 Review
Review

Affiliation:
INSERM U.1193, Faculté des Sciences d’Orsay, Université Paris Saclay, 91400 Orsay, France
ORCID: https://orcid.org/0000-0003-1230-5971
Affiliation:
INSERM U.1193, Faculté des Sciences d’Orsay, Université Paris Saclay, 91400 Orsay, France
ORCID: https://orcid.org/0000-0002-2163-8455
Affiliation:
INSERM U.1193, Faculté des Sciences d’Orsay, Université Paris Saclay, 91400 Orsay, France
Affiliation:
INSERM U.1193, Faculté des Sciences d’Orsay, Université Paris Saclay, 91400 Orsay, France
Affiliation:
INSERM U.1193, Faculté des Sciences d’Orsay, Université Paris Saclay, 91400 Orsay, France
Email: thierry.tordjmann@universite-paris-saclay.fr
ORCID: https://orcid.org/0000-0001-8703-6898
Explor Dig Dis. 2022;1:154–169 DOI: https://doi.org/10.37349/edd.2022.00011
Received: June 09, 2022 Accepted: September 15, 2022 Published: December 30, 2022
Academic Editor: Jose J. G. Marin, Institute for Biomedical Research of Salamanca (IBSAL), Spain
The article belongs to the special issue CHOLESTASIS
During liver injury and cholestasis, the mechanisms allowing the organ to protect itself with the aim of maintaining biliary homeostasis are not completely understood. Central to their biological roles, bile acids (BAs) and their receptors constitute a signaling network with multiple molecular and cellular impacts on both liver repair and protection from BA overload. BA signal through nuclear [mainly farnesoid X receptor (FXR)] and membrane [mainly G protein-coupled BA receptor 1 (GPBAR-1), aka Takeda G protein-coupled receptor 5 (TGR5)] receptors, in which activation elicits a wide array of biological responses. So far, most of the studies have been focused on FXR signaling as hepato-protective, TGR5 being less explored to this regard. While the liver faces massive and potentially harmful BA overload during cholestasis, it is crucial to understand that BAs induce also protective responses contributing not only to reduce the inflammatory burden, but also to spare liver cells and their repair capacities. Based on the available literature, the TGR5 BA receptor protects the liver in the cholestatic context and counteracts BA overload with the aim of restoring biliary homeostasis mainly through the control of inflammatory processes, biliary epithelial barrier permeability, and BA pool composition. Mouse experimental models of cholestasis reveal that the lack of TGR5 was associated with exacerbated inflammation and necrosis, leaky biliary epithelium, and excessive BA pool hydrophobicity, resulting in biliary cell and parenchymal insult, and compromising optimal restoration of biliary homeostasis and liver repair. There are thus widely opened translational perspectives with the aim of targeting TGR5-related signaling or biological responses to trigger protection of the cholestatic liver.
Bile acids (BAs) are synthesized from cholesterol (CT) in hepatocytes (primary BA), transported in the bile canaliculus lumen, flow through the biliary tree towards the duodenum, progress in the intestine, and are finally massively reabsorbed in the ileum, going back to the liver through the portal circulation, accomplishing the enterohepatic cycle (EHC). A small percentage of BAs (2–5%) escape from reabsorption and keep on flowing through the colon where they are transformed by the gut microbiota into more hydrophobic secondary BA (more toxic) as compared with primary BA. Thanks to their high hydrophobicity, secondary BAs passively cross the colon epithelium, joining other BA in the so-called “BA pool”. Integrity of the EHC is central to biliary homeostasis during which BAs are almost confined in the gut-liver axis, with only BA traces spilling over into the general circulation [1, 2]. If ileal BA reabsorption would be defective, exacerbated BA loss in the feces would occur. On the opposite, during cholestasis in case of insufficient trans-hepatocyte BA flow (reduced sinusoidal and/or canalicular BA transport) or when bile duct obstruction occurs, BA overload (hepatic and systemic) will appear. In the biliary tree, the gallbladder (GB) in addition to its physiological role of bile reservoir, also has a critical impact on bile composition [3], and is particularly enriched in the BA Takeda G protein-coupled receptor 5 (TGR5) receptor [4, 5]. Precise mechanisms by which BAs induce hepatic injury remain controversial, with reports on both direct and indirect BA cytotoxicity [6–8]. Direct BA-induced cell death mechanisms include plasma membrane damage (for high concentrations of hydrophobic BA), mitochondrial injury [9], and reactive oxygen species (ROS) production [10], as well as induction of apoptosis [11]. Reported indirect mechanisms mainly focus on the impact of BA on inflammatory processes [12], but the precise machinery linking BA and inflammation is still difficult to precisely depict, with BA species and targeted cell types having a crucial impact [7].
When bile secretion or bile flow is defective (i.e. in case of cholestasis), the organism is overloaded with BA which cannot be secreted or excreted in bile. Such BA overload also takes place when the balance between BA return from the gut and BA uptake in the liver is altered. This type of imbalance is observed when part of the liver is either removed [partial hepatectomy (PH)] or injured [13, 14]. In that non-cholestatic setting, BA secretion is normal or even increased, while the remnant hepatocyte mass is too reduced to cope with the important BA load coming back from the gut: this results in a significant accumulation of BA in the systemic blood and consequently in the liver and in the whole organism (referred as a “BA spillover” or “BA overload”). More precisely, systemic BA concentration will rise from 2–5 μmol/L in basal conditions, to 200–500 μmol/L or even more during cholestasis [15] or in the first minutes and hours after PH as reported in mice and rats [13, 16, 17], but also in humans [2, 13]. With that said, it can be envisioned to consider BA overload as a possible consequence of any type of significant liver injury, and not only as a specific hallmark of cholestasis per se [2, 18, 19].
Another important aspect of BA overload is that it surprisingly has not only deleterious but also positive influence on liver repair processes [14, 19–21]. Thus, BA should be envisioned as double edge swords, signaling in hepatocytes for their protection and favoring their proliferation on the one hand, whilst being deleterious for those cells on the other hand [14].
Since specific nuclear and membrane BA receptors have been discovered, BA signaling is known to be coupled mainly with the nuclear receptor farnesoid X receptor (FXR), and the G protein-coupled BA receptor 1 (GPBAR-1, or TGR5) [22]. Although FXR has been amply studied in the cholestatic and hepatoprotection contexts, this is not the case for TGR5 which has been less explored; only recently TGR5 has proved to be hepatoprotective [14, 16, 23, 24]. As a whole, TGR5 has been more reported in non-hepatic cells and tissues than in the liver [25, 26], in which its functions are relatively less described [23, 27]. Importantly, secondary BAs [deoxycholic acid (DCA) and lithocholic acid (LCA)] either conjugated or not to taurine or glycine, exhibit the highest affinity for TGR5 [median effective concentrations (EC50): 0.5–1 μmol/L], over the primary BA cholic acid (CA) and chenodeoxycholic acids (CDCAs) [25, 28]. In line, hydrophilic BAs like ursodeoxycholic acid (UDCA) display very weak affinities for TGR5 (EC50: 36 μmol/L) [25, 29, 30]. Interestingly, TGR5 is also activated by hyocholic acid (HCA) in the pig, with potential therapeutic impact on glucose metabolism [31]. When activated, TGR5 classically induces cyclic adenosine monophosphate (cAMP) production, although it can also interfere with intracellular calcium signaling [32, 33]. A number of studies showed an association of TGR5 activation with epidermal growth factor receptor (EGFR) transactivation [34–37] and ROS production [32, 38], providing evidence that TGR5 is able to modulate cell proliferation and apoptosis [39]. TGR5 is also reported as a regulator of energy homeostasis and inflammation, and as such has the potential to be a therapeutic target in the context of metabolic syndrome, with a particular interest in diabetes and obesity-related non-alcoholic steatohepatitis (NASH) [40–44]. As to TGR5 expression in the liver, it is weak or not significant in hepatocytes at least in rodents, but TGR5 has repeatedly been reported as highly concentrated in the GB and biliary tract [5, 45], as well as in endothelial and Kupffer cells [46].
The role of TGR5 on bile secretion is not really established, i.e. the TGR5 impact on transport of BA, phospholipid, and ion in bile has not been fully explored (Figure 1) [47–49]. Authors including us reported in TGR5-knockout (KO) as compared with wild-type (WT) mice a smaller BA pool size, a more hydrophobic BA composition, and a slightly reduced bile flow; intriguingly the effects of TGR5 agonists on bile flow were only slight or not obvious [16, 47–49]. In the rat, TGR5 stimulation was choleretic through BA-independent effects [50] (Figure 2). In mice, we did not find any direct effect of TGR5 stimulation on bile flow [4]. Noteworthy, neither BA synthesis nor canalicular bile secretion is expected to be directly impacted by TGR5 because this receptor expression is very weak in hepatocytes. Slight TGR5 hepatocyte expression has been recently reported [45], with a possible functional impact on glucose metabolism, but consequences on bile secretion have not been specifically explored. We recently found that in the lack of TGR5, the expression of BA synthesis enzymes and BA transporters messenger RNAs (mRNAs) was not changed as compared with WT mice [16]. In the same line, hepatocyte-specific TGR5-KO and WT mice exhibited similar responses to PH [4], while BA overload was exacerbated, and bile secretion adaptation was altered post-PH in total TGR5-KO mice [16]. Conversely, as TGR5 expression in cholangiocytes is high, this points to a possible impact of this receptor and related signaling on the ductular contribution to bile production. Indeed, TGR5 regulates Cl– secretion in human GB [51] and other epithelial cells through an impact on cystic fibrosis transmembrane regulator (CFTR) (Figure 1) [52]. However, since these reports, no study quantifying the physiological contribution of TGR5 to ductular bile secretion has been published.
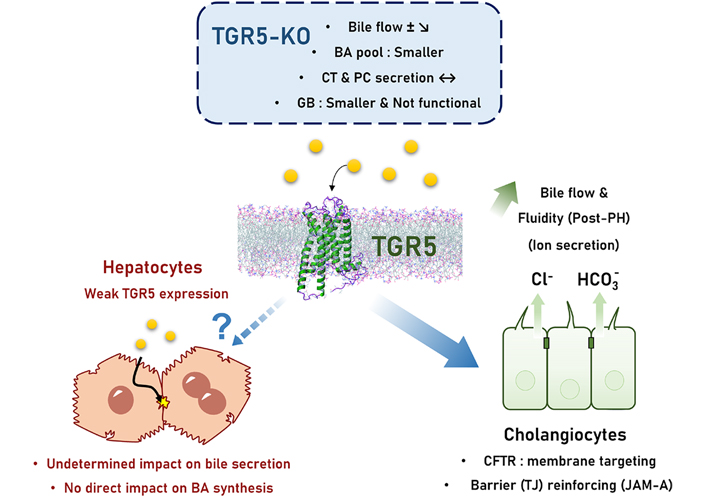
TGR5 control on bile secretion. Bile secretion in TGR5-KO mice exhibit only mild abnormalities, including a discrete (if any) bile flow reduction, a smaller BA pool, and a smaller and non-functional GB. Direct effects related to TGR5 on hepatocyte bile secretion are undetermined although unlikely because of a very weak expression of this receptor. TGR5 mainly operates its control on bile secretion/bile flow through the regulation of cholangiocyte processes: ion (chloride and bicarbonate) secretion, and tight junction (TJ) barrier reinforcing. JAM-A: junctional adhesion molecule-A; PC: phosphatidylcholine; ±: still discussed; ?: unknown mechanism; full line arrow: documented effect; dashed line arrow: uncertain effect
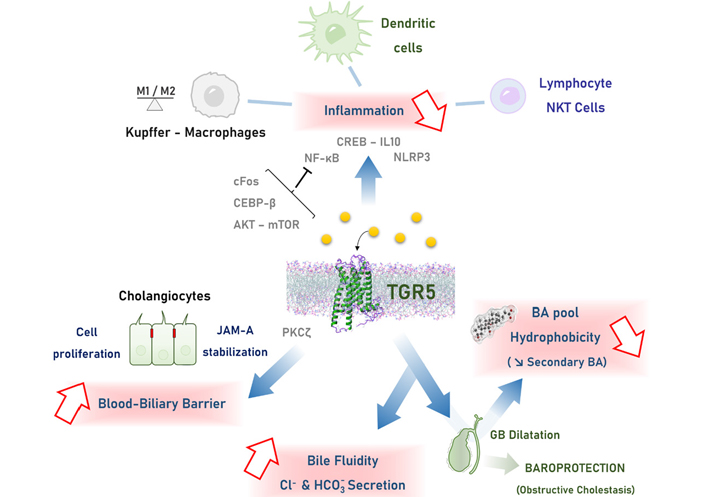
Mechanisms of TGR5-mediated hepato-protection. TGR5 engagement is mainly associated with: 1. reduction of inflammation in the liver, through multiple signaling pathways in monocytes-macrophages, lymphocytes and dendritic cells; 2. biliary epithelial barrier reinforcing by promoting cholangiocyte proliferation and TJ strengthening through stabilization of the JAM-A protein; 3. modulation of bile composition, through 2 main mechanisms converging on making a less toxic bile: a. stimulation of chloride and bicarbonate transport in bile, reducing BA protonation and protecting cells from BA toxicity; b. GB dilatation, favoring BA reabsorption reported as the cholecysto-hepatic shunt, reducing the amount of hydrophobic secondary BA. CEBP-β: CCAAT/enhancer-binding protein beta; CREB: cAMP response element-binding protein; IL10: interleukin 10; mTOR: mechanistic target of rapamycin; NF-κB: nuclear factor-kappa B; NKT: natural killer T; NLRP3: Nod-like receptor family pyrin domain containing 3; PKCζ: protein kinase C-zeta
During cholestasis, but also after liver injury or immediately after PH [19, 20], it has been well described that hepatocytes handle excessively raised intracellular BA concentrations by using so-called “adaptive reactions” [53, 54] that are at least in part FXR-dependent. This adaptation is performed by the modulation of gene transcription for membrane BA transporters, as well as for BA synthesis and conjugation enzymes [16, 54], with aim of counteracting intracellular BA overload [53]. In case of significant parenchymal loss in the context of cholestasis, hepatocyte cell cycle progression can also be boosted through FXR-dependent mechanisms [20], contributing to replenish a functional hepatocyte mass that will progressively restore a full BA handling capacity to the liver. Also in enterocytes of the ileum, fibroblast growth factor 15 (FGF15) induction through BA-induced FXR activation has been identified as an important suppressor of hepatocyte BA synthesis [55], which is protective during BA overload and cholestasis [56]. Besides these FXR-dependent processes during cholestasis, TGR5-elicited hepato-protective pathways have been also reported, as described in the following (Figure 2).
One of the most frequently reported mechanisms of TGR5-mediated liver protection involves its anti-inflammatory properties (Figure 2), and the bulk of related studies in this field is growing [57]. As pointed out by several papers, BAs induce pro-inflammatory effects and this can be considered as crucial for mechanisms of liver injury during cholestasis [7, 12, 58, 59]. During cholestasis, a primary event would be that BAs activate cytokine production in hepatocytes, and neutrophils would secondarily contribute to liver injury [59]. Although the role of macrophages and Kupffer cells during cholestasis remains controversial, the anti-inflammatory action of TGR5 during cholestasis likely occurs at least in part in these cells, downstream an initiating effect of BA in hepatocytes (Figure 1) [5, 45, 46, 60]. In line, early and recent studies reported that TGR5 activation reduces the effect of lipopolysaccharide (LPS) on cytokine gene induction in rat Kupffer cells and mouse macrophages [60, 61]. Consequently, anti-inflammatory effects of TGR5 have been reported as protective in several rodent experimental models (see below) [40, 61–65]. From a signaling perspective in macrophages, TGR5 stimulation blunts NF-κB activation through mechanisms depending on cAMP production, involving an inhibition of Ikappa beta (IκB) phosphorylation and p65 nuclear translocation [61]. Anti-inflammatory effects of TGR5 engagement also involve impact on the AKT-mTOR complex 1 pathway, on CEBP-β induction, and on a complex modulation of the NLRP3 inflammasome activity [66–69]. We previously observed that cytokine induction was exacerbated in TGR5-KO as compared to WT mice after either PH or bile duct ligation (BDL), and that it enhanced cholestasis [70], favored hepatocyte necrosis, and delayed regeneration [16]. This was later confirmed by others [39, 71, 72], and reproduced by us [4, 5] in the BDL model. In line, TGR5-KO mice harbored exacerbated sensitivity to LPS-induced hepatic inflammation, while treatment with a TGR5 agonist improved WT mice [61]. In the more chronic Mdr2-KO mice model of primary sclerosing cholangitis (PSC), specific TGR5 stimulation alone did not improve the phenotype, in contrast with a dual FXR/TGR5 agonist [49]. As a whole in the lack of TGR5, mice harbored an excessive susceptibility to pro-inflammatory stimuli, not only in the liver but also outside this organ [64].
As a whole, TGR5 engagement during cholestasis or after liver injury likely tunes Kupffer cells and macrophage activation, with an impact on hepatocyte protection and proliferation allowing compensation of parenchymal cell loss [73, 74]. In line, it has been reported that TGR5 stimulation stabilized the M1/M2 ratio of macrophage populations towards an anti-inflammatory phenotype [75, 76]. Importantly, translational data suggested that such TGR5-dependent anti-inflammatory responses may also occur in human patients with liver failure, with an impact on clinical outcome [77]. In the same line, it has been recently reported that during PSC in humans, as well as in the Mdr2-KO murine model, TGR5 downregulation in biliary epithelial cells contributed to the pro-inflammatory phenotype observed in cholangiocytes during this disease [78].
During cholestatic liver diseases, especially when obstructive components occur within their pathophysiological course, the biliary epithelium integrity is obviously key to protect parenchymal cells (hepatocytes) from BA-induced injury. This may involve not only the replacement of dead cells but also the strengthening of barrier function [79]. In fact, the “blood-biliary barrier (BBB)” [80], namely the separation between blood and bile, is mainly formed by both epithelial liver cell types, hepatocytes and cholangiocytes, and especially involves their respective TJs that seal adjacent cells (Figure 1). Hepatocytes, due to their physiological position along the biliary path, are obviously frontally exposed to BA, but they share this frontline position in the BBB with the downstream layer of cholangiocytes that shields parenchyma from BA-induced damage. In the biliary epithelium as well as in any lining epithelial sheet, TJs constitute a regulated barrier to the diffusion of molecules in the paracellular space, allowing selective exchanges of small molecules (especially ions) between apical and basolateral microenvironments [81]. TJs are composed of occludin, claudins, and JAMs, which constitute the main core of plasma membrane proteins which associate with cytoplasmic actin-binding proteins [including zonula occludens (ZO) proteins] [81, 82]. In contiguous cells, extracellular domains of the transmembrane proteins interact with their counterparts and seal plasma membranes. Paracellular permeability is obviously a regulated process, although the mechanisms involved still remain poorly explored. It is reported that occludin, JAM-A, and claudin-4 phosphorylation can modulate paracellular permeability, although this aspect has been poorly studied in liver epithelial cells [83, 84]. In the liver, the role of selected TJ proteins expressed in hepatocytes in the regulation of bile secretion has been explored [85–87], in contrast with TJ proteins expressed in cholangiocytes of which pathophysiological impact is not strongly and specifically delineated. It is hypothesized that if inter-cholangiocyte TJs would be altered, bile duct walls would leak, resulting in bile-induced parenchymal injury. However, strong evidence confirming this pathophysiological view is still lacking [88–90], with a few exceptions. Indeed, severe liver disease is observed in patients with neonatal ichtyosis and sclerosing cholangitis (NISCH) syndrome, a primary (mutational) claudin-1 defect [91, 92], or with the more recently described mutations in the TJ-associated protein ZO-2 gene [93]. In line, TJP2 inactivation in both hepatocytes and cholangiocytes in mice resulted in altered biliary homeostasis, with exacerbated cholestatic features upon CA diet [94]. But still, the balanced influence of inter-cholangiocyte and inter-hepatocyte TJ protein defects during these diseases is not defined, in part because TJ protein repertoires differ in hepatocytes and cholangiocytes [95]. In mice also, double inactivation of ZO-1 and ZO-2 genes in hepatocytes resulted in profound alteration in hepatic tissue architecture and functions leading to lethality by 6 weeks of age [96]. During inflammatory cholestatic diseases, hepatic TJ can also be secondarily altered [88, 89, 97] by cytokines and endotoxin [97]. Interestingly, double deletion of β- and γ-catenins genes in hepatocytes in mice resulted in complete disorganization of TJ proteins and structure. Those mice displayed a complete loss of hepatocyte BBB, and as a consequence severe cholestasis-induced parenchymal injury [98]. Crucial impact of TJ integrity for liver recovery after cholestatic injury in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) mouse model has also been reported, pointing out that vaso-intestinal peptide (VIP) secreted by periportal mesenchymal cells and cholangiocyte VIP receptor 1 were involved in a crucial crosstalk for TJP expression in this experimental setting [99]. As a whole, the literature points to a crucial hepatoprotective role of TJ in the liver, although it is better documented in mice than in human patients, and more specifically addressed in hepatocytes than in cholangiocytes.
The regulatory role of BA on paracellular permeability is mostly known as deleterious, because BAs enhance paracellular passage in intestinal [100] and respiratory epithelial cells [101], but signal transduction mechanisms and in particular the BA receptors likely involved were not reported. However, other reports established that TGR5-dependent BA signaling reinforces barriers in several murine tissues, i.e. intestinal, endothelial, retinal, and blood-brain barriers [102–105]. Presumably, BAs exert their effects on paracellular permeability through multiple pathways, possibly enhancing or reducing it as a function of the pathophysiological context and depending on the interested cell types. As the biliary tract is naturally exposed to high concentrations of BA, searching for an impact of BA signaling on cholangiocyte paracellular permeability in physiological and cholestatic conditions is thus highly relevant. In a recent study, we found that BA-induced TGR5 engagement protected the liver parenchyme against BA overload during cholestasis through a reduction of paracellular permeability [5]. More precisely TGR5 activation induces the PKCζ-dependent phosphorylation of the TJ protein JAM-A, which results in a protective modulation of the TJ barrier function (Figure 1). This was shown in two mice experimental models of cholestasis [BDL and 1 naphthyl isothiocyanate (ANIT) intoxication], and importantly, we provided data from patients with primary biliary cirrhosis (PBC) and PSC suggesting that the TJ protein JAM-A may be protective in the biliary epithelium during human cholestatic diseases, rising the therapeutic challenge of targeting this protein and TGR5 related pathways in the future.
Although scarce literature is available, it is suggested that the impact of TGR5 on bile composition operates mainly through cholangiocyte-dependent processes (Figure 2). As evoked above, the TGR5 effect on both CFTR and the anion exchanger 2 (AE2) mediates chloride/bicarbonate exchange across the apical membrane, promoting the formation of the so-called “bicarbonate umbrella”. Consequently, the alkaline environment enables the reduction of BA protonation and thus protects the cholangiocytes (bile ducts) and hepatocytes (liver parenchyma) from BA cytotoxicity in liver injury and cholestatic contexts [49, 106].
This might constitute an adaptive mechanism enhancing bile secretion and fluidity through which TGR5 would protect the liver from BA toxicity during repair [16] or cholestasis. TGR5 is also expressed on cholangiocyte cilia and may regulate bile flow through an effect on this organelle [107, 108], via a coupling with HCO3– secretion in bile [109]. Finally, TGR5 may modulate water handling in the biliary epithelium, although no direct evidence has been reported yet, while further studies would be necessary to eventually extend data published in kidney epithelial cells [110, 111].
It is also emphasized that the BA pool composition itself including its hydrophobicity is crucial to liver homeostasis [112, 113]. Excessive BA pool hydrophobicity is indeed reported to be noxious for liver repair and cholestatic contexts [56, 114–118]. This is best exemplified in the BSEP/abcb11–/– mice, which exhibit non-progressive mild cholestasis [119], contrasting with the severe human progressive familial intrahepatic cholestasis type 2 (PFIC2, severe progressive cholestasis in children) phenotype. This discrepancy is, at least in part, due to the less hydrophobic (less toxic) BA pool observed in Bsep–/– mice, as it is enriched in hyperhydroxylated BA [120]. These BA pool features are viewed as protective against cholestatic liver injury not only in mice [119], but also in children with progressive intrahepatic cholestasis [121]. Nice proof of concept of this protection has been reported in double Mdr2–/– and Bsep–/– mice, in which hyper-hydrophilic BA prevented liver damage caused by the lack of biliary phospholipids [122]. Recent studies also pointed out that an efficient activation of the BA receptors (entirely dependent on the BA pool composition) had a strong impact on alcoholic and non-alcoholic liver diseases [65, 123]. In line, Cyp8b1 inhibition, reducing BA pool hydrophobicity, prevented steatosis in high-fat diet-fed mice [124]. It has also been emphasized in a murine NASH preclinical model that the BA pool composition was dramatically altered, with poor signaling power on both FXR and TGR5, and that DCA diet complementation prevented liver disorders in this model [125]. Recently, the BA metabolizing enzyme Cyp2c70 which converts CDCA (a hydrophobic primary BA) into the more hydrophilic muricholic acid (MCA), has been identified as a master regulator of the BA pool composition in mice [126]. This enzyme is lacking in humans, explaining in part why the BA pool is more hydrophobic in humans than in mice. Interestingly, Cyp2c70–/– mice exhibit a human-like more hydrophobic BA pool as compared with control animals, associated with liver inflammation [127] and altered FXR signaling [128]. More recently, it was reported that a cholangiopathy with biliary fibrosis developed in female Cyp2c70–/– mice, a phenotype reversed upon UDCA treatment [129], reinforcing the fact that BA pool hydrophobicity had a crucial pathophysiological impact on liver disease outcome.
Others and we reported that BA pool composition was more hydrophobic in TGR5-KO than in WT mice [4, 16, 47, 48, 130], as measured in bile, plasma, liver, and feces [4, 16]. More precisely, MCA the most hydrophilic primary BA in mice, and the MCA/CA ratio, were strongly reduced, whereas secondary (more hydrophobic) BAs were significantly overrepresented in TGR5-KO as compared with WT mice [16]. Our recent study explored different mechanistic hypotheses to support this phenotype, linking TGR5 with BA pool composition [4]. First, we did not find any significant direct impact of TGR5 on BA synthesis. Second, based on gut microbiota transfer experiments, we provided data showing that despite the lack of TGR5 was associated with an intestinal dysbiosis, this latest was not responsible for the observed more hydrophobic BA pool. This is in contrast with data indicating that FXR-mediated modification of the gut microbiota resulted in reshaping the BA pool (LCA enrichment) towards TGR5 stimulation and improved glucose tolerance through intestinal glucagon-like peptide-1 (GLP-1) secretion [131]. We finally focused on TGR5-mediated impact on the GB, as it is reported as the tissue displaying the highest TGR5 expression, at least in mice [132], suggesting that this receptor may have a physiological impact on GB functions, including GB relaxation as reported [48] and potentially BA reabsorption also known as the cholecysto-hepatic shunt [3]. This shunt is expected to allow a short circuit by which BAs from the GB return directly to the liver via the portal circulation without passing through the intestine [3], thereby restricting the amount of toxic secondary BA in the pool. Importantly, we found that in the lack of TGR5, defective GB dilatation resulted in reduced cholecysto-hepatic shunt, building a more hydrophobic BA pool. We also put forward TGR5-dependent hepato-protective properties of GB dilatation in the setting of obstructive cholestasis [4]. We showed that after cholecystectomy (as compared with spared GB), BDL mice had more severe necrotico-inflammatory liver injury, more bile duct dilatation, and reduced survival rate. Importantly this phenotype was not found in TGR5-KO mice, suggesting that TGR5-dependent GB dilatation was crucial in hepatoprotection upon obstructive cholestasis [4].
In liver diseases in general, BAs as well as their analogues and derivatives begin to be viewed as potential or patent therapeutic agents. Indeed, BAs are now well known for their signaling properties on nuclear or transmembrane receptors that modulate several cellular functions as explained above [28, 133, 134]. In this perspective, FXR-activating therapies have been developed for their benefit in non-alcoholic fatty liver disease (NAFLD)/NASH, metabolic syndrome-related liver diseases, and cholestatic liver diseases (i.e. in PBC) [133]. Besides these approaches, preclinical mouse experimental data also suggest that TGR5 might be a potential target for liver disease treatments. Unfortunately, encouraging mice data have not yet been translated towards human clinics. The multi-organ TGR5 expression and the related potential of unwanted intra- and extra-hepatic effects are challenging liver researchers and pharmaceutical industry in their quest for TGR5-mediated hepato-protective therapies.
BAs are particularly powerful molecules because during the course of cholestasis and liver injury, they contribute both to injure liver cells and to launch protection and repair reactions. Central to this duality, BAs generate corresponding signaling pathways through the binding to two main receptors, FXR and TGR5. Compiled literature strongly supports a hepato-protective role of TGR5 in mice during cholestasis and liver injury, through a combination of several regulatory actions of this receptor on inflammation, cholangiocyte secretion, biliary epithelial barrier permeability, and BA pool composition. Targeting these pathways should be considered, expecting for direct or indirect reduction of BA-induced tissue injury while optimizing liver regeneration potential. Future studies should delineate in depth TGR5-dependent mechanisms controlling these processes in the biliary epithelium and other liver and non-liver cells, with the aim of setting-up TGR5-based therapies in hepato-biliary medicine.
BAs: bile acids
BBB: blood-biliary barrier
BDL: bile duct ligation
CA: cholic acid
cAMP: cyclic adenosine monophosphate
CFTR: cystic fibrosis transmembrane regulator
FXR: farnesoid X receptor
GB: gallbladder
JAM-A: junctional adhesion molecule-A
KO: knockout
MCA: muricholic acid
NASH: non-alcoholic steatohepatitis
PH: partial hepatectomy
PSC: primary sclerosing cholangitis
TGR5: Takeda G protein-coupled receptor 5
TJ: tight junction
WT: wild-type
ZO: zonula occludens
TT: Writing—original draft. GM, VBJ, ID and IG: Writing—review & editing.
The authors declare that there is no conflict of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Our studies are funded by INSERM (Institut National de la Santé et de la Recherche Médicale) and ANR (Agence Nationale de la Recherche, grant number: ANR-15-CE14-0007-01). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2022.
Copyright: © The Author(s) 2022. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 12643
Download: 84
Times Cited: 0
Maitane Asensio ... Jose J. G. Marin
Ricardo Espinosa-Escudero ... Maria J. Monte
Beatriz Sanchez de Blas ... Marta R. Romero
Carola Dröge ... Verena Keitel
Vasiliy Ivanovich Reshetnyak, Igor Veniaminovich Maev
Jose M. Pinazo-Bandera ... Miren García-Cortés
Elias Kouroumalis ... Argyro Voumvouraki
