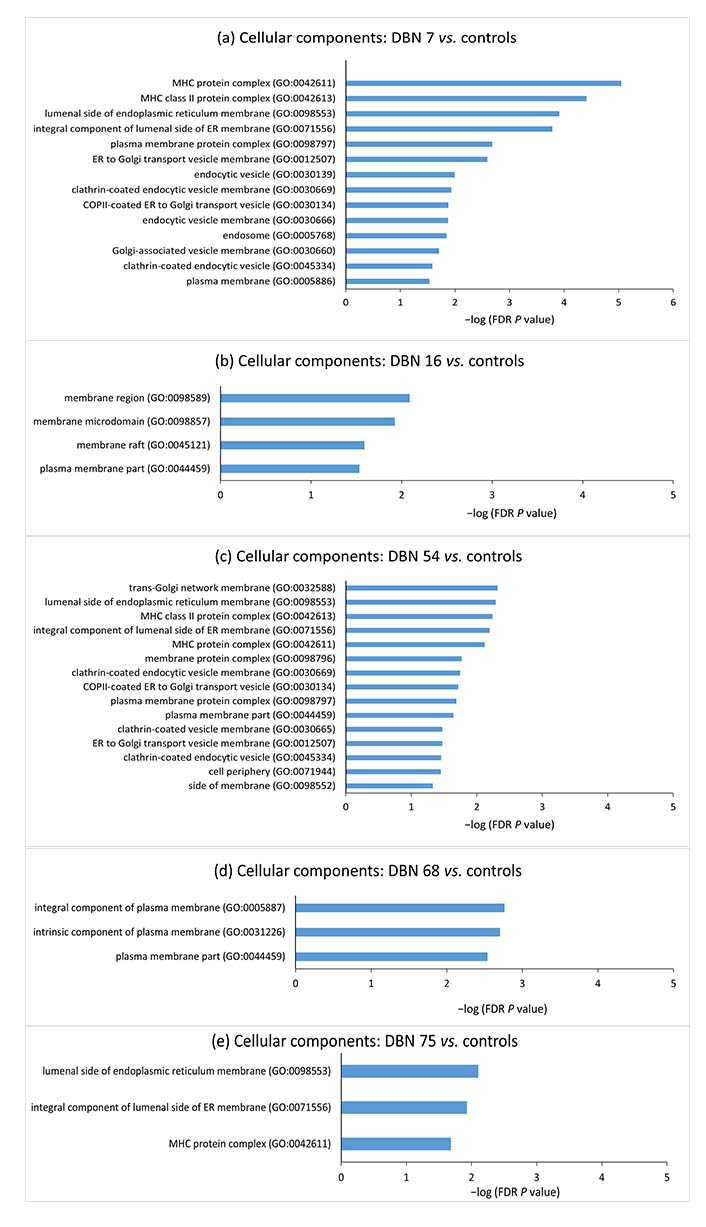
Gene set enrichment analyses for the aberrantly methylated genes belonging to cellular components between individual hyperprolactinemia cases and the controls. The terms were sorted according to the −log (FDR P value). DBN: data base number; FDR: false discovery rate; MHC: major histocompatibility complex; ER: endoplasmic reticulum; COP II: coat protein complex II
The study showed that 48.19% (40/83) of hyperprolactinemia cases had significantly changed global DNA methylation. Of these, 66.67% (6/9) hypomethylated and 12.9% (4/31) hypermethylated hyperprolactinemia cases were on antipsychotic drugs. Earlier studies have observed an association of global DNA methylation in schizophrenia patients on antipsychotics [31]. Differential changes in methylation in our cases may be due to different combinations of antipsychotic drugs these patients are taking, which might be mediating their actions via different pathways, either by direct hypomethylation of prolactin promoter gene or indirectly, by hypermethylation of promoter regions of prolactin inhibitory factors such as dopamine. Decrease in D2R in the pituitary occurs by increased methylation of D2R gene promoter, which is accompanied by an increase in levels of DNA methylating gene mRNAs DNA methyltransferase 1 (DNMT1), DNMT3b, methyl CpG binding protein 2 (MeCP2), and histone modifying gene mRNAs histone deacetylase 2 (HDAC2), HDAC4, and G9a. The reduced D2R prevents dopamine from regulating prolactin, resulting in elevated prolactin levels and hyperprolactinemia [16].
Whether these changes in DNA methylation are part of the etiology/cause of psychiatric disorders or the effect of antipsychotic drugs used to treat psychiatric disorders is a major question to address. However, this cannot be commented upon as drug withdrawal was not possible. Studies, however, have associated psychiatric disorders with alteration in gene expression [32], potentiating the role of regulatory mechanisms such as DNA methylation in psychiatric disorders [33]. Various approaches have shown the role of DNA methylation in developing psychiatric disorders, including animal models [34], monozygotic twins [35], and post-mortem human brains [36]. A study conducted on medication-free schizophrenia subjects shows that changes in DNA methylation are part of the pathophysiology of the disease [37], acting via dopaminergic or non-dopaminergic pathways [38, 39]. On the other hand, studies on both human and animal models have given evidence that antipsychotic drugs may also mediate their action via DNA methylation [31, 38–42]. Significant hypomethylation was observed in schizophrenia patients on antipsychotics compared to those not on medication [38]. Studies on monozygotic twins with major depressive disorders on medication and their comparison with unaffected co-twins show similar effects of antipsychotics on DNA methylation patterns rather than a cause or effect of psychiatric conditions [43]. Antipsychotic drugs of dibenzepine derivatives (e.g., clozapine, quetiapine, olanzapine) induce chromatin remodeling changes and activate DNA demethylation of gamma amino butyric acidergic (GABAergic) gene promoters [40], which is well known to regulate prolactin secretion [44]. Methionine is related to increased s-adenosyl methionine (SAM) levels in the brain [42, 45–47], which is known to restore prolactin receptors in the brain [48]. Methionine is also related to elevated DNMT1 activity, hypermethylation of GAD67 promoters (an enzyme catalyzing the decarboxylation of glutamate to GABA), and alteration in physiological and behavioral responses [42, 45–47]. This suggests that changes in DNA methylation play a role in the development of psychiatric conditions and are also involved in the action of antipsychotic drugs [49, 50]. However, the effect of antipsychotics on DNA methylation may be direct or indirect via inhibition of HDAC [51], as is shown by the effect of valproate, an HDAC inhibitor, on methylation changes [52, 53]. Inhibition of HDAC might be interfering with the normal epigenetic prolactin regulatory mechanism of activation of D2R, causing histone deacetylation and recruitment of mSin3A/HDAC corepressor complex to the prolactin gene promoter [8]. Whether anti-acidity drugs also have a similar epigenetic role while causing their effect and side effects must be probed further.
Another concern in methylation studies is whether the changes in methylation are tissue-specific. The present study shows global DNA methylation changes in peripheral blood. Others have shown similar differential methylation patterns in distinct brain regions—hippocampus and cerebellum as an effect of antipsychotics which vary significantly in other regions like the liver [34]. Even blood and brain samples from schizophrenia patients had similar differences in DNA methylation compared to controls [36].
Among the hyperprolactinemia cases caused by pituitary adenoma, 66.67% (12/18) showed a significant change in methylation, and all these 12 cases were hypermethylated. Pituitary adenoma causes hyperprolactinemia by restricting dopamine from reaching D2R on the anterior pituitary lactotroph cells to regulate prolactin secretion. Hypermethylation in 66.67% of cases with hyperprolactinemia caused by pituitary adenoma stresses upon the role of epigenetics in pituitary micro/macro adenomas causing hyperprolactinemia which might occur by increasing methylation of D2R promoter resulting in reduced D2R expression on lactotroph cells, poor dopamine inhibition, and elevated prolactin levels as is seen in case of fetal alcohol exposure [16], However, further experiments are needed to verify this.
Since no significant genetic cause was found in idiopathic hyperprolactinemia cases (unpublished data), here also, epigenetics might be playing a role in the etiopathology of hyperprolactinemic condition as 50% (14/28) idiopathic hyperprolactinemia cases showed significant change in methylation compared to control mean, out of which 12 cases showed hypermethylation and 2 showed hypomethylation. The absence of heritability and lack of familial cases with hyperprolactinemia also emphasize the role of epigenetics in the manifestation of the condition.
Since epigenetic methylation pattern varies from individual to individual and targeted therapy would focus on the methylation difference in the individual case, DNA methylation difference in individual hyperprolactinemia case from mean methylation of controls was analyzed by selecting CpG sites showing more than 50% deviation in methylation from the control mean. GO analysis was conducted to probe further into each case’s biological functions of aberrantly methylated genes. Gene enrichment analysis of the hypermethylated and hypomethylated CpG site-associated genes demonstrated that “major histocompatibility complex” (MHC) protein classes, “MHC protein complex”, “luminal side of endoplasmic reticulum (ER) membrane”, “clathrin-coated endocytic vesicle” and “plasma membrane protein complex” cellular components were significantly enriched in the majority of cases.
Prolactin is known to upregulate MHC expression on dendritic cell surfaces [54]. Zinc-α2-glycoprotein (ZAG), an MHC class I-like molecule, accommodates prolactin-induced protein (PIP). PIP has structural similarity with the light chain [β2-microglobulin (β2M)] of MHC class I molecule [55]. β2M is homologous to the third constant H chain (CH3) domain of immunoglobulin [56–58]. MHC I proteins are also involved in immune tolerance during pregnancy [59, 60]. MHC I refers to human leukocyte antigen (HLA). The expression of HLA-C (human classical MHC I) on the surface of trophoblast villi increases as they invade the endometrium and become more exposed to the maternal immune system [61]. The communication between HLA-C on trophoblast villi and killer cell immunoglobulin-like receptor 2D (KIR2D) receptors on uterine natural killer (uNK) cells maintains the blood supply to the trophoblast [62, 63]. The non-classical MHC I, HLA-E, HLA-F, and HLA-G are expressed on villous and extravillous trophoblast. These are responsible for both immune tolerance and support of pregnancy [62, 64, 65]. The immunological changes during pregnancy are regulated by changes in concentrations of various hormones, including prolactin [66]. The decidualized endometrial cells of the uterus (placental decidua) and uterine myometrium secrete large amounts of prolactin during pregnancy [67]. Thus, the increased prolactin during physiological pregnancy could regulate MHC I expression by epigenetic alterations and help maintain immune tolerance. Pathologically, raised prolactin may disrupt the immune balance and promote autoreactivity, resulting in autoimmune conditions. Immunity and defense-related protein classes were significantly enriched in the case of DBN 7, further strengthening the correlation between hyperprolactinemia and the immune system.
The significantly enriched cellular components included the ER and the Golgi complex. ER and Golgi are involved in the prolactin secretory pathway [68–70]. Components of plasma membrane and clathrin-coated vesicles were also significantly enriched in cellular components. Regarding the association of plasma membrane and clathrin-coated vesicles with prolactin, a study has related changes in protein trafficking with establishing plasma membrane polarity in mammary epithelial cells [71]. They also found that annexin VI, which probably acts as a sorting signal in prolactin transport, is increased in membrane fractions having excess endocytic clathrin-coated vesicles [71]. This suggests that epigenetic alterations may contribute to changes in the packaging and secretion of prolactin molecules in hyperprolactinemia.
No study has looked for methylation changes, specifically in hyperprolactinemia cases. A study on congenital hypopituitarism (CH) found 51 CpG sites significantly different in CH patients compared to controls [72]. Another study reported six genes in prolactin-secreting pituitary adenoma to be differentially methylated compared to controls [73]. Genome-wide significant DNA methylation patterns were found in association with prolactin level in a study on polycystic ovary syndrome (PCOS) women [74]. The ten significantly enriched functional pathways identified in the study were involved in immune function and immune-mediated inflammatory conditions suggesting a strong association of the immune system, in specific MHC region, in epigenetic regulation of prolactin in PCOS patients. Prolactin has a well-defined role in the immune system in maintaining immune tolerance and balance between apoptosis and proliferation of immune cells under normal conditions [75]. Since there is no association between hyperprolactinemia and PCOS [76], the changes in DNA methylation found in the study of Li et al. [74] can be considered specific to the hyperprolactinemia condition. On the contrary, Li et al. [74] suggested the association between methylation and prolactin levels to be specific to PCOS patients as no CpG site was found to be significantly associated with prolactin levels in their study on 30 healthy controls.
GO analysis did not yield statistically significant results for site-level or region-level differential methylation analysis for all comparisons. This could be due to some limitations of the study. The study had a very small sample size. The CpG methylation status was studied in whole blood DNA, and its consistency with prolactin-secreting cells of the pituitary is mainly unknown.
In conclusion, antipsychotic-induced changes in DNA methylation may mediate the efficacy as well as side effects of these drugs via variable pathways. Hypermethylation in 67% of hyperprolactinemia cases with pituitary adenoma suggests a role of epigenetics in pituitary micro/macro adenomas. About 50% of idiopathic hyperprolactinemia cases with a significant change in methylation also point towards the involvement of epigenetics in the etiopathology of hyperprolactinemia. Genome-wide methylation level analysis in hyperprolactinemia cases directed towards the association of hyperprolactinemia with the immune system, as MHC-related protein classes and cellular components were significantly enriched.
Certain limitations of the present study were that it studied 5-mC percentage, however, changes in 5-hydroxymethylation, histone acetylation, and miRNAs might also be involved in epigenetic causes of hyperprolactinemia. DNA methylation was studied only in the peripheral blood of a relatively small number of samples. Also, the limited number of cases selected for GWMA belonged to something other than a homogenous group of patients.
Insight into case-specific epigenetic aberrations occurring during antipsychotic drug treatment would help know the genes and pathways modified and predict side-effects of drugs in each case which can be reversed as is shown by the epigenetic involvement in response of dopamine agonist—cabergoline, used in the treatment of hyperprolactinemia. This may lead to new pharmacological targets and drugs that aim at effective and targeted personalized therapies helping in methylation-based classification and treatment of individual patients. The MHC class of proteins is involved in immune tolerance during pregnancy. The increased prolactin during physiological pregnancy could regulate MHC I expression by epigenetic alterations and help maintain immune tolerance. Studies on methylation changes in pregnant females are expected to provide insight into the mechanisms involved in MHC-related immune tolerance during pregnancy, a state of physiological hyperprolactinemia.
5-mC: 5-methylcytosine
CpG: 5-cytosine-phosphate-guanine-3
D2R: dopamine D2 receptor
DBN: data base number
DNMT1: DNA methyltransferase 1
ER: endoplasmic reticulum
FDR: false discovery rate
GO: gene ontology
GWMA: genome-wide methylation analysis
HDAC2: histone deacetylase 2
HLA: human leukocyte antigen
MHC: major histocompatibility complex
OD: optical density
PCOS: polycystic ovary syndrome
AKK: Data curation, Formal analysis, Investigation, Software, Validation, Visualization, Writing—original draft, Writing—review & editing. AH: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing—review & editing. MJ: Methodology, Project administration, Resources, Supervision, Validation. JBS: Resources, Supervision, Validation. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Ethical clearance for the study was obtained from the Institute Ethics Committee, AIIMS, New Delhi, India (IESC/T-63/21.01.2015, RT-39/01.04.2015). All procedures performed in the study involving human participants were in accordance with the ethical standards of the institutional ethical committee. The study complies with the Declaration of Helsinki.
Informed consent to participate in the study was obtained from all participants.
Informed consent to publication was obtained from relevant participants.
Requests for accessing the datasets should be directed to [Amanpreet Kaur Kalsi, kalsi.amanpreetkaur@gmail.com].
This work was supported by the Department of Reproductive Biology, AIIMS. AKK was supported by the Council of Scientific & Industrial Research-University Grants Commission (CSIR-UGC) fellowship [23/06/2013(i)EU-V]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Stefan R. Bornstein ... Waldemar Kanczkowski
Eva B. van Dijk ... Lara E. Graves
Waldemar Kanczkowski ... George P. Chrousos
John Milton, Alexander Churilov
Laura C. A. van der Zwet ... Tom Deboer
